

Unraveling the Kaposi Sarcoma-Associated Herpesvirus (KSHV) Lifecycle: An Overview of Latency, Lytic Replication, and KSHV-Associated Diseases

Abstract
:1. Introduction
2. Virion Structure, Viral Genome, and KSHV Entry
2.1. Virion Structure
2.2. KSHV Genome
2.3. KSHV Entry
3. Latency: Quiescent Phase of the Lifecycle
3.1. Establishment of Latency
3.2. KSHV Latency-Associated Nuclear Antigen (LANA)
3.3. KSHV Viral Cyclin (vCyclin)
3.4. KSHV Viral FLICE-Inhibitory Protein (vFLIP)
3.5. KSHV Kaposins
3.6. LANA2/Viral Interferon Regulatory Factor 3 (vIRF3)
3.7. microRNAs: Tiny Regulators, Big Impact
4. KSHV Signaling Proteins
4.1. KSHV K1
4.2. KSHV K2/vIL-6
4.3. KSHV K15
5. Lytic Replication
5.1. KSHV RTA: Master Regulator of the Lytic Switch
5.2. KSHV K8
5.3. KSHV ORF57, Delayed Early Protein
5.4. Viral DNA Replication and Late Gene Expression
6. Modulation of Cell Pathways by Lytic Proteins
6.1. K3 and K5: Modulators of Immune Recognition
6.2. KSHV IRFs: v-IRF1, 2, and 4
6.3. Viral Chemokines: vCCL1, 2, and 3
6.4. Lytic Signaling Proteins: vGPCR and vPK
6.5. KSHV vBcl-2
6.6. Contribution of Lytic Proteins to KSHV Pathogenesis
7. KSHV-Associated Diseases
7.1. Kaposi Sarcoma (KS)
7.2. Primary Effusion Lymphoma (PEL)
7.3. Multicentric Castleman Disease (MCD)
7.4. KSHV Inflammatory Cytokine Syndrome (KICS)
8. Therapeutics
8.1. Current Therapeutic Approaches

{kind=link}
{kind=link}
| Name | Type | Target | Disease | Reference |
|---|---|---|---|---|
| BV | Chemotherapeutic regimen | DNA/Topoisomerase II, Microtubules | KS | [309] |
| ABV | KS | |||
| PLD | Topoisomerase II | KS | ||
| CHOP | DNA/Topoisomerase II, Microtubules, Glucocorticoid receptor | PEL | [298] | |
| EPOCH | PEL | |||
| Paclitaxel | Chemotherapeutic agent | Microtubules | KS | [310] |
| Ganciclovir | Antiviral drug | DNA polymerase | KS | [312,313] |
| Cidofovir | PEL | [314] | ||
| Siltuximab | Antibody therapy | IL-6 | MCD | [315] |
| Tocilizumab | IL-6 receptor | MCD | [316] | |
| Rituximab | CD-20 | MCD | [317] | |
| Pembrolizumab and nivolumab | Immune checkpoint blockade therapy | PD-1 and PD-L1 | KS | [318,319] |
| Nivolumab and ipilimumab | PD-1 and CTLA-4 | KS | [320] | |
| Pomalidomide and lenalidomide | Immunomodulatory imide drugs (IMiDs) | Ubiquitin E3 ligase substrate adapter Cereblon | KS/PEL | [321] |
| Interleukin-12 therapy | Cytokine | IL-12 | KS | [322] |
| Rapamycin | Macrocyclic immunosuppressive drug | mTOR pathway | KS/PEL | [323,324] |
| Azidothymidine and interferon alpha | Nucleoside analog reverse transcriptase inhibitor (NRTI) and immunomodulator | NF-κB | PEL | [309] |
| Imatinib | Kinase inhibitor | Tyrosine kinase inhibitor | KS | [325] |
| Bortezomib | Protease inhibitor | 26S proteasome | KS | [326] |
| Bevacizumab | Antibody therapy | VEGF inhibitor | KS | [327] |
8.2. Targeted and Emerging Therapies
8.3. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef]
- Moore, P.S.; Gao, S.J.; Dominguez, G.; Cesarman, E.; Lungu, O.; Knowles, D.M.; Garber, R.; Pellett, P.E.; McGeoch, D.J.; Chang, Y. Primary characterization of a herpesvirus agent associated with Kaposi’s sarcomae. J. Virol. 1996, 70, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Slavin, G.; Cameron, H.M.; Forbes, C.; Mitchell, R.M. Kaposi’s sarcoma in East African children: A report of 51 cases. J. Pathol. 1970, 100, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Jenner, R.G.; Alba, M.M.; Boshoff, C.; Kellam, P. Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 2001, 75, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Jary, A.; Veyri, M.; Gothland, A.; Leducq, V.; Calvez, V.; Marcelin, A.G. Kaposi’s Sarcoma-Associated Herpesvirus, the Etiological Agent of All Epidemiological Forms of Kaposi’s Sarcoma. Cancers 2021, 13, 6208. [Google Scholar] [CrossRef]
- Broussard, G.; Damania, B. Regulation of KSHV Latency and Lytic Reactivation. Viruses 2020, 12, 1034. [Google Scholar] [CrossRef] [PubMed]
- Lange, P.; Damania, B. Kaposi Sarcoma-Associated Herpesvirus (KSHV). Trends Microbiol. 2020, 28, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Nador, R.G.; Cesarman, E.; Chadburn, A.; Dawson, D.B.; Ansari, M.Q.; Sald, J.; Knowles, D.M. Primary effusion lymphoma: A distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood 1996, 88, 645–656. [Google Scholar] [CrossRef]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Uldrick, T.S.; Wang, V.; O’Mahony, D.; Aleman, K.; Wyvill, K.M.; Marshall, V.; Steinberg, S.M.; Pittaluga, S.; Maric, I.; Whitby, D.; et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin. Infect. Dis. 2010, 51, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Gong, D.; Lim, H.; Jih, J.; Wu, T.T.; Sun, R.; Zhou, Z.H. Structure and mutagenesis reveal essential capsid protein interactions for KSHV replication. Nature 2018, 553, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Nealon, K.; Newcomb, W.W.; Pray, T.R.; Craik, C.S.; Brown, J.C.; Kedes, D.H. Lytic replication of Kaposi’s sarcoma-associated herpesvirus results in the formation of multiple capsid species: Isolation and molecular characterization of A, B, and C capsids from a gammaherpesvirus. J. Virol. 2001, 75, 2866–2878. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Kedes, D.H. Mass spectrometric analyses of purified rhesus monkey rhadinovirus reveal 33 virion-associated proteins. J. Virol. 2006, 80, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.X.; Chong, J.M.; Wu, L.; Yuan, Y. Virion proteins of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 800–811. [Google Scholar] [CrossRef]
- Bechtel, J.T.; Winant, R.C.; Ganem, D. Host and viral proteins in the virion of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 4952–4964. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, J.; Grundhoff, A.; Ganem, D. RNAs in the virion of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 10138–10146. [Google Scholar] [CrossRef]
- Gong, D.; Dai, X.; Jih, J.; Liu, Y.T.; Bi, G.Q.; Sun, R.; Zhou, Z.H. DNA-Packing Portal and Capsid-Associated Tegument Complexes in the Tumor Herpesvirus KSHV. Cell 2019, 178, 1329–1343. [Google Scholar] [CrossRef]
- Zhen, J.; Chen, J.; Huang, H.; Liao, S.; Liu, S.; Yuan, Y.; Sun, R.; Longnecker, R.; Wu, T.T.; Zhou, Z.H. Structures of Epstein-Barr virus and Kaposi’s sarcoma-associated herpesvirus virions reveal species-specific tegument and envelope features. J. Virol. 2024, 98, e0119424. [Google Scholar] [CrossRef]
- Dai, X.; Gong, D.; Wu, T.T.; Sun, R.; Zhou, Z.H. Organization of capsid-associated tegument components in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2014, 88, 12694–12702. [Google Scholar] [CrossRef] [PubMed]
- Sathish, N.; Wang, X.; Yuan, Y. Tegument Proteins of Kaposi’s Sarcoma-Associated Herpesvirus and Related Gamma-Herpesviruses. Front. Microbiol. 2012, 3, 98. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Sarid, R.; Flore, O.; Bohenzky, R.A.; Chang, Y.; Moore, P.S. Transcription mapping of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 1998, 72, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef] [PubMed]
- Prazsak, I.; Tombacz, D.; Fulop, A.; Torma, G.; Gulyas, G.; Dormo, A.; Kakuk, B.; McKenzie Spires, L.; Toth, Z.; Boldogkoi, Z. KSHV 3.0: A state-of-the-art annotation of the Kaposi’s sarcoma-associated herpesvirus transcriptome using cross-platform sequencing. mSystems 2024, 9, e0100723. [Google Scholar] [CrossRef]
- Wang, F.Z.; Akula, S.M.; Pramod, N.P.; Zeng, L.; Chandran, B. Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J. Virol. 2001, 75, 7517–7527. [Google Scholar] [CrossRef]
- Akula, S.M.; Wang, F.Z.; Vieira, J.; Chandran, B. Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 2001, 282, 245–255. [Google Scholar] [CrossRef]
- Birkmann, A.; Mahr, K.; Ensser, A.; Yaguboglu, S.; Titgemeyer, F.; Fleckenstein, B.; Neipel, F. Cell surface heparan sulfate is a receptor for human herpesvirus 8 and interacts with envelope glycoprotein K8.1. J. Virol. 2001, 75, 11583–11593. [Google Scholar] [CrossRef] [PubMed]
- Mark, L.; Lee, W.H.; Spiller, O.B.; Villoutreix, B.O.; Blom, A.M. The Kaposi’s sarcoma-associated herpesvirus complement control protein (KCP) binds to heparin and cell surfaces via positively charged amino acids in CCP1-2. Mol. Immunol. 2006, 43, 1665–1675. [Google Scholar] [CrossRef]
- Akula, S.M.; Pramod, N.P.; Wang, F.Z.; Chandran, B. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 2002, 108, 407–419. [Google Scholar] [CrossRef]
- Wang, F.Z.; Akula, S.M.; Sharma-Walia, N.; Zeng, L.; Chandran, B. Human herpesvirus 8 envelope glycoprotein B mediates cell adhesion via its RGD sequence. J. Virol. 2003, 77, 3131–3147. [Google Scholar] [CrossRef] [PubMed]
- Rappocciolo, G.; Jenkins, F.J.; Hensler, H.R.; Piazza, P.; Jais, M.; Borowski, L.; Watkins, S.C.; Rinaldo, C.R., Jr. DC-SIGN is a receptor for human herpesvirus 8 on dendritic cells and macrophages. J. Immunol. 2006, 176, 1741–1749. [Google Scholar] [CrossRef]
- Kaleeba, J.A.; Berger, E.A. Kaposi’s sarcoma-associated herpesvirus fusion-entry receptor: Cystine transporter xCT. Science 2006, 311, 1921–1924. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Samols, M.A.; Plaisance, K.B.; Boss, I.W.; Riva, A.; Lopez, M.C.; Baker, H.V.; Renne, R. Kaposi’s sarcoma-associated herpesvirus encodes an ortholog of miR-155. J. Virol. 2007, 81, 12836–12845. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Freitas, E.; Sullivan, R.; Mohan, S.; Bacelieri, R.; Branch, D.; Romano, M.; Kearney, P.; Oates, J.; Plaisance, K.; et al. Upregulation of xCT by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in an environment of oxidative stress. PLoS Pathog. 2010, 6, e1000742. [Google Scholar] [CrossRef] [PubMed]
- Veettil, M.V.; Sadagopan, S.; Sharma-Walia, N.; Wang, F.Z.; Raghu, H.; Varga, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus forms a multimolecular complex of integrins (αVβ5, αVβ3, and α3β1) and CD98-xCT during infection of human dermal microvascular endothelial cells, and CD98-xCT is essential for the postentry stage of infection. J. Virol. 2008, 82, 12126–12144. [Google Scholar] [CrossRef]
- Hahn, A.S.; Kaufmann, J.K.; Wies, E.; Naschberger, E.; Panteleev-Ivlev, J.; Schmidt, K.; Holzer, A.; Schmidt, M.; Chen, J.; Konig, S.; et al. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus. Nat. Med. 2012, 18, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, X.; Schaller, S.; Jardetzky, T.S.; Longnecker, R. Ephrin Receptor A4 is a New Kaposi’s Sarcoma-Associated Herpesvirus Virus Entry Receptor. MBio 2019, 10, e02892-18. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.S.; Desrosiers, R.C. Binding of the Kaposi’s sarcoma-associated herpesvirus to the ephrin binding surface of the EphA2 receptor and its inhibition by a small molecule. J. Virol. 2014, 88, 8724–8734. [Google Scholar] [CrossRef]
- Krishnan, H.H.; Sharma-Walia, N.; Streblow, D.N.; Naranatt, P.P.; Chandran, B. Focal adhesion kinase is critical for entry of Kaposi’s sarcoma-associated herpesvirus into target cells. J. Virol. 2006, 80, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Naranatt, P.P.; Smith, M.S.; Zeng, L.; Bloomer, C.; Chandran, B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 2004, 78, 3601–3620. [Google Scholar] [CrossRef] [PubMed]
- Speck, S.H.; Ganem, D. Viral latency and its regulation: Lessons from the gamma-herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Lagunoff, M.; Renne, R.; Staskus, K.; Haase, A.; Ganem, D. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1998, 72, 8309–8315. [Google Scholar] [CrossRef]
- Sarid, R.; Wiezorek, J.S.; Moore, P.S.; Chang, Y. Characterization and cell cycle regulation of the major Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J. Virol. 1999, 73, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Talbot, S.J.; Weiss, R.A.; Kellam, P.; Boshoff, C. Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology 1999, 257, 84–94. [Google Scholar] [CrossRef]
- Li, H.; Komatsu, T.; Dezube, B.J.; Kaye, K.M. The Kaposi’s sarcoma-associated herpesvirus K12 transcript from a primary effusion lymphoma contains complex repeat elements, is spliced, and initiates from a novel promoter. J. Virol. 2002, 76, 11880–11888. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.; Matsumura, S.; Wilson, A.C. Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi’s sarcoma-associated herpesvirus originate from a common promoter. J. Virol. 2005, 79, 14457–14464. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Verma, S.C.; Sharma, N.; Murakami, M.; Robertson, E.S. Induction of Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: A novel mechanism for establishment of latency. J. Virol. 2005, 79, 7453–7465. [Google Scholar] [CrossRef]
- Matsumura, S.; Fujita, Y.; Gomez, E.; Tanese, N.; Wilson, A.C. Activation of the Kaposi’s sarcoma-associated herpesvirus major latency locus by the lytic switch protein RTA (ORF50). J. Virol. 2005, 79, 8493–8505. [Google Scholar] [CrossRef] [PubMed]
- Staudt, M.R.; Dittmer, D.P. Promoter switching allows simultaneous transcription of LANA and K14/vGPCR of Kaposi’s sarcoma-associated herpesvirus. Virology 2006, 350, 192–205. [Google Scholar] [CrossRef]
- Rivas, C.; Thlick, A.E.; Parravicini, C.; Moore, P.S.; Chang, Y. Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 2001, 75, 429–438. [Google Scholar] [CrossRef]
- Chandriani, S.; Ganem, D. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2010, 84, 5565–5573. [Google Scholar] [CrossRef] [PubMed]
- Sharp, T.V.; Wang, H.W.; Koumi, A.; Hollyman, D.; Endo, Y.; Ye, H.; Du, M.Q.; Boshoff, C. K15 protein of Kaposi’s sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J. Virol. 2002, 76, 802–816. [Google Scholar] [CrossRef]
- Samols, M.A.; Hu, J.; Skalsky, R.L.; Renne, R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 9301–9305. [Google Scholar] [CrossRef]
- Cai, X.; Cullen, B.R. Transcriptional origin of Kaposi’s sarcoma-associated herpesvirus microRNAs. J. Virol. 2006, 80, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Rainbow, L.; Platt, G.M.; Simpson, G.R.; Sarid, R.; Gao, S.J.; Stoiber, H.; Herrington, C.S.; Moore, P.S.; Schulz, T.F. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J. Virol. 1997, 71, 5915–5921. [Google Scholar] [CrossRef]
- Kedes, D.H.; Lagunoff, M.; Renne, R.; Ganem, D. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi’s sarcoma-associated herpesvirus. J. Clin. Investig. 1997, 100, 2606–2610. [Google Scholar] [CrossRef]
- Kellam, P.; Boshoff, C.; Whitby, D.; Matthews, S.; Weiss, R.A.; Talbot, S.J. Identification of a major latent nuclear antigen, LNA-1, in the human herpesvirus 8 genome. J. Hum. Virol. 1997, 1, 19–29. [Google Scholar] [PubMed]
- Ballestas, M.E.; Chatis, P.A.; Kaye, K.M. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 1999, 284, 641–644. [Google Scholar] [CrossRef]
- Cotter, M.A., 2nd; Robertson, E.S. The latency-associated nuclear antigen tethers the Kaposi’s sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 1999, 264, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Ballestas, M.E.; Kaye, K.M. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J. Virol. 2001, 75, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Cotter, M.A., 2nd; Subramanian, C.; Robertson, E.S. The Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy-terminus. Virology 2001, 291, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.C.; Hu, J.; Renne, R. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 2002, 277, 27401–27411. [Google Scholar] [CrossRef]
- Piolot, T.; Tramier, M.; Coppey, M.; Nicolas, J.C.; Marechal, V. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J. Virol. 2001, 75, 3948–3959. [Google Scholar] [CrossRef]
- Grant, M.J.; Loftus, M.S.; Stoja, A.P.; Kedes, D.H.; Smith, M.M. Superresolution microscopy reveals structural mechanisms driving the nanoarchitecture of a viral chromatin tether. Proc. Natl. Acad. Sci. USA 2018, 115, 4992–4997. [Google Scholar] [CrossRef] [PubMed]
- Stedman, W.; Deng, Z.; Lu, F.; Lieberman, P.M. ORC, MCM, and histone hyperacetylation at the Kaposi’s sarcoma-associated herpesvirus latent replication origin. J. Virol. 2004, 78, 12566–12575. [Google Scholar] [CrossRef]
- Verma, S.C.; Choudhuri, T.; Kaul, R.; Robertson, E.S. Latency-associated nuclear antigen (LANA) of Kaposi’s sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 2006, 80, 2243–2256. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Jha, H.C.; Robertson, E.S. Bub1 in Complex with LANA Recruits PCNA To Regulate Kaposi’s Sarcoma-Associated Herpesvirus Latent Replication and DNA Translesion Synthesis. J. Virol. 2015, 89, 10206–10218. [Google Scholar] [CrossRef]
- Hu, J.; Garber, A.C.; Renne, R. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 2002, 76, 11677–11687. [Google Scholar] [CrossRef]
- Grundhoff, A.; Ganem, D. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J. Virol. 2003, 77, 2779–2783. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Choudhuri, T.; Robertson, E.S. The minimal replicator element of the Kaposi’s sarcoma-associated herpesvirus terminal repeat supports replication in a semiconservative and cell-cycle-dependent manner. J. Virol. 2007, 81, 3402–3413. [Google Scholar] [CrossRef]
- Verma, S.C.; Lan, K.; Choudhuri, T.; Cotter, M.A.; Robertson, E.S. An autonomous replicating element within the KSHV genome. Cell Host Microbe 2007, 2, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.Y.; Matchett, G.A.; Wilson, A.C. Transcriptional activation by the Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen is facilitated by an N-terminal chromatin-binding motif. J. Virol. 2004, 78, 10074–10085. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Barry, C.; Dittmer, D.; Compitello, N.; Brown, P.O.; Ganem, D. Modulation of cellular and viral gene expression by the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2001, 75, 458–468. [Google Scholar] [CrossRef]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jha, H.C.; Verma, S.C.; Sun, Z.; Banerjee, S.; Dzeng, R.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded LANA contributes to viral latent replication by activating phosphorylation of survivin. J. Virol. 2014, 88, 4204–4217. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, Z.; Xia, T.; Li, X.; Liang, D.; Lin, X.; Wen, H.; Lan, K. Latency-associated nuclear antigen of Kaposi sarcoma-associated herpesvirus promotes angiogenesis through targeting notch signaling effector Hey1. Cancer Res. 2014, 74, 2026–2037. [Google Scholar] [CrossRef] [PubMed]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Wu, F.Y.; ApRhys, C.; Kajumbula, H.; Young, D.B.; Hayward, G.S.; Hayward, S.D. A novel viral mechanism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat. Med. 2003, 9, 300–306. [Google Scholar] [CrossRef]
- Bubman, D.; Guasparri, I.; Cesarman, E. Deregulation of c-Myc in primary effusion lymphoma by Kaposi’s sarcoma herpesvirus latency-associated nuclear antigen. Oncogene 2007, 26, 4979–4986. [Google Scholar] [CrossRef]
- Liu, J.; Martin, H.J.; Liao, G.; Hayward, S.D. The Kaposi’s sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J. Virol. 2007, 81, 10451–10459. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Sun, Z.; Pei, Y.; Singh, R.K.; Jha, H.C.; Robertson, E.S. Shugoshin 1 is dislocated by KSHV-encoded LANA inducing aneuploidy. PLoS Pathog. 2018, 14, e1007253. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 2005, 79, 3468–3478. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Murakami, M.; Choudhuri, T.; Kuppers, D.A.; Robertson, E.S. Intracellular-activated Notch1 can reactivate Kaposi’s sarcoma-associated herpesvirus from latency. Virology 2006, 351, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Platt, G.M.; Simpson, G.R.; Mittnacht, S.; Schulz, T.F. Latent nuclear antigen of Kaposi’s sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 1999, 73, 9789–9795. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Lee, D.; Seo, T.; Choi, C.; Choe, J. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus functionally interacts with heterochromatin protein 1. J. Biol. Chem. 2003, 278, 7397–7405. [Google Scholar] [CrossRef]
- Hu, J.; Yang, Y.; Turner, P.C.; Jain, V.; McIntyre, L.M.; Renne, R. LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLoS Pathog. 2014, 10, e1004240. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.A.; Naiman, N.E.; Wang, V.; Shrestha, P.; Haque, M.; Hu, D.; Anagho, H.A.; Carey, R.F.; Davidoff, K.S.; Yarchoan, R. Identification of Caspase Cleavage Sites in KSHV Latency-Associated Nuclear Antigen and Their Effects on Caspase-Related Host Defense Responses. PLoS Pathog. 2015, 11, e1005064. [Google Scholar] [CrossRef] [PubMed]
- Toptan, T.; Fonseca, L.; Kwun, H.J.; Chang, Y.; Moore, P.S. Complex alternative cytoplasmic protein isoforms of the Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 generated through noncanonical translation initiation. J. Virol. 2013, 87, 2744–2755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Chan, B.; Samarina, N.; Abere, B.; Weidner-Glunde, M.; Buch, A.; Pich, A.; Brinkmann, M.M.; Schulz, T.F. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl. Acad. Sci. USA 2016, 113, E1034–E1043. [Google Scholar] [CrossRef] [PubMed]
- Mariggio, G.; Koch, S.; Zhang, G.; Weidner-Glunde, M.; Ruckert, J.; Kati, S.; Santag, S.; Schulz, T.F. Kaposi Sarcoma Herpesvirus (KSHV) Latency-Associated Nuclear Antigen (LANA) recruits components of the MRN (Mre11-Rad50-NBS1) repair complex to modulate an innate immune signaling pathway and viral latency. PLoS Pathog. 2017, 13, e1006335. [Google Scholar] [CrossRef]
- Chang, Y.; Moore, P.S.; Talbot, S.J.; Boshoff, C.H.; Zarkowska, T.; Godden, K.; Paterson, H.; Weiss, R.A.; Mittnacht, S. Cyclin encoded by KS herpesvirus. Nature 1996, 382, 410. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lee, H.; Yoon, D.W.; Albrecht, J.C.; Fleckenstein, B.; Neipel, F.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus encodes a functional cyclin. J. Virol. 1997, 71, 1984–1991. [Google Scholar] [CrossRef]
- Sarek, G.; Jarviluoma, A.; Moore, H.M.; Tojkander, S.; Vartia, S.; Biberfeld, P.; Laiho, M.; Ojala, P.M. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathog. 2010, 6, e1000818. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C.; Mann, D.J.; Fleckenstein, B.; Neipel, F.; Peters, G.; Jones, N. Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature 1997, 390, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.; Ramos da Silva, S.; Bedolla, R.; Ye, F.; Zhou, F.; Gao, S.J. Viral cyclin promotes KSHV-induced cellular transformation and tumorigenesis by overriding contact inhibition. Cell Cycle 2014, 13, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Ojala, P.M.; Tiainen, M.; Salven, P.; Veikkola, T.; Castanos-Velez, E.; Sarid, R.; Biberfeld, P.; Makela, T.P. Kaposi’s sarcoma-associated herpesvirus-encoded v-cyclin triggers apoptosis in cells with high levels of cyclin-dependent kinase 6. Cancer Res. 1999, 59, 4984–4989. [Google Scholar] [PubMed]
- Cuomo, M.E.; Knebel, A.; Morrice, N.; Paterson, H.; Cohen, P.; Mittnacht, S. p53-Driven apoptosis limits centrosome amplification and genomic instability downstream of NPM1 phosphorylation. Nat. Cell Biol. 2008, 10, 723–730. [Google Scholar] [CrossRef]
- Ojala, P.M.; Yamamoto, K.; Castanos-Velez, E.; Biberfeld, P.; Korsmeyer, S.J.; Makela, T.P. The apoptotic v-cyclin-CDK6 complex phosphorylates and inactivates Bcl-2. Nat. Cell Biol. 2000, 2, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Li, M. Kaposi’s sarcoma-associated herpesvirus K-cyclin interacts with Cdk9 and stimulates Cdk9-mediated phosphorylation of p53 tumor suppressor. J. Virol. 2008, 82, 278–290. [Google Scholar] [CrossRef]
- Koopal, S.; Furuhjelm, J.H.; Jarviluoma, A.; Jaamaa, S.; Pyakurel, P.; Pussinen, C.; Wirzenius, M.; Biberfeld, P.; Alitalo, K.; Laiho, M.; et al. Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog. 2007, 3, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.L.; Schroter, M.; et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Belanger, C.; Gravel, A.; Tomoiu, A.; Janelle, M.E.; Gosselin, J.; Tremblay, M.J.; Flamand, L. Human herpesvirus 8 viral FLICE-inhibitory protein inhibits Fas-mediated apoptosis through binding and prevention of procaspase-8 maturation. J. Hum. Virol. 2001, 4, 62–73. [Google Scholar]
- Chugh, P.; Matta, H.; Schamus, S.; Zachariah, S.; Kumar, A.; Richardson, J.A.; Smith, A.L.; Chaudhary, P.M. Constitutive NF-kappaB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc. Natl. Acad. Sci. USA 2005, 102, 12885–12890. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Jasmin, A.; Eby, M.T.; Hood, L. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 1999, 18, 5738–5746. [Google Scholar] [CrossRef]
- Guasparri, I.; Wu, H.; Cesarman, E. The KSHV oncoprotein vFLIP contains a TRAF-interacting motif and requires TRAF2 and TRAF3 for signalling. EMBO Rep. 2006, 7, 114–119. [Google Scholar] [CrossRef]
- Tolani, B.; Matta, H.; Gopalakrishnan, R.; Punj, V.; Chaudhary, P.M. NEMO is essential for Kaposi’s sarcoma-associated herpesvirus-encoded vFLIP K13-induced gene expression and protection against death receptor-induced cell death, and its N-terminal 251 residues are sufficient for this process. J. Virol. 2014, 88, 6345–6354. [Google Scholar] [CrossRef] [PubMed]
- Matta, H.; Chaudhary, P.M. Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP). Proc. Natl. Acad. Sci. USA 2004, 101, 9399–9404. [Google Scholar] [CrossRef]
- Sun, Q.; Zachariah, S.; Chaudhary, P.M. The human herpes virus 8-encoded viral FLICE-inhibitory protein induces cellular transformation via NF-kappaB activation. J. Biol. Chem. 2003, 278, 52437–52445. [Google Scholar] [CrossRef] [PubMed]
- Ballon, G.; Chen, K.; Perez, R.; Tam, W.; Cesarman, E. Kaposi sarcoma herpesvirus (KSHV) vFLIP oncoprotein induces B cell transdifferentiation and tumorigenesis in mice. J. Clin. Investig. 2011, 121, 1141–1153. [Google Scholar] [CrossRef]
- Godfrey, A.; Anderson, J.; Papanastasiou, A.; Takeuchi, Y.; Boshoff, C. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 2005, 105, 2510–2518. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.B.; Meng, F.R.; Fang, Y.X.; Wu, X.; Zhang, C.W.; Liu, Y.; Liu, D.; Li, G.Q.; Feng, F.B.; Qiu, H.Y. Inhibition of NF-kappaB signaling pathway induces apoptosis and suppresses proliferation and angiogenesis of human fibroblast-like synovial cells in rheumatoid arthritis. Medicine 2018, 97, e10920. [Google Scholar] [CrossRef] [PubMed]
- Sadler, R.; Wu, L.; Forghani, B.; Renne, R.; Zhong, W.; Herndier, B.; Ganem, D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5722–5730. [Google Scholar] [CrossRef]
- Tomkowicz, B.; Singh, S.P.; Cartas, M.; Srinivasan, A. Human herpesvirus-8 encoded Kaposin: Subcellular localization using immunofluorescence and biochemical approaches. DNA Cell Biol. 2002, 21, 151–162. [Google Scholar] [CrossRef]
- Muralidhar, S.; Pumfery, A.M.; Hassani, M.; Sadaie, M.R.; Kishishita, M.; Brady, J.N.; Doniger, J.; Medveczky, P.; Rosenthal, L.J. Identification of kaposin (open reading frame K12) as a human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) transforming gene. J. Virol. 1998, 72, 4980–4988. [Google Scholar] [CrossRef]
- Kliche, S.; Nagel, W.; Kremmer, E.; Atzler, C.; Ege, A.; Knorr, T.; Koszinowski, U.; Kolanus, W.; Haas, J. Signaling by human herpesvirus 8 kaposin A through direct membrane recruitment of cytohesin-1. Mol. Cell 2001, 7, 833–843. [Google Scholar] [CrossRef]
- McCormick, C.; Ganem, D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 2005, 307, 739–741. [Google Scholar] [CrossRef]
- McCormick, C.; Ganem, D. Phosphorylation and function of the kaposin B direct repeats of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2006, 80, 6165–6170. [Google Scholar] [CrossRef]
- Yoo, J.; Kang, J.; Lee, H.N.; Aguilar, B.; Kafka, D.; Lee, S.; Choi, I.; Lee, J.; Ramu, S.; Haas, J.; et al. Kaposin-B enhances the PROX1 mRNA stability during lymphatic reprogramming of vascular endothelial cells by Kaposi’s sarcoma herpes virus. PLoS Pathog. 2010, 6, e1001046. [Google Scholar] [CrossRef]
- Lubyova, B.; Pitha, P.M. Characterization of a novel human herpesvirus 8-encoded protein, vIRF-3, that shows homology to viral and cellular interferon regulatory factors. J. Virol. 2000, 74, 8194–8201. [Google Scholar] [CrossRef]
- Dittmer, D.P. Transcription profile of Kaposi’s sarcoma-associated herpesvirus in primary Kaposi’s sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 2003, 63, 2010–2015. [Google Scholar]
- Laura, M.V.; de la Cruz-Herrera, C.F.; Ferreiros, A.; Baz-Martinez, M.; Lang, V.; Vidal, A.; Munoz-Fontela, C.; Rodriguez, M.S.; Collado, M.; Rivas, C. KSHV latent protein LANA2 inhibits sumo2 modification of p53. Cell Cycle 2015, 14, 277–282. [Google Scholar] [CrossRef]
- Joo, C.H.; Shin, Y.C.; Gack, M.; Wu, L.; Levy, D.; Jung, J.U. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 2007, 81, 8282–8292. [Google Scholar] [CrossRef] [PubMed]
- Wies, E.; Hahn, A.S.; Schmidt, K.; Viebahn, C.; Rohland, N.; Lux, A.; Schellhorn, T.; Holzer, A.; Jung, J.U.; Neipel, F. The Kaposi’s Sarcoma-associated Herpesvirus-encoded vIRF-3 Inhibits Cellular IRF-5. J. Biol. Chem. 2009, 284, 8525–8538. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Ju, H.; Li, Q.; Mei, S.C.; Chen, D.; Choi, Y.B.; Nicholas, J. Human Herpesvirus 8 Interferon Regulatory Factors 1 and 3 Mediate Replication and Latency Activities via Interactions with USP7 Deubiquitinase. J. Virol. 2018, 92, e02003-17. [Google Scholar] [CrossRef]
- Wies, E.; Mori, Y.; Hahn, A.; Kremmer, E.; Sturzl, M.; Fleckenstein, B.; Neipel, F. The viral interferon-regulatory factor-3 is required for the survival of KSHV-infected primary effusion lymphoma cells. Blood 2008, 111, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Joo, C.H.; Gack, M.U.; Lee, H.R.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res. 2008, 68, 1751–1759. [Google Scholar] [CrossRef]
- Lee, H.R.; Li, F.; Choi, U.Y.; Yu, H.R.; Aldrovandi, G.M.; Feng, P.; Gao, S.J.; Hong, Y.K.; Jung, J.U. Deregulation of HDAC5 by Viral Interferon Regulatory Factor 3 Plays an Essential Role in Kaposi’s Sarcoma-Associated Herpesvirus-Induced Lymphangiogenesis. MBio 2018, 9, e02217-17. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lu, S.; Zhang, Z.; Gonzalez, C.M.; Damania, B.; Cullen, B.R. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5570–5575. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef]
- Grundhoff, A.; Sullivan, C.S.; Ganem, D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA 2006, 12, 733–750. [Google Scholar] [CrossRef]
- O’Hara, A.J.; Wang, L.; Dezube, B.J.; Harrington, W.J., Jr.; Damania, B.; Dittmer, D.P. Tumor suppressor microRNAs are underrepresented in primary effusion lymphoma and Kaposi sarcoma. Blood 2009, 113, 5938–5941. [Google Scholar] [CrossRef] [PubMed]
- Samols, M.A.; Skalsky, R.L.; Maldonado, A.M.; Riva, A.; Lopez, M.C.; Baker, H.V.; Renne, R. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007, 3, e65. [Google Scholar] [CrossRef]
- Gay, L.A.; Sethuraman, S.; Thomas, M.; Turner, P.C.; Renne, R. Modified Cross-Linking, Ligation, and Sequencing of Hybrids (qCLASH) Identifies Kaposi’s Sarcoma-Associated Herpesvirus MicroRNA Targets in Endothelial Cells. J. Virol. 2018, 92, e02138-17. [Google Scholar] [CrossRef]
- Hansen, A.; Henderson, S.; Lagos, D.; Nikitenko, L.; Coulter, E.; Roberts, S.; Gratrix, F.; Plaisance, K.; Renne, R.; Bower, M.; et al. KSHV-encoded miRNAs target MAF to induce endothelial cell reprogramming. Genes Dev 2010, 24, 195–205. [Google Scholar] [CrossRef]
- Bellare, P.; Ganem, D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: An evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 2009, 6, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Stedman, W.; Yousef, M.; Renne, R.; Lieberman, P.M. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J. Virol. 2010, 84, 2697–2706. [Google Scholar] [CrossRef]
- Lei, X.; Bai, Z.; Ye, F.; Xie, J.; Kim, C.G.; Huang, Y.; Gao, S.J. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 2010, 12, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Plaisance-Bonstaff, K.; Choi, H.S.; Beals, T.; Krueger, B.J.; Boss, I.W.; Gay, L.A.; Haecker, I.; Hu, J.; Renne, R. KSHV miRNAs decrease expression of lytic genes in latently infected PEL and endothelial cells by targeting host transcription factors. Viruses 2014, 6, 4005–4023. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Henderson, S.; Hayes, M.J.; Marelli, S.S.; Ofir-Birin, Y.; Regev-Rudzki, N.; Herrero, J.; Enver, T. Herpesviruses shape tumour microenvironment through exosomal transfer of viral microRNAs. PLoS Pathog. 2017, 13, e1006524. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Yoo, S.M.; Choi, H.S.; Mun, J.Y.; Kang, H.G.; Lee, J.; Park, J.; Gao, S.J.; Lee, M.S. Extracellular vesicles from KSHV-infected endothelial cells activate the complement system. Oncotarget 2017, 8, 99841–99860. [Google Scholar] [CrossRef]
- Toptan, T.; Abere, B.; Nalesnik, M.A.; Swerdlow, S.H.; Ranganathan, S.; Lee, N.; Shair, K.H.; Moore, P.S.; Chang, Y. Circular DNA tumor viruses make circular RNAs. Proc. Natl. Acad. Sci. USA 2018, 115, E8737–E8745. [Google Scholar] [CrossRef]
- Tagawa, T.; Gao, S.; Koparde, V.N.; Gonzalez, M.; Spouge, J.L.; Serquina, A.P.; Lurain, K.; Ramaswami, R.; Uldrick, T.S.; Yarchoan, R.; et al. Discovery of Kaposi’s sarcoma herpesvirus-encoded circular RNAs and a human antiviral circular RNA. Proc. Natl. Acad. Sci. USA 2018, 115, 12805–12810. [Google Scholar] [CrossRef] [PubMed]
- Ungerleider, N.A.; Jain, V.; Wang, Y.; Maness, N.J.; Blair, R.V.; Alvarez, X.; Midkiff, C.; Kolson, D.; Bai, S.; Roberts, C.; et al. Comparative Analysis of Gammaherpesvirus Circular RNA Repertoires: Conserved and Unique Viral Circular RNAs. J. Virol. 2019, 93, e01952-18. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, T.; Oh, D.; Dremel, S.; Mahesh, G.; Koparde, V.N.; Duncan, G.; Andresson, T.; Ziegelbauer, J.M. A virus-induced circular RNA maintains latent infection of Kaposi’s sarcoma herpesvirus. Proc. Natl. Acad. Sci. USA 2023, 120, e2212864120. [Google Scholar] [CrossRef]
- Bowser, B.S.; DeWire, S.M.; Damania, B. Transcriptional regulation of the K1 gene product of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2002, 76, 12574–12583. [Google Scholar] [CrossRef] [PubMed]
- Abere, B.; Mamo, T.M.; Hartmann, S.; Samarina, N.; Hage, E.; Ruckert, J.; Hotop, S.K.; Busche, G.; Schulz, T.F. The Kaposi’s sarcoma-associated herpesvirus (KSHV) non-structural membrane protein K15 is required for viral lytic replication and may represent a therapeutic target. PLoS Pathog. 2017, 13, e1006639. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Guo, J.; Li, M.; Choi, J.K.; DeMaria, M.; Rosenzweig, M.; Jung, J.U. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi’s sarcoma-associated herpesvirus. Mol. Cell Biol. 1998, 18, 5219–5228. [Google Scholar] [CrossRef]
- Tomlinson, C.C.; Damania, B. The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J. Virol. 2004, 78, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Lee, S.H.; Feng, P.; Chang, H.; Cho, N.H.; Jung, J.U. Characterization of the Kaposi’s sarcoma-associated herpesvirus K1 signalosome. J. Virol. 2005, 79, 12173–12184. [Google Scholar] [CrossRef]
- Anders, P.M.; Zhang, Z.; Bhende, P.M.; Giffin, L.; Damania, B. The KSHV K1 Protein Modulates AMPK Function to Enhance Cell Survival. PLoS Pathog. 2016, 12, e1005985. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.W.; Damania, B. Hsp90 and Hsp40/Erdj3 are required for the expression and anti-apoptotic function of KSHV K1. Oncogene 2010, 29, 3532–3544. [Google Scholar] [CrossRef]
- Lagunoff, M.; Majeti, R.; Weiss, A.; Ganem, D. Deregulated signal transduction by the K1 gene product of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1999, 96, 5704–5709. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Veazey, R.; Williams, K.; Li, M.; Guo, J.; Neipel, F.; Fleckenstein, B.; Lackner, A.; Desrosiers, R.C.; Jung, J.U. Deregulation of cell growth by the K1 gene of Kaposi’s sarcoma-associated herpesvirus. Nat. Med. 1998, 4, 435–440. [Google Scholar] [CrossRef]
- Wang, L.; Wakisaka, N.; Tomlinson, C.C.; DeWire, S.M.; Krall, S.; Pagano, J.S.; Damania, B. The Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) K1 protein induces expression of angiogenic and invasion factors. Cancer Res. 2004, 64, 2774–2781. [Google Scholar] [CrossRef]
- Wang, L.; Dittmer, D.P.; Tomlinson, C.C.; Fakhari, F.D.; Damania, B. Immortalization of primary endothelial cells by the K1 protein of Kaposi’s sarcoma-associated herpesvirus. Cancer Res. 2006, 66, 3658–3666. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, S.; Maeng, H.; Young, D.P.; Prakash, O.; Fayad, L.E.; Younes, A.; Samaniego, F. K1 protein of human herpesvirus 8 suppresses lymphoma cell Fas-mediated apoptosis. Blood 2007, 109, 2174–2182. [Google Scholar] [CrossRef]
- Lagunoff, M.; Lukac, D.M.; Ganem, D. Immunoreceptor tyrosine-based activation motif-dependent signaling by Kaposi’s sarcoma-associated herpesvirus K1 protein: Effects on lytic viral replication. J. Virol. 2001, 75, 5891–5898. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Paulose-Murphy, M.; Chung, Y.H.; Connlole, M.; Zeichner, S.; Jung, J.U. Suppression of tetradecanoyl phorbol acetate-induced lytic reactivation of Kaposi’s sarcoma-associated herpesvirus by K1 signal transduction. J. Virol. 2002, 76, 12185–12199. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, W.; Sanders, M.K.; Brulois, K.F.; Dittmer, D.P.; Damania, B. The K1 Protein of Kaposi’s Sarcoma-Associated Herpesvirus Augments Viral Lytic Replication. J. Virol. 2016, 90, 7657–7666. [Google Scholar] [CrossRef]
- Hu, F.; Nicholas, J. Signal transduction by human herpesvirus 8 viral interleukin-6 (vIL-6) is modulated by the nonsignaling gp80 subunit of the IL-6 receptor complex and is distinct from signaling induced by human IL-6. J. Virol. 2006, 80, 10874–10878. [Google Scholar] [CrossRef]
- Giffin, L.; West, J.A.; Damania, B. Kaposi’s Sarcoma-Associated Herpesvirus Interleukin-6 Modulates Endothelial Cell Movement by Upregulating Cellular Genes Involved in Migration. MBio 2015, 6, e01499-15. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Jaffe, E.S.; Chang, Y.; Jones, K.; Teruya-Feldstein, J.; Moore, P.S.; Tosato, G. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood 1999, 93, 4034–4043. [Google Scholar] [CrossRef]
- Meggetto, F.; Cesarman, E.; Mourey, L.; Massip, P.; Delsol, G.; Brousset, P. Detection and characterization of human herpesvirus-8-infected cells in bone marrow biopsies of human immunodeficiency virus-positive patients. Hum. Pathol. 2001, 32, 288–291. [Google Scholar] [CrossRef]
- Vart, R.J.; Nikitenko, L.L.; Lagos, D.; Trotter, M.W.; Cannon, M.; Bourboulia, D.; Gratrix, F.; Takeuchi, Y.; Boshoff, C. Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6 and G-protein-coupled receptor regulate angiopoietin-2 expression in lymphatic endothelial cells. Cancer Res. 2007, 67, 4042–4051. [Google Scholar] [CrossRef]
- Rivera-Soto, R.; Dissinger, N.J.; Damania, B. Kaposi’s Sarcoma-Associated Herpesvirus Viral Interleukin-6 Signaling Upregulates Integrin beta3 Levels and Is Dependent on STAT3. J. Virol. 2020, 94, e01384-19. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.D.; Aoki, Y.; Chang, Y.; Moore, P.S.; Yarchoan, R.; Tosato, G. Involvement of interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi’s sarcoma herpesvirus-associated infected primary effusion lymphoma cells. Blood 1999, 94, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Sandford, G.; Nicholas, J. Intracellular signaling mechanisms and activities of human herpesvirus 8 interleukin-6. J. Virol. 2009, 83, 722–733. [Google Scholar] [CrossRef]
- Glenn, M.; Rainbow, L.; Aurade, F.; Davison, A.; Schulz, T.F. Identification of a spliced gene from Kaposi’s sarcoma-associated herpesvirus encoding a protein with similarities to latent membrane proteins 1 and 2A of Epstein-Barr virus. J. Virol. 1999, 73, 6953–6963. [Google Scholar] [CrossRef]
- Choi, J.K.; Lee, B.S.; Shim, S.N.; Li, M.; Jung, J.U. Identification of the novel K15 gene at the rightmost end of the Kaposi’s sarcoma-associated herpesvirus genome. J. Virol. 2000, 74, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M.M.; Glenn, M.; Rainbow, L.; Kieser, A.; Henke-Gendo, C.; Schulz, T.F. Activation of mitogen-activated protein kinase and NF-kappaB pathways by a Kaposi’s sarcoma-associated herpesvirus K15 membrane protein. J. Virol. 2003, 77, 9346–9358. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M.M.; Pietrek, M.; Dittrich-Breiholz, O.; Kracht, M.; Schulz, T.F. Modulation of host gene expression by the K15 protein of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2007, 81, 42–58. [Google Scholar] [CrossRef] [PubMed]
- Pietrek, M.; Brinkmann, M.M.; Glowacka, I.; Enlund, A.; Havemeier, A.; Dittrich-Breiholz, O.; Kracht, M.; Lewitzky, M.; Saksela, K.; Feller, S.M.; et al. Role of the Kaposi’s sarcoma-associated herpesvirus K15 SH3 binding site in inflammatory signaling and B-cell activation. J. Virol. 2010, 84, 8231–8240. [Google Scholar] [CrossRef]
- Bala, K.; Bosco, R.; Gramolelli, S.; Haas, D.A.; Kati, S.; Pietrek, M.; Havemeier, A.; Yakushko, Y.; Singh, V.V.; Dittrich-Breiholz, O.; et al. Kaposi’s sarcoma herpesvirus K15 protein contributes to virus-induced angiogenesis by recruiting PLCgamma1 and activating NFAT1-dependent RCAN1 expression. PLoS Pathog. 2012, 8, e1002927. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef]
- Lukac, D.M.; Kirshner, J.R.; Ganem, D. Transcriptional activation by the product of open reading frame 50 of Kaposi’s sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 1999, 73, 9348–9361. [Google Scholar] [CrossRef]
- Xu, Y.; AuCoin, D.P.; Huete, A.R.; Cei, S.A.; Hanson, L.J.; Pari, G.S. A Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 2005, 79, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Damania, B.; Jeong, J.H.; Bowser, B.S.; DeWire, S.M.; Staudt, M.R.; Dittmer, D.P. Comparison of the Rta/Orf50 transactivator proteins of gamma-2-herpesviruses. J. Virol. 2004, 78, 5491–5499. [Google Scholar] [CrossRef]
- Bu, W.; Carroll, K.D.; Palmeri, D.; Lukac, D.M. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta lytic switch protein functions as a tetramer. J. Virol. 2007, 81, 5788–5806. [Google Scholar] [CrossRef]
- Chen, J.; Ye, F.; Xie, J.; Kuhne, K.; Gao, S.J. Genome-wide identification of binding sites for Kaposi’s sarcoma-associated herpesvirus lytic switch protein, RTA. Virology 2009, 386, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Brown, H.J.; Wu, T.T.; Sun, R. Transcription activation of polyadenylated nuclear rna by rta in human herpesvirus 8/Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2001, 75, 3129–3140. [Google Scholar] [CrossRef]
- Chang, P.J.; Shedd, D.; Gradoville, L.; Cho, M.S.; Chen, L.W.; Chang, J.; Miller, G. Open reading frame 50 protein of Kaposi’s sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol. 2002, 76, 3168–3178. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Deng, H.; Sun, R. Comparative study of regulation of RTA-responsive genes in Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 2003, 77, 9451–9462. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chang, J.; Lynch, S.J.; Lukac, D.M.; Ganem, D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev 2002, 16, 1977–1989. [Google Scholar] [CrossRef]
- Wang, S.E.; Wu, F.Y.; Fujimuro, M.; Zong, J.; Hayward, S.D.; Hayward, G.S. Role of CCAAT/enhancer-binding protein alpha (C/EBPalpha) in activation of the Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 2003, 77, 600–623. [Google Scholar] [CrossRef]
- Carroll, K.D.; Khadim, F.; Spadavecchia, S.; Palmeri, D.; Lukac, D.M. Direct interactions of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta protein with the cellular protein octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J. Virol. 2007, 81, 8451–8467. [Google Scholar] [CrossRef]
- Gwack, Y.; Hwang, S.; Lim, C.; Won, Y.S.; Lee, C.H.; Choe, J. Kaposi’s Sarcoma-associated herpesvirus open reading frame 50 stimulates the transcriptional activity of STAT3. J. Biol. Chem. 2002, 277, 6438–6442. [Google Scholar] [CrossRef]
- Gwack, Y.; Byun, H.; Hwang, S.; Lim, C.; Choe, J. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi’s sarcoma-associated herpesvirus open reading frame 50. J. Virol. 2001, 75, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Baek, H.J.; Nakamura, H.; Lee, S.H.; Meisterernst, M.; Roeder, R.G.; Jung, J.U. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi’s sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol. Cell Biol. 2003, 23, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Nakamura, H.; Lee, S.H.; Souvlis, J.; Yustein, J.T.; Gygi, S.; Kung, H.J.; Jung, J.U. Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication. Mol. Cell Biol. 2003, 23, 8282–8294. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yan, Z.; Wood, C. Kaposi’s sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 2008, 82, 3590–3603. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Liu, Y.; Liang, D.; Wang, Z.; Robertson, E.S.; Lan, K. Cellular corepressor TLE2 inhibits replication-and-transcription- activator-mediated transactivation and lytic reactivation of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2010, 84, 2047–2062. [Google Scholar] [CrossRef] [PubMed]
- Combs, L.R.; Spires, L.M.; Alonso, J.D.; Papp, B.; Toth, Z. KSHV RTA Induces Degradation of the Host Transcription Repressor ID2 To Promote the Viral Lytic Cycle. J. Virol. 2022, 96, e0010122. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hayward, G.S. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 2010, 33, 863–877. [Google Scholar] [CrossRef]
- Meyer, F.; Ehlers, E.; Steadman, A.; Waterbury, T.; Cao, M.; Zhang, L. TLR-TRIF pathway enhances the expression of KSHV replication and transcription activator. J. Biol. Chem. 2013, 288, 20435–20442. [Google Scholar] [CrossRef] [PubMed]
- Lingel, A.; Ehlers, E.; Wang, Q.; Cao, M.; Wood, C.; Lin, R.; Zhang, L. Kaposi’s Sarcoma-Associated Herpesvirus Reduces Cellular Myeloid Differentiation Primary-Response Gene 88 (MyD88) Expression via Modulation of Its RNA. J. Virol. 2016, 90, 180–188. [Google Scholar] [CrossRef]
- Gould, F.; Harrison, S.M.; Hewitt, E.W.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 2009, 83, 6727–6738. [Google Scholar] [CrossRef]
- Ehrlich, E.S.; Chmura, J.C.; Smith, J.C.; Kalu, N.N.; Hayward, G.S. KSHV RTA abolishes NFkappaB responsive gene expression during lytic reactivation by targeting vFLIP for degradation via the proteasome. PLoS ONE 2014, 9, e91359. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Zhang, D.; Gui, C.; Huang, L.; Chang, S.; Dong, L.; Bai, L.; Wu, S.; Lan, K. KSHV RTA antagonizes SMC5/6 complex-induced viral chromatin compaction by hijacking the ubiquitin-proteasome system. PLoS Pathog. 2022, 18, e1010744. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, Z.; Gu, F.; Xu, Y.; Li, Y.; Sun, W.; Rao, W.; Du, S.; Zhu, C.; Wang, Y.; et al. Degradation of TRIM32 is induced by RTA for Kaposi’s sarcoma-associated herpesvirus lytic replication. J. Virol. 2024, 98, e0000524. [Google Scholar] [CrossRef]
- Causey, A.; Constantine, M.; Oswald, J.; Dellomo, A.; Masters, B.; Omorogbe, E.; Admon, A.; Garzino-Demo, A.; Ehrlich, E. Analysis of the ubiquitin-modified proteome identifies novel host factors in Kaposi’s sarcoma herpesvirus lytic reactivation. BioRxiv 2024. [Google Scholar] [CrossRef]
- Zhu, F.X.; Cusano, T.; Yuan, Y. Identification of the immediate-early transcripts of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5556–5567. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chong, O.T.; Yuan, Y. Differential regulation of K8 gene expression in immediate-early and delayed-early stages of Kaposi’s sarcoma-associated herpesvirus. Virology 2004, 325, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.Y.; Wang, S.E.; Tang, Q.Q.; Fujimuro, M.; Chiou, C.J.; Zheng, Q.; Chen, H.; Hayward, S.D.; Lane, M.D.; Hayward, G.S. Cell cycle arrest by Kaposi’s sarcoma-associated herpesvirus replication-associated protein is mediated at both the transcriptional and posttranslational levels by binding to CCAAT/enhancer-binding protein alpha and p21(CIP-1). J. Virol. 2003, 77, 8893–8914. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Gwack, Y.; Byun, H.; Lim, C.; Choe, J. The Kaposi’s sarcoma-associated herpesvirus K8 protein interacts with CREB-binding protein (CBP) and represses CBP-mediated transcription. J. Virol. 2001, 75, 9509–9516. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Ellison, T.J.; Yeh, E.T.; Jung, J.U.; Luciw, P.A.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus K-bZIP represses gene transcription via SUMO modification. J. Virol. 2005, 79, 9912–9925. [Google Scholar] [CrossRef]
- Kaul, R.; Purushothaman, P.; Uppal, T.; Verma, S.C. KSHV lytic proteins K-RTA and K8 bind to cellular and viral chromatin to modulate gene expression. PLoS ONE 2019, 14, e0215394. [Google Scholar] [CrossRef]
- Liao, W.; Tang, Y.; Lin, S.F.; Kung, H.J.; Giam, C.Z. K-bZIP of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) binds KSHV/HHV-8 Rta and represses Rta-mediated transactivation. J. Virol. 2003, 77, 3809–3815. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, Y.; Yuan, Y. Kaposi’s Sarcoma-Associated Herpesvirus K8 Is an RNA Binding Protein That Regulates Viral DNA Replication in Coordination with a Noncoding RNA. J. Virol. 2018, 92, e02177-17. [Google Scholar] [CrossRef]
- Kirshner, J.R.; Lukac, D.M.; Chang, J.; Ganem, D. Kaposi’s sarcoma-associated herpesvirus open reading frame 57 encodes a posttranscriptional regulator with multiple distinct activities. J. Virol. 2000, 74, 3586–3597. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Blackbourn, D.J.; Clements, J.B. The evolutionarily conserved Kaposi’s sarcoma-associated herpesvirus ORF57 protein interacts with REF protein and acts as an RNA export factor. J. Biol. Chem. 2004, 279, 33001–33011. [Google Scholar] [CrossRef]
- Majerciak, V.; Yamanegi, K.; Allemand, E.; Kruhlak, M.; Krainer, A.R.; Zheng, Z.M. Kaposi’s sarcoma-associated herpesvirus ORF57 functions as a viral splicing factor and promotes expression of intron-containing viral lytic genes in spliceosome-mediated RNA splicing. J. Virol. 2008, 82, 2792–2801. [Google Scholar] [CrossRef] [PubMed]
- Boyne, J.R.; Jackson, B.R.; Taylor, A.; Macnab, S.A.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus ORF57 protein interacts with PYM to enhance translation of viral intronless mRNAs. EMBO J. 2010, 29, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.C.; Hunter, O.V.; Conrad, N.K. Kaposi’s sarcoma-associated herpesvirus ORF57 protein protects viral transcripts from specific nuclear RNA decay pathways by preventing hMTR4 recruitment. PLoS Pathog. 2019, 15, e1007596. [Google Scholar] [CrossRef]
- Sahin, B.B.; Patel, D.; Conrad, N.K. Kaposi’s sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog. 2010, 6, e1000799. [Google Scholar] [CrossRef]
- Palmeri, D.; Spadavecchia, S.; Carroll, K.D.; Lukac, D.M. Promoter- and cell-specific transcriptional transactivation by the Kaposi’s sarcoma-associated herpesvirus ORF57/Mta protein. J. Virol. 2007, 81, 13299–13314. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Pripuzova, N.; McCoy, J.P.; Gao, S.J.; Zheng, Z.M. Targeted disruption of Kaposi’s sarcoma-associated herpesvirus ORF57 in the viral genome is detrimental for the expression of ORF59, K8alpha, and K8.1 and the production of infectious virus. J. Virol. 2007, 81, 1062–1071. [Google Scholar] [CrossRef]
- Wu, F.Y.; Ahn, J.H.; Alcendor, D.J.; Jang, W.J.; Xiao, J.; Hayward, S.D.; Hayward, G.S. Origin-independent assembly of Kaposi’s sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J. Virol. 2001, 75, 1487–1506. [Google Scholar] [CrossRef]
- Lin, C.L.; Li, H.; Wang, Y.; Zhu, F.X.; Kudchodkar, S.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus lytic origin (ori-Lyt)-dependent DNA replication: Identification of the ori-Lyt and association of K8 bZip protein with the origin. J. Virol. 2003, 77, 5578–5588. [Google Scholar] [CrossRef] [PubMed]
- AuCoin, D.P.; Colletti, K.S.; Cei, S.A.; Papouskova, I.; Tarrant, M.; Pari, G.S. Amplification of the Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 2004, 318, 542–555. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Chan, M.Y.; Zhu, F.X.; Lukac, D.M.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: Cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 2004, 78, 8615–8629. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, Y.; Watanabe, T.; Hanajiri, M.; Sekine, Y.; Fujimuro, M. Kaposi’s Sarcoma-Associated Herpesvirus ORF7 Is Essential for Virus Production. Microorganisms 2021, 9, 1169. [Google Scholar] [CrossRef] [PubMed]
- Dunn-Kittenplon, D.D.; Kalt, I.; Lellouche, J.M.; Sarid, R. The KSHV portal protein ORF43 is essential for the production of infectious viral particles. Virology 2019, 529, 205–215. [Google Scholar] [CrossRef]
- Sanchez, D.J.; Coscoy, L.; Ganem, D. Functional organization of MIR2, a novel viral regulator of selective endocytosis. J. Biol. Chem. 2002, 277, 6124–6130. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, P.G.; Efstathiou, S.; Doherty, P.C.; Lehner, P.J. Inhibition of MHC class I-restricted antigen presentation by gamma 2-herpesviruses. Proc. Natl. Acad. Sci. USA 2000, 97, 8455–8460. [Google Scholar] [CrossRef]
- Ishido, S.; Wang, C.; Lee, B.S.; Cohen, G.B.; Jung, J.U. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 2000, 74, 5300–5309. [Google Scholar] [CrossRef]
- Duncan, L.M.; Piper, S.; Dodd, R.B.; Saville, M.K.; Sanderson, C.M.; Luzio, J.P.; Lehner, P.J. Lysine-63-linked ubiquitination is required for endolysosomal degradation of class I molecules. EMBO J. 2006, 25, 1635–1645. [Google Scholar] [CrossRef]
- Li, Q.; Means, R.; Lang, S.; Jung, J.U. Downregulation of gamma interferon receptor 1 by Kaposi’s sarcoma-associated herpesvirus K3 and K5. J. Virol. 2007, 81, 2117–2127. [Google Scholar] [CrossRef] [PubMed]
- Ishido, S.; Choi, J.K.; Lee, B.S.; Wang, C.; DeMaria, M.; Johnson, R.P.; Cohen, G.B.; Jung, J.U. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi’s sarcoma-associated herpesvirus K5 protein. Immunity 2000, 13, 365–374. [Google Scholar] [CrossRef]
- Coscoy, L.; Ganem, D. A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J. Clin. Investig. 2001, 107, 1599–1606. [Google Scholar] [CrossRef]
- Nanes, B.A.; Grimsley-Myers, C.M.; Cadwell, C.M.; Robinson, B.S.; Lowery, A.M.; Vincent, P.A.; Mosunjac, M.; Fruh, K.; Kowalczyk, A.P. p120-catenin regulates VE-cadherin endocytosis and degradation induced by the Kaposi sarcoma-associated ubiquitin ligase K5. Mol. Biol. Cell 2017, 28, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Brulois, K.; Toth, Z.; Wong, L.Y.; Feng, P.; Gao, S.J.; Ensser, A.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus K3 and K5 ubiquitin E3 ligases have stage-specific immune evasion roles during lytic replication. J. Virol. 2014, 88, 9335–9349. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.J.; Boshoff, C.; Jayachandra, S.; Weiss, R.A.; Chang, Y.; Moore, P.S. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene 1997, 15, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Damania, B.; Alvarez, X.; Ogryzko, V.; Ozato, K.; Jung, J.U. Inhibition of p300 histone acetyltransferase by viral interferon regulatory factor. Mol. Cell Biol. 2000, 20, 8254–8263. [Google Scholar] [CrossRef]
- Lin, R.; Genin, P.; Mamane, Y.; Sgarbanti, M.; Battistini, A.; Harrington, W.J., Jr.; Barber, G.N.; Hiscott, J. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 2001, 20, 800–811. [Google Scholar] [CrossRef]
- Jacobs, S.R.; Stopford, C.M.; West, J.A.; Bennett, C.L.; Giffin, L.; Damania, B. Kaposi’s Sarcoma-Associated Herpesvirus Viral Interferon Regulatory Factor 1 Interacts with a Member of the Interferon-Stimulated Gene 15 Pathway. J. Virol. 2015, 89, 11572–11583. [Google Scholar] [CrossRef]
- Hwang, K.Y.; Choi, Y.B. Modulation of Mitochondrial Antiviral Signaling by Human Herpesvirus 8 Interferon Regulatory Factor 1. J. Virol. 2016, 90, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef] [PubMed]
- Fuld, S.; Cunningham, C.; Klucher, K.; Davison, A.J.; Blackbourn, D.J. Inhibition of interferon signaling by the Kaposi’s sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. J. Virol. 2006, 80, 3092–3097. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.W.; Kim, D.; Jung, J.U.; Lee, H.R. KSHV-encoded viral interferon regulatory factor 4 (vIRF4) interacts with IRF7 and inhibits interferon alpha production. Biochem. Biophys. Res. Commun. 2017, 486, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Doganay, S.; Chung, B.; Toth, Z.; Brulois, K.; Lee, S.; Kanketayeva, Z.; Feng, P.; Ha, T.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 4 (vIRF4) targets expression of cellular IRF4 and the Myc gene to facilitate lytic replication. J. Virol. 2014, 88, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Li, M.; Zarycki, J.; Jung, J.U. Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J. Virol. 2001, 75, 7572–7582. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.B.; Nicholas, J. Bim nuclear translocation and inactivation by viral interferon regulatory factor. PLoS Pathog. 2010, 6, e1001031. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Damania, B. The viral interferon regulatory factors of KSHV: Immunosuppressors or oncogenes? Front. Immunol. 2011, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, J. Human gammaherpesvirus cytokines and chemokine receptors. J. Interferon Cytokine Res. 2005, 25, 373–383. [Google Scholar] [CrossRef]
- Stine, J.T.; Wood, C.; Hill, M.; Epp, A.; Raport, C.J.; Schweickart, V.L.; Endo, Y.; Sasaki, T.; Simmons, G.; Boshoff, C.; et al. KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood 2000, 95, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.S.; Grone, H.J.; Rocken, M.; Klier, C.; Gu, S.; Wank, R.; Proudfoot, A.E.; Nelson, P.J.; Weber, C. Selective recruitment of Th2-type cells and evasion from a cytotoxic immune response mediated by viral macrophage inhibitory protein-II. Eur. J. Immunol. 2001, 31, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.B.; Nicholas, J. Autocrine and paracrine promotion of cell survival and virus replication by human herpesvirus 8 chemokines. J. Virol. 2008, 82, 6501–6513. [Google Scholar] [CrossRef]
- Liu, C.; Okruzhnov, Y.; Li, H.; Nicholas, J. Human herpesvirus 8 (HHV-8)-encoded cytokines induce expression of and autocrine signaling by vascular endothelial growth factor (VEGF) in HHV-8-infected primary-effusion lymphoma cell lines and mediate VEGF-independent antiapoptotic effects. J. Virol. 2001, 75, 10933–10940. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, L.; Geras-Raaka, E.; Varma, A.; Gershengorn, M.C.; Cesarman, E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 1997, 385, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Gershengorn, M.C.; Geras-Raaka, E.; Varma, A.; Clark-Lewis, I. Chemokines activate Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor in mammalian cells in culture. J. Clin. Investig. 1998, 102, 1469–1472. [Google Scholar] [CrossRef]
- Montaner, S.; Sodhi, A.; Pece, S.; Mesri, E.A.; Gutkind, J.S. The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 2001, 61, 2641–2648. [Google Scholar]
- Smit, M.J.; Verzijl, D.; Casarosa, P.; Navis, M.; Timmerman, H.; Leurs, R. Kaposi’s sarcoma-associated herpesvirus-encoded G protein-coupled receptor ORF74 constitutively activates p44/p42 MAPK and Akt via G(i) and phospholipase C-dependent signaling pathways. J. Virol. 2002, 76, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Bais, C.; Van Geelen, A.; Eroles, P.; Mutlu, A.; Chiozzini, C.; Dias, S.; Silverstein, R.L.; Rafii, S.; Mesri, E.A. Kaposi’s sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/ KDR. Cancer Cell 2003, 3, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, A.; Chaisuparat, R.; Hu, J.; Ramsdell, A.K.; Manning, B.D.; Sausville, E.A.; Sawai, E.T.; Molinolo, A.; Gutkind, J.S.; Montaner, S. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell 2006, 10, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Montaner, S.; Sodhi, A.; Servitja, J.M.; Ramsdell, A.K.; Barac, A.; Sawai, E.T.; Gutkind, J.S. The small GTPase Rac1 links the Kaposi sarcoma-associated herpesvirus vGPCR to cytokine secretion and paracrine neoplasia. Blood 2004, 104, 2903–2911. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yu, F.X.; Kim, Y.C.; Meng, Z.; Naipauer, J.; Looney, D.J.; Liu, X.; Gutkind, J.S.; Mesri, E.A.; Guan, K.L. Kaposi sarcoma-associated herpesvirus promotes tumorigenesis by modulating the Hippo pathway. Oncogene 2015, 34, 3536–3546. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, A.D.; Cavallin, L.E.; Vincent, L.; Chiozzini, C.; Eroles, P.; Duran, E.M.; Asgari, Z.; Hooper, A.T.; La Perle, K.M.; Hilsher, C.; et al. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: A cell and animal model of virally induced Kaposi’s sarcoma. Cancer Cell 2007, 11, 245–258. [Google Scholar] [CrossRef]
- Sandford, G.; Choi, Y.B.; Nicholas, J. Role of ORF74-encoded viral G protein-coupled receptor in human herpesvirus 8 lytic replication. J. Virol. 2009, 83, 13009–13014. [Google Scholar] [CrossRef]
- Haque, M.; Wang, V.; Davis, D.A.; Zheng, Z.M.; Yarchoan, R. Genetic organization and hypoxic activation of the Kaposi’s sarcoma-associated herpesvirus ORF34-37 gene cluster. J. Virol. 2006, 80, 7037–7051. [Google Scholar] [CrossRef] [PubMed]
- Hamza, M.S.; Reyes, R.A.; Izumiya, Y.; Wisdom, R.; Kung, H.J.; Luciw, P.A. ORF36 protein kinase of Kaposi’s sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 2004, 279, 38325–38330. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, J.; Xie, Z.; Liao, G.; Liu, J.; Chen, M.R.; Hu, S.; Woodard, C.; Lin, J.; Taverna, S.D.; et al. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 2011, 10, 390–400. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Wong, J.P.; Weinberg, M.S.; Host, K.M.; Giffin, L.C.; Buijnink, J.; van Dijk, E.; Izumiya, Y.; Kung, H.J.; Temple, B.R.; et al. A viral kinase mimics S6 kinase to enhance cell proliferation. Proc. Natl. Acad. Sci. USA 2016, 113, 7876–7881. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.L.; Sandhu, P.K.; Wong, J.P.; Bhatt, A.P.; Liu, X.; Buhrlage, S.J.; Temple, B.R.S.; Major, M.B.; Damania, B. KSHV Viral Protein Kinase Interacts with USP9X to Modulate the Viral Lifecycle. J. Virol. 2023, 97, e0176322. [Google Scholar] [CrossRef] [PubMed]
- Anders, P.M.; Montgomery, N.D.; Montgomery, S.A.; Bhatt, A.P.; Dittmer, D.P.; Damania, B. Human herpesvirus-encoded kinase induces B cell lymphomas in vivo. J. Clin. Investig. 2018, 128, 2519–2534. [Google Scholar] [CrossRef] [PubMed]
- Sarid, R.; Sato, T.; Bohenzky, R.A.; Russo, J.J.; Chang, Y. Kaposi’s sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nat. Med. 1997, 3, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H.; Nicholas, J.; Bellows, D.S.; Hayward, G.S.; Guo, H.G.; Reitz, M.S.; Hardwick, J.M. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. USA 1997, 94, 690–694. [Google Scholar] [CrossRef]
- Gelgor, A.; Kalt, I.; Bergson, S.; Brulois, K.F.; Jung, J.U.; Sarid, R. Viral Bcl-2 Encoded by the Kaposi’s Sarcoma-Associated Herpesvirus Is Vital for Virus Reactivation. J. Virol. 2015, 89, 5298–5307. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Lampe, M.; Gunther, T.; Brune, W. The Viral Bcl-2 Homologs of Kaposi’s Sarcoma-Associated Herpesvirus and Rhesus Rhadinovirus Share an Essential Role for Viral Replication. J. Virol. 2017, 91, e01875-16. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Chang, B.; Lee, P.; Brulois, K.F.; Ge, J.; Shi, M.; Rodgers, M.A.; Feng, P.; Oh, B.H.; Liang, C.; et al. Identification of the Essential Role of Viral Bcl-2 for Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication. J. Virol. 2015, 89, 5308–5317. [Google Scholar] [CrossRef]
- Broussard, G.; Damania, B. KSHV: Immune Modulation and Immunotherapy. Front. Immunol. 2019, 10, 3084. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Soto, R.; Damania, B. Modulation of Angiogenic Processes by the Human Gammaherpesviruses, Epstein-Barr Virus and Kaposi’s Sarcoma-Associated Herpesvirus. Front. Microbiol. 2019, 10, 1544. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Primers 2019, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Damania, B.; Dittmer, D.P. Today’s Kaposi sarcoma is not the same as it was 40 years ago, or is it? J. Med. Virol. 2023, 95, e28773. [Google Scholar] [CrossRef] [PubMed]
- Onunu, A.N.; Okoduwa, C.; Eze, E.U.; Adeyekun, A.A.; Kubeyinje, E.P.; Schwartz, R.A. Kaposi’s sarcoma in Nigeria. Int. J. Dermatol. 2007, 46, 264–267. [Google Scholar] [CrossRef]
- Friedman-Kien, A.E.; Laubenstein, L.J.; Rubinstein, P.; Buimovici-Klein, E.; Marmor, M.; Stahl, R.; Spigland, I.; Kim, K.S.; Zolla-Pazner, S. Disseminated Kaposi’s sarcoma in homosexual men. Ann. Intern. Med. 1982, 96, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Harwood, A.R.; Osoba, D.; Hofstader, S.L.; Goldstein, M.B.; Cardella, C.J.; Holecek, M.J.; Kunynetz, R.; Giammarco, R.A. Kaposi’s sarcoma in recipients of renal transplants. Am. J. Med. 1979, 67, 759–765. [Google Scholar] [CrossRef]
- Denis, D.; Seta, V.; Regnier-Rosencher, E.; Kramkimel, N.; Chanal, J.; Avril, M.F.; Dupin, N. A fifth subtype of Kaposi’s sarcoma, classic Kaposi’s sarcoma in men who have sex with men: A cohort study in Paris. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 1377–1384. [Google Scholar] [CrossRef]
- Regezi, J.A.; MacPhail, L.A.; Daniels, T.E.; DeSouza, Y.G.; Greenspan, J.S.; Greenspan, D. Human immunodeficiency virus-associated oral Kaposi’s sarcoma. A heterogeneous cell population dominated by spindle-shaped endothelial cells. Am. J. Pathol. 1993, 143, 240–249. [Google Scholar]
- O’Hara, A.J.; Chugh, P.; Wang, L.; Netto, E.M.; Luz, E.; Harrington, W.J.; Dezube, B.J.; Damania, B.; Dittmer, D.P. Pre-micro RNA signatures delineate stages of endothelial cell transformation in Kaposi sarcoma. PLoS Pathog. 2009, 5, e1000389. [Google Scholar] [CrossRef] [PubMed]
- Hosseinipour, M.C.; Sweet, K.M.; Xiong, J.; Namarika, D.; Mwafongo, A.; Nyirenda, M.; Chiwoko, L.; Kamwendo, D.; Hoffman, I.; Lee, J.; et al. Viral profiling identifies multiple subtypes of Kaposi’s sarcoma. MBio 2014, 5, e01633-14. [Google Scholar] [CrossRef]
- Hong, Y.K.; Foreman, K.; Shin, J.W.; Hirakawa, S.; Curry, C.L.; Sage, D.R.; Libermann, T.; Dezube, B.J.; Fingeroth, J.D.; Detmar, M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 2004, 36, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Trotter, M.W.; Lagos, D.; Bourboulia, D.; Henderson, S.; Makinen, T.; Elliman, S.; Flanagan, A.M.; Alitalo, K.; Boshoff, C. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 2004, 36, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.A.; Punjabi, A.S.; Lagunoff, M. Activation of Akt through gp130 receptor signaling is required for Kaposi’s sarcoma-associated herpesvirus-induced lymphatic reprogramming of endothelial cells. J. Virol. 2008, 82, 8771–8779. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Damania, B. Kaposi’s sarcoma-associated herpesvirus confers a survival advantage to endothelial cells. Cancer Res. 2008, 68, 4640–4648. [Google Scholar] [CrossRef]
- Davis, D.A.; Rinderknecht, A.S.; Zoeteweij, J.P.; Aoki, Y.; Read-Connole, E.L.; Tosato, G.; Blauvelt, A.; Yarchoan, R. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 2001, 97, 3244–3250. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, M.; Rose, P.P.; Moses, A.V.; Fruh, K. Remodeling of endothelial adherens junctions by Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2008, 82, 9615–9628. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.W.; Greene, W.; Ye, F.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus disrupts adherens junctions and increases endothelial permeability by inducing degradation of VE-cadherin. J. Virol. 2008, 82, 11902–11912. [Google Scholar] [CrossRef] [PubMed]
- Klepfish, A.; Sarid, R.; Shtalrid, M.; Shvidel, L.; Berrebi, A.; Schattner, A. Primary effusion lymphoma (PEL) in HIV-negative patients--a distinct clinical entity. Leuk. Lymphoma 2001, 41, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Chadburn, A.; Hyjek, E.; Mathew, S.; Cesarman, E.; Said, J.; Knowles, D.M. KSHV-positive solid lymphomas represent an extra-cavitary variant of primary effusion lymphoma. Am. J. Surg. Pathol. 2004, 28, 1401–1416. [Google Scholar] [CrossRef] [PubMed]
- Guillet, S.; Gerard, L.; Meignin, V.; Agbalika, F.; Cuccini, W.; Denis, B.; Katlama, C.; Galicier, L.; Oksenhendler, E. Classic and extracavitary primary effusion lymphoma in 51 HIV-infected patients from a single institution. Am. J. Hematol. 2016, 91, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Goto, H.; Yotsumoto, M. Current status of treatment for primary effusion lymphoma. Intractable Rare Dis. Res. 2014, 3, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, M.G.; Nador, R.G.; Chadburn, A.; Hyjek, E.M.; Inghirami, G.; Knowles, D.M.; Cesarman, E. Epstein-Barr virus latent gene expression in primary effusion lymphomas containing Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8. Blood 1997, 90, 1186–1191. [Google Scholar] [CrossRef]
- Mack, A.A.; Sugden, B. EBV is necessary for proliferation of dually infected primary effusion lymphoma cells. Cancer Res. 2008, 68, 6963–6968. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Caduff, N.; Barros, M.H.M.; Ramer, P.C.; Raykova, A.; Murer, A.; Landtwing, V.; Quast, I.; Styles, C.T.; Spohn, M.; et al. Persistent KSHV Infection Increases EBV-Associated Tumor Formation In Vivo via Enhanced EBV Lytic Gene Expression. Cell Host Microbe 2017, 22, 61–73.E7. [Google Scholar] [CrossRef]
- Panaampon, J.; Okada, S. Promising immunotherapeutic approaches for primary effusion lymphoma. Explor. Target. Antitumor Ther. 2024, 5, 699–713. [Google Scholar] [CrossRef]
- Hu, Z.; Pan, Z.; Chen, W.; Shi, Y.; Wang, W.; Yuan, J.; Wang, E.; Zhang, S.; Kurt, H.; Mai, B.; et al. Primary Effusion Lymphoma: A Clinicopathological Study of 70 Cases. Cancers 2021, 13, 878. [Google Scholar] [CrossRef] [PubMed]
- Dunleavy, K.; Wilson, W.H. How I treat HIV-associated lymphoma. Blood 2012, 119, 3245–3255. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chadburn, A.; Rubinstein, P.G. KSHV/HHV8-mediated hematologic diseases. Blood 2022, 139, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Parravicini, C.; Chandran, B.; Corbellino, M.; Berti, E.; Paulli, M.; Moore, P.S.; Chang, Y. Differential viral protein expression in Kaposi’s sarcoma-associated herpesvirus-infected diseases: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Am. J. Pathol. 2000, 156, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, E.; Gerard, L.; Gabarre, J.; Molina, J.M.; Rapp, C.; Abino, J.F.; Cadranel, J.; Chevret, S.; Oksenhendler, E. Prognostic factors and outcome of human herpesvirus 8-associated primary effusion lymphoma in patients with AIDS. J. Clin. Oncol. 2005, 23, 4372–4380. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Fajgenbaum, D.C. Overview of Castleman disease. Blood 2020, 135, 1353–1364. [Google Scholar] [CrossRef]
- Oksenhendler, E.; Boutboul, D.; Fajgenbaum, D.; Mirouse, A.; Fieschi, C.; Malphettes, M.; Vercellino, L.; Meignin, V.; Gerard, L.; Galicier, L. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br. J. Haematol. 2018, 180, 206–216. [Google Scholar] [CrossRef]
- Du, M.Q.; Liu, H.; Diss, T.C.; Ye, H.; Hamoudi, R.A.; Dupin, N.; Meignin, V.; Oksenhendler, E.; Boshoff, C.; Isaacson, P.G. Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in Castleman disease and associated lymphoproliferative disorders. Blood 2001, 97, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Parravicini, C.; Corbellino, M.; Paulli, M.; Magrini, U.; Lazzarino, M.; Moore, P.S.; Chang, Y. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman’s disease. Am. J. Pathol. 1997, 151, 1517–1522. [Google Scholar]
- Caro-Vegas, C.; Sellers, S.; Host, K.M.; Seltzer, J.; Landis, J.; Fischer, W.A., 2nd; Damania, B.; Dittmer, D.P. Runaway Kaposi Sarcoma-associated herpesvirus replication correlates with systemic IL-10 levels. Virology 2020, 539, 18–25. [Google Scholar] [CrossRef]
- Polizzotto, M.N.; Uldrick, T.S.; Wyvill, K.M.; Aleman, K.; Marshall, V.; Wang, V.; Whitby, D.; Pittaluga, S.; Jaffe, E.S.; Millo, C.; et al. Clinical Features and Outcomes of Patients With Symptomatic Kaposi Sarcoma Herpesvirus (KSHV)-associated Inflammation: Prospective Characterization of KSHV Inflammatory Cytokine Syndrome (KICS). Clin. Infect. Dis. 2016, 62, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.; Jablonowski, H.; Goebel, F.D.; Arasteh, K.; Spittle, M.; Rios, A.; Aboulafia, D.; Galleshaw, J.; Dezube, B.J. Randomized comparative trial of pegylated liposomal doxorubicin versus bleomycin and vincristine in the treatment of AIDS-related Kaposi’s sarcoma. International Pegylated Liposomal Doxorubicin Study Group. J. Clin. Oncol. 1998, 16, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Cianfrocca, M.; Lee, S.; Von Roenn, J.; Tulpule, A.; Dezube, B.J.; Aboulafia, D.M.; Ambinder, R.F.; Lee, J.Y.; Krown, S.E.; Sparano, J.A. Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma: Evidence of symptom palliation from chemotherapy. Cancer 2010, 116, 3969–3977. [Google Scholar] [CrossRef]
- Simonelli, C.; Spina, M.; Cinelli, R.; Talamini, R.; Tedeschi, R.; Gloghini, A.; Vaccher, E.; Carbone, A.; Tirelli, U. Clinical features and outcome of primary effusion lymphoma in HIV-infected patients: A single-institution study. J. Clin. Oncol. 2003, 21, 3948–3954. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Kuppermann, B.D.; Wolitz, R.A.; Palestine, A.G.; Li, H.; Robinson, C.A. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N. Engl. J. Med. 1999, 340, 1063–1070. [Google Scholar] [CrossRef]
- Casper, C.; Krantz, E.M.; Corey, L.; Kuntz, S.R.; Wang, J.; Selke, S.; Hamilton, S.; Huang, M.L.; Wald, A. Valganciclovir for suppression of human herpesvirus-8 replication: A randomized, double-blind, placebo-controlled, crossover trial. J. Infect. Dis. 2008, 198, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Moyo, T.K.; Richards, K.L.; Damania, B. Use of cidofovir for the treatment of HIV-negative human herpes virus-8-associated primary effusion lymphoma. Clin. Adv. Hematol. Oncol. 2010, 8, 372–374. [Google Scholar]
- Nishimoto, N.; Kanakura, Y.; Aozasa, K.; Johkoh, T.; Nakamura, M.; Nakano, S.; Nakano, N.; Ikeda, Y.; Sasaki, T.; Nishioka, K.; et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 2005, 106, 2627–2632. [Google Scholar] [CrossRef] [PubMed]
- Corbellino, M.; Bestetti, G.; Scalamogna, C.; Calattini, S.; Galazzi, M.; Meroni, L.; Manganaro, D.; Fasan, M.; Moroni, M.; Galli, M.; et al. Long-term remission of Kaposi sarcoma-associated herpesvirus-related multicentric Castleman disease with anti-CD20 monoclonal antibody therapy. Blood 2001, 98, 3473–3475. [Google Scholar] [CrossRef]
- Yu, L.; Tu, M.; Cortes, J.; Xu-Monette, Z.Y.; Miranda, R.N.; Zhang, J.; Orlowski, R.Z.; Neelapu, S.; Boddu, P.C.; Akosile, M.A.; et al. Clinical and pathological characteristics of HIV- and HHV-8-negative Castleman disease. Blood 2017, 129, 1658–1668. [Google Scholar] [CrossRef] [PubMed]
- Galanina, N.; Goodman, A.M.; Cohen, P.R.; Frampton, G.M.; Kurzrock, R. Successful Treatment of HIV-Associated Kaposi Sarcoma with Immune Checkpoint Blockade. Cancer Immunol. Res. 2018, 6, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Uldrick, T.S.; Goncalves, P.H.; Abdul-Hay, M.; Claeys, A.J.; Emu, B.; Ernstoff, M.S.; Fling, S.P.; Fong, L.; Kaiser, J.C.; Lacroix, A.M.; et al. Assessment of the Safety of Pembrolizumab in Patients With HIV and Advanced Cancer-A Phase 1 Study. JAMA Oncol. 2019, 5, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Tabata, M.M.; Novoa, R.A.; Bui, N.Q.; Zaba, L.C. Successful treatment of HIV-negative Kaposi sarcoma with ipilimumab and nivolumab and concurrent management of baseline psoriasis and bullous pemphigoid. JAAD Case Rep. 2020, 6, 447–449. [Google Scholar] [CrossRef]
- Polizzotto, M.N.; Uldrick, T.S.; Wyvill, K.M.; Aleman, K.; Peer, C.J.; Bevans, M.; Sereti, I.; Maldarelli, F.; Whitby, D.; Marshall, V.; et al. Pomalidomide for Symptomatic Kaposi’s Sarcoma in People With and Without HIV Infection: A Phase I/II Study. J. Clin. Oncol. 2016, 34, 4125–4131. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, R.; Pluda, J.M.; Wyvill, K.M.; Aleman, K.; Rodriguez-Chavez, I.R.; Tosato, G.; Catanzaro, A.T.; Steinberg, S.M.; Little, R.F. Treatment of AIDS-related Kaposi’s sarcoma with interleukin-12: Rationale and preliminary evidence of clinical activity. Crit. Rev. Immunol. 2007, 27, 401–414. [Google Scholar] [CrossRef]
- Caro-Vegas, C.; Bailey, A.; Bigi, R.; Damania, B.; Dittmer, D.P. Targeting mTOR with MLN0128 Overcomes Rapamycin and Chemoresistant Primary Effusion Lymphoma. MBio 2019, 10, e02871-18. [Google Scholar] [CrossRef] [PubMed]
- Campistol, J.M.; Gutierrez-Dalmau, A.; Torregrosa, J.V. Conversion to sirolimus: A successful treatment for posttransplantation Kaposi’s sarcoma. Transplantation 2004, 77, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.B.; Krown, S.E.; Lee, J.Y.; Honda, K.; Rapisuwon, S.; Wang, Z.; Aboulafia, D.; Reid, E.G.; Rudek, M.A.; Dezube, B.J.; et al. Phase II trial of imatinib in AIDS-associated Kaposi’s sarcoma: AIDS Malignancy Consortium Protocol 042. J. Clin. Oncol. 2014, 32, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Reid, E.G.; Suazo, A.; Lensing, S.Y.; Dittmer, D.P.; Ambinder, R.F.; Maldarelli, F.; Gorelick, R.J.; Aboulafia, D.; Mitsuyasu, R.; Dickson, M.A.; et al. Pilot Trial AMC-063: Safety and Efficacy of Bortezomib in AIDS-associated Kaposi Sarcoma. Clin. Cancer Res. 2020, 26, 558–565. [Google Scholar] [CrossRef]
- Uldrick, T.S.; Wyvill, K.M.; Kumar, P.; O’Mahony, D.; Bernstein, W.; Aleman, K.; Polizzotto, M.N.; Steinberg, S.M.; Pittaluga, S.; Marshall, V.; et al. Phase II study of bevacizumab in patients with HIV-associated Kaposi’s sarcoma receiving antiretroviral therapy. J. Clin. Oncol. 2012, 30, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.A.; Mishra, S.; Anagho, H.A.; Aisabor, A.I.; Shrestha, P.; Wang, V.; Takamatsu, Y.; Maeda, K.; Mitsuya, H.; Zeldis, J.B.; et al. Restoration of immune surface molecules in Kaposi sarcoma-associated herpes virus infected cells by lenalidomide and pomalidomide. Oncotarget 2017, 8, 50342–50358. [Google Scholar] [CrossRef]
- Bhatt, S.; Ashlock, B.M.; Toomey, N.L.; Diaz, L.A.; Mesri, E.A.; Lossos, I.S.; Ramos, J.C. Efficacious proteasome/HDAC inhibitor combination therapy for primary effusion lymphoma. J. Clin. Investig. 2013, 123, 2616–2628. [Google Scholar] [CrossRef]
- Yiakoumis, X.; Pangalis, G.A.; Kyrtsonis, M.C.; Vassilakopoulos, T.P.; Kontopidou, F.N.; Kalpadakis, C.; Korkolopoulou, P.; Levidou, G.; Androulaki, A.; Siakantaris, M.P.; et al. Primary effusion lymphoma in two HIV-negative patients successfully treated with pleurodesis as first-line therapy. Anticancer Res. 2010, 30, 271–276. [Google Scholar]
- Gantt, S.; Cattamanchi, A.; Krantz, E.; Magaret, A.; Selke, S.; Kuntz, S.R.; Huang, M.L.; Corey, L.; Wald, A.; Casper, C. Reduced human herpesvirus-8 oropharyngeal shedding associated with protease inhibitor-based antiretroviral therapy. J. Clin. Virol. 2014, 60, 127–132. [Google Scholar] [CrossRef]

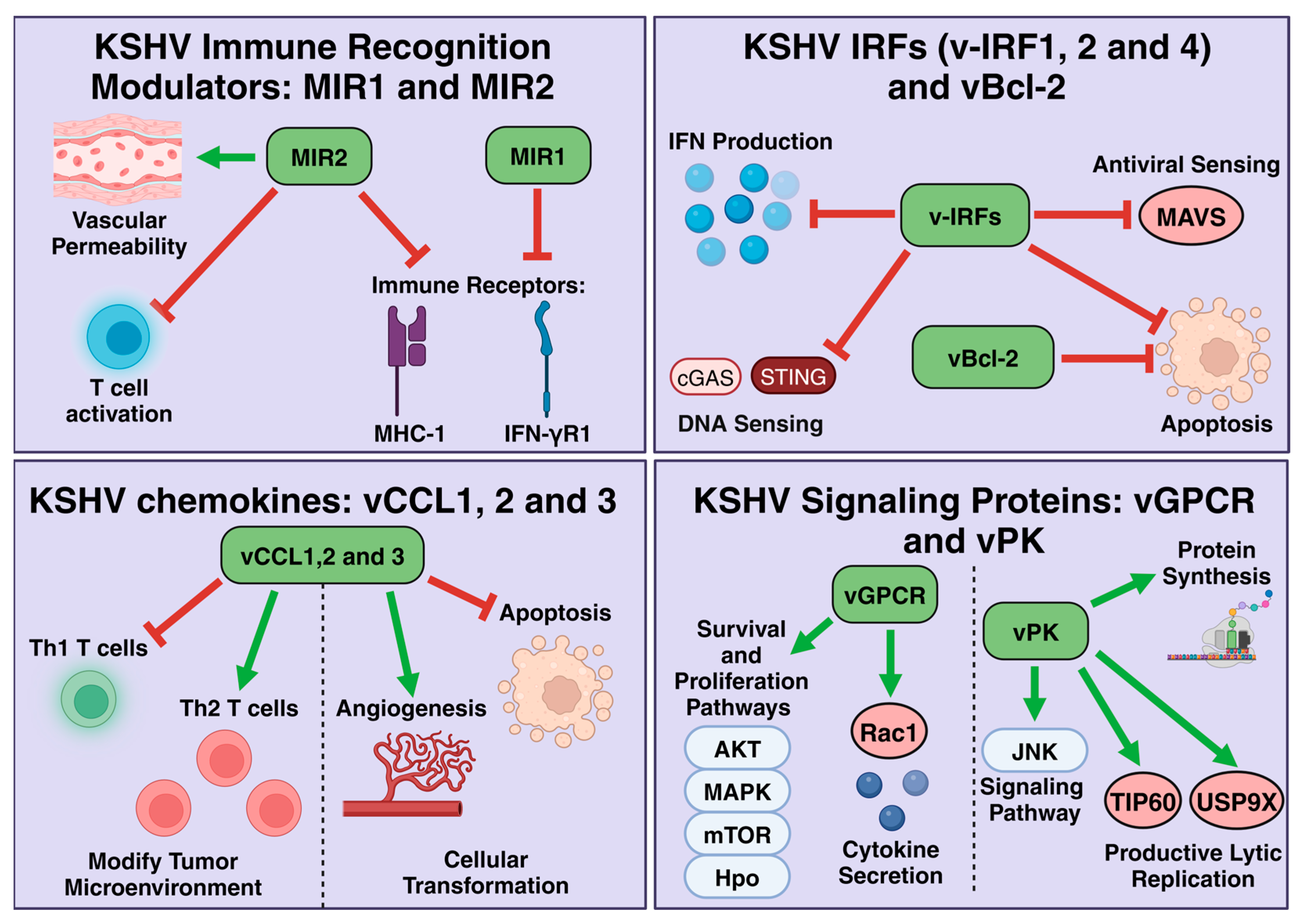
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losay, V.A.; Damania, B. Unraveling the Kaposi Sarcoma-Associated Herpesvirus (KSHV) Lifecycle: An Overview of Latency, Lytic Replication, and KSHV-Associated Diseases. Viruses 2025, 17, 177. https://doi.org/10.3390/v17020177
Losay VA, Damania B. Unraveling the Kaposi Sarcoma-Associated Herpesvirus (KSHV) Lifecycle: An Overview of Latency, Lytic Replication, and KSHV-Associated Diseases. Viruses. 2025; 17(2):177. https://doi.org/10.3390/v17020177
Chicago/Turabian StyleLosay, Victor A., and Blossom Damania. 2025. "Unraveling the Kaposi Sarcoma-Associated Herpesvirus (KSHV) Lifecycle: An Overview of Latency, Lytic Replication, and KSHV-Associated Diseases" Viruses 17, no. 2: 177. https://doi.org/10.3390/v17020177
APA StyleLosay, V. A., & Damania, B. (2025). Unraveling the Kaposi Sarcoma-Associated Herpesvirus (KSHV) Lifecycle: An Overview of Latency, Lytic Replication, and KSHV-Associated Diseases. Viruses, 17(2), 177. https://doi.org/10.3390/v17020177