
Viruses and the Brain—A Relationship Prone to Trouble

Abstract
:1. Introduction
2. Getting into the Brain Is Not So Easy
3. What Makes a Virus Capable of Neuroinvasion?
3.1. Neuronal Route of Infection
3.2. Hematogenous Route of Infection
3.2.1. Neuroinvasion from the Blood Stream
3.2.2. Trojan Horse Delivery
3.2.3. Hematogenous Route of Neuroinvasion, Conclusions
4. Once the Barriers Protecting the CNS Have Been Breached
4.1. The Disease Scenario When Viruses Infect the Human Brain
4.1.1. Flavivirus Infections of the CNS
4.1.2. HIV Infection of the CNS
4.1.3. SARS-CoV-2 Infection of the CNS
4.2. Chronic Neurodegenerative Diseases and Viral Infections
4.2.1. Parkinson’s Disease (PD)
4.2.2. Alzheimer’s Disease (AD) and Other Major Neurocognitive Disorders (MNDs)
4.2.3. Multiple Sclerosis (MS) and Other Demyelinating Diseases
4.2.4. Amyotrophic Lateral Sclerosis (ALS)
4.2.5. Human Endogenous Retroviruses and Their Association with Neurodegenerative Syndromes
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Daneman, R.; Prat, A. The Blood-Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Alahmari, A. Blood-Brain Barrier Overview: Structural and Functional Correlation. Neural Plast. 2021, 2021, 6564585. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. The Continued Threat of Emerging Flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.C.; Liu, H.; Mohamud, Y.; Bahreyni, A.; Zhang, J.; Cashman, N.R.; Luo, H. Sublethal Enteroviral Infection Exacerbates Disease Progression in an ALS Mouse Model. J. Neuroinflammation 2022, 19, 16. [Google Scholar] [CrossRef] [PubMed]
- Adler, G.L.; Le, K.; Fu, Y.H.; Kim, W.S. Human Endogenous Retroviruses in Neurodegenerative Diseases. Genes 2024, 15, 745. [Google Scholar] [CrossRef]
- Dotiwala, A.K.; McCausland, C.; Samra, N.S. Anatomy, Head and Neck: Blood Brain Barrier; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chen, Z.; Li, G. Immune Response and Blood–Brain Barrier Dysfunction during Viral Neuroinvasion. Innate Immun. 2021, 27, 109. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Johnson, A.M.; Keep, R.F.; Andjelkovic, A.V. Junctional Proteins of the Blood-Brain Barrier: New Insights into Function and Dysfunction. Tissue Barriers 2016, 4, e1154641. [Google Scholar] [CrossRef] [PubMed]
- Duong, C.N.; Vestweber, D. Mechanisms Ensuring Endothelial Junction Integrity Beyond VE-Cadherin. Front. Physiol. 2020, 11, 519. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, Z.; Zhang, L.; Wei, X.; Li, L. Tight Junction in Blood-Brain Barrier: An Overview of Structure, Regulation, and Regulator Substances. CNS Neurosci. Ther. 2012, 18, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef]
- De Bock, M.; De Smet, M.; Verwaerde, S.; Tahiri, H.; Schumacher, S.; Van Haver, V.; Witschas, K.; Steinhäuser, C.; Rouach, N.; Vandenbroucke, R.E.; et al. Targeting Gliovascular Connexins Prevents Inflammatory Blood-Brain Barrier Leakage and Astrogliosis. JCI Insight 2022, 7, e135263. [Google Scholar] [CrossRef] [PubMed]
- Kameritsch, P.; Pogoda, K.; Pohl, U. Channel-Independent Influence of Connexin 43 on Cell Migration. Biochim. Biophys. Acta 2012, 1818, 1993–2001. [Google Scholar] [CrossRef]
- Noe, C.R.; Noe-Letschnig, M.; Handschuh, P.; Noe, C.A.; Lanzenberger, R. Dysfunction of the Blood-Brain Barrier—A Key Step in Neurodegeneration and Dementia. Front. Aging Neurosci. 2020, 12, 185. [Google Scholar] [CrossRef]
- De Vlieger, L.; Vandenbroucke, R.E.; Van Hoecke, L. Recent Insights into Viral Infections as a Trigger and Accelerator in Alzheimer’s Disease. Drug Discov. Today 2022, 27, 103340. [Google Scholar] [CrossRef] [PubMed]
- Krey, L.; Huber, M.K.; Höglinger, G.U.; Wegner, F. Can SARS-CoV-2 Infection Lead to Neurodegeneration and Parkinson’s Disease? Brain Sci. 2021, 11, 1654. [Google Scholar] [CrossRef]
- Saboori, P.; Sadegh, A. Histology and Morphology of the Brain Subarachnoid Trabeculae. Anat. Res. Int. 2015, 2015, 279814. [Google Scholar] [CrossRef] [PubMed]
- Laterra, J.; Keep, R.; Betz, L.A.; Goldstein, G.W. Blood—Brain—Cerebrospinal Fluid Barriers. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Lippincott-Raven: Philadelphia, PA, USA, 1999. [Google Scholar]
- Kaiser, K.; Bryja, V. Choroid Plexus: The Orchestrator of Long-Range Signalling Within the CNS. Int. J. Mol. Sci. 2020, 21, 4760. [Google Scholar] [CrossRef]
- Sanchez-Covarrubias, L.; Slosky, L.M.; Thompson, B.J.; Davis, T.P.; Ronaldson, P.T. Transporters at CNS Barrier Sites: Obstacles or Opportunities for Drug Delivery? Curr. Pharm. Des. 2014, 20, 1422. [Google Scholar] [CrossRef]
- Ayub, M.; Jin, H.K.; Bae, J.-S. The Blood Cerebrospinal Fluid Barrier Orchestrates Immunosurveillance, Immunoprotection, and Immunopathology in the Central Nervous System. BMB Rep. 2021, 54, 196. [Google Scholar] [CrossRef] [PubMed]
- Dabbagh, F.; Schroten, H.; Schwerk, C. In Vitro Models of the Blood–Cerebrospinal Fluid Barrier and Their Applications in the Development and Research of (Neuro)Pharmaceuticals. Pharmaceutics 2022, 14, 1729. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.L.; Kassem, N.A.; Redzic, Z.B.; Chen, C.P.C.; Segal, M.B.; Preston, J.E. Age-Related Changes in Choroid Plexus and Blood-Cerebrospinal Fluid Barrier Function in the Sheep. Exp. Gerontol. 2009, 44, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Proulx, S.T.; Engelhardt, B. Central Nervous System Zoning: How Brain Barriers Establish Subdivisions for CNS Immune Privilege and Immune Surveillance. J. Intern. Med. 2022, 292, 47. [Google Scholar] [CrossRef] [PubMed]
- Muldoon, L.L.; Alvarez, J.I.; Begley, D.J.; Boado, R.J.; Del Zoppo, G.J.; Doolittle, N.D.; Engelhardt, B.; Hallenbeck, J.M.; Lonser, R.R.; Ohlfest, J.R.; et al. Immunologic Privilege in the Central Nervous System and the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 33, 13. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS Immune Privilege: Hiding in Plain Sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Daniels, B.P.; Klein, R.S. Knocking on Closed Doors: Host Interferons Dynamically Regulate Blood-Brain Barrier Function during Viral Infections of the Central Nervous System. PLoS Pathog. 2015, 11, e1005096. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Long, Y.; Sun, J.; Gong, L. Towards a Comprehensive View of the Herpes B Virus. Front. Immunol. 2023, 14, 1281384. [Google Scholar] [CrossRef]
- Sakulwira, K.; Theamboonlers, A.; Charoonrut, P.; Ratanakorn, P.; Poovorawan, Y. Serological Evidence of Herpesvirus Infection in Gibbons. BMC Microbiol. 2002, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- de Matos, R.; Russell, D.; Van Alstine, W.; Miller, A. Spontaneous Fatal Human Herpesvirus 1 Encephalitis in Two Domestic Rabbits (Oryctolagus cuniculus). J. Vet. Diagn. Investig. 2014, 26, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Xue, T.; Zhao, X.; Zou, J.; Pu, H.; Hu, X.; Tian, Z. Pseudorabies Virus Associations in Wild Animals: Review of Potential Reservoirs for Cross-Host Transmission. Viruses 2022, 14, 2254. [Google Scholar] [CrossRef] [PubMed]
- Sehl, J.; Teifke, J.P. Comparative Pathology of Pseudorabies in Different Naturally and Experimentally Infected Species-A Review. Pathogens 2020, 9, 633. [Google Scholar] [CrossRef] [PubMed]
- Wisner, T.W.; Sugimoto, K.; Howard, P.W.; Kawaguchi, Y.; Johnson, D.C. Anterograde Transport of Herpes Simplex Virus Capsids in Neurons by Both Separate and Married Mechanisms. J. Virol. 2011, 85, 5919. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Farías, M.A.; Álvarez, D.M.; Bueno, S.M.; Riedel, C.A.; González, P.A. Herpes Simplex Virus Type 1 Infection of the Central Nervous System: Insights into Proposed Interrelationships with Neurodegenerative Disorders. Front. Cell Neurosci. 2019, 13, 46. [Google Scholar] [CrossRef]
- Bai, F.; Ashley Thompson, E.; Vig, P.J.S.; Arturo Leis, A. Current Understanding of West Nile Virus Clinical Manifestations, Immune Responses, Neuroinvasion, and Immunotherapeutic Implications. Pathogens 2019, 8, 193. [Google Scholar] [CrossRef]
- Samuel, M.A.; Wang, H.; Siddharthan, V.; Morrey, J.D.; Diamond, M.S. Axonal Transport Mediates West Nile Virus Entry into the Central Nervous System and Induces Acute Flaccid Paralysis. Proc. Natl. Acad. Sci. USA 2007, 104, 17140–17145. [Google Scholar] [CrossRef]
- Fooks, A.R.; Cliquet, F.; Finke, S.; Freuling, C.; Hemachudha, T.; Mani, R.S.; Müller, T.; Nadin-Davis, S.; Picard-Meyer, E.; Wilde, H.; et al. Rabies. Nat. Rev. Dis. Primers 2017, 3, 17091. [Google Scholar] [CrossRef]
- Le Blanc, I.; Luyet, P.P.; Pons, V.; Ferguson, C.; Emans, N.; Petiot, A.; Mayran, N.; Demaurex, N.; Fauré, J.; Sadoul, R.; et al. Endosome-to-Cytosol Transport of Viral Nucleocapsids. Nat. Cell Biol. 2005, 7, 653. [Google Scholar] [CrossRef]
- Piccinotti, S.; Kirchhausen, T.; Whelan, S.P.J. Uptake of Rabies Virus into Epithelial Cells by Clathrin-Mediated Endocytosis Depends upon Actin. J. Virol. 2013, 87, 11637. [Google Scholar] [CrossRef] [PubMed]
- Tsiang, H.; Ceccaldi, P.E.; Lycke, E. Rabies Virus Infection and Transport in Human Sensory Dorsal Root Ganglia Neurons. J. Gen. Virol. 1991, 72, 1191–1194. [Google Scholar] [CrossRef]
- Bauer, A.; Nolden, T.; Nemitz, S.; Perlson, E.; Finke, S. A Dynein Light Chain 1 Binding Motif in Rabies Virus Polymerase L Protein Plays a Role in Microtubule Reorganization and Viral Primary Transcription. J. Virol. 2015, 89, 9591. [Google Scholar] [CrossRef] [PubMed]
- Chailangkarn, T.; Tanwattana, N.; Jaemthaworn, T.; Sriswasdi, S.; Wanasen, N.; Tangphatsornruang, S.; Leetanasaksakul, K.; Jantraphakorn, Y.; Nawae, W.; Chankeeree, P.; et al. Establishment of Human-Induced Pluripotent Stem Cell-Derived Neurons—A Promising in Vitro Model for a Molecular Study of Rabies Virus and Host Interaction. Int. J. Mol. Sci. 2021, 22, 11986. [Google Scholar] [CrossRef] [PubMed]
- Rieder, M.; Conzelmann, K.K. Interferon in Rabies Virus Infection. Adv. Virus Res. 2011, 79, 91–114. [Google Scholar] [CrossRef] [PubMed]
- Brzózka, K.; Finke, S.; Conzelmann, K.-K. Identification of the Rabies Virus Alpha/Beta Interferon Antagonist: Phosphoprotein P Interferes with Phosphorylation of Interferon Regulatory Factor 3. J. Virol. 2005, 79, 7673. [Google Scholar] [CrossRef]
- Kiflu, A.B. The Immune Escape Strategy of Rabies Virus and Its Pathogenicity Mechanisms. Viruses 2024, 16, 1774. [Google Scholar] [CrossRef]
- Ito, N.; Moseley, G.W.; Sugiyama, M. The Importance of Immune Evasion in the Pathogenesis of Rabies Virus: Veterinary Science Award Winner’s (No.116) Commemorative Review. J. Vet. Med. Sci. 2016, 78, 1089. [Google Scholar] [CrossRef]
- Korie, G.C.; Sallau, A.B.; Kanu, B.; Kia, G.S.N.; Kwaga, J.K.P. Rabies Virus Infection Is Associated with Variations in Calbindin D-28K and Calretinin MRNA Expression Levels in Mouse Brain Tissue. Arch. Virol. 2023, 168, 143. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, V.; Green, D.; Locke, K.; O’Brien, C.M.; Dearnley, M.; Bingham, J. Novel Role of SARM1 Mediated Axonal Degeneration in the Pathogenesis of Rabies. PLoS Pathog. 2020, 16, e1008343. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nawaz, Z.; Guo, C.; Ali, S.; Naeem, M.A.; Jamil, T.; Ahmad, W.; Siddiq, M.U.; Ahmed, S.; Asif Idrees, M.; et al. Rabies Virus Exploits Cytoskeleton Network to Cause Early Disease Progression and Cellular Dysfunction. Front. Vet. Sci. 2022, 9, 889873. [Google Scholar] [CrossRef]
- Kim, S.; Larrous, F.; Varet, H.; Legendre, R.; Feige, L.; Dumas, G.; Matsas, R.; Kouroupi, G.; Grailhe, R.; Bourhy, H. Early Transcriptional Changes in Rabies Virus-Infected Neurons and Their Impact on Neuronal Functions. Front. Microbiol. 2021, 12, 730892. [Google Scholar] [CrossRef]
- Davis, B.M.; Rall, G.F.; Schnell, M.J. Everything You Always Wanted to Know about Rabies Virus (but Were Afraid to Ask). Annu. Rev. Virol. 2015, 2, 451. [Google Scholar] [CrossRef]
- Feige, L.; Zaeck, L.M.; Sehl-Ewert, J.; Finke, S.; Bourhy, H. Innate Immune Signaling and Role of Glial Cells in Herpes Simplex Virus- and Rabies Virus-Induced Encephalitis. Viruses 2021, 13, 2364. [Google Scholar] [CrossRef]
- Potratz, M.; Zaeck, L.; Christen, M.; Te Kamp, V.; Klein, A.; Nolden, T.; Freuling, C.M.; Müller, T.; Finke, S. Astrocyte Infection during Rabies Encephalitis Depends on the Virus Strain and Infection Route as Demonstrated by Novel Quantitative 3D Analysis of Cell Tropism. Cells 2020, 9, 412. [Google Scholar] [CrossRef]
- Potratz, M.; Zaeck, L.M.; Weigel, C.; Klein, A.; Freuling, C.M.; Müller, T.; Finke, S. Neuroglia Infection by Rabies Virus after Anterograde Virus Spread in Peripheral Neurons. Acta Neuropathol. Commun. 2020, 8, 199. [Google Scholar] [CrossRef] [PubMed]
- Sivasubramanian, M.K.; Monteiro, R.; Harrison, K.S.; Plakkot, B.; Subramanian, M.; Jones, C. Herpes Simplex Virus Type 1 Preferentially Enhances Neuro-Inflammation and Senescence in Brainstem of Female Mice. J. Virol. 2022, 96, e0108122. [Google Scholar] [CrossRef] [PubMed]
- Gnann, J.W.; Whitley, R.J. Herpes Simplex Encephalitis: An Update. Curr. Infect. Dis. Rep. 2017, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Rohani, H.; Arjmand, R.; Mozhgani, S.H.; Shafiee, A.; Javad Amini, M.; Forghani-Ramandi, M.M. The Worldwide Prevalence of Herpes Simplex Virus Encephalitis and Meningitis: A Systematic Review and Meta-Analysis. Turk. Arch. Pediatr. 2023, 58, 580. [Google Scholar] [CrossRef]
- Quist-Paulsen, E.; Kran, A.M.B.; Lindland, E.S.; Ellefsen, K.; Sandvik, L.; Dunlop, O.; Ormaasen, V. To What Extent Can Clinical Characteristics Be Used to Distinguish Encephalitis from Encephalopathy of Other Causes? Results from a Prospective Observational Study. BMC Infect. Dis. 2019, 19, 80. [Google Scholar] [CrossRef]
- Verzosa, A.L.; McGeever, L.A.; Bhark, S.J.; Delgado, T.; Salazar, N.; Sanchez, E.L. Herpes Simplex Virus 1 Infection of Neuronal and Non-Neuronal Cells Elicits Specific Innate Immune Responses and Immune Evasion Mechanisms. Front. Immunol. 2021, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.K.; Seppänen, M.; Hautala, T.; Ciancanelli, M.J.; Itan, Y.; Lafaille, F.G.; Dell, W.; Lorenzo, L.; Byun, M.; Pauwels, E.; et al. TLR3 Deficiency in Herpes Simplex Encephalitis: High Allelic Heterogeneity and Recurrence Risk. Neurology 2014, 83, 1888–1897. [Google Scholar] [CrossRef]
- Casanova, J.L.; Abel, L. From Rare Disorders of Immunity to Common Determinants of Infection: Following the Mechanistic Thread. Cell 2022, 185, 3086. [Google Scholar] [CrossRef]
- Schnell, M.J.; McGettigan, J.P.; Wirblich, C.; Papaneri, A. The Cell Biology of Rabies Virus: Using Stealth to Reach the Brain. Nat. Rev. Microbiol. 2009, 8, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Weed, D.J.; Nicola, A.V. Herpes Simplex Virus Membrane Fusion. Adv. Anat. Embryol. Cell Biol. 2017, 223, 29. [Google Scholar] [CrossRef]
- Gaudin, Y.; Ruigrok, R.W.H.; Tuffereau, C.; Knossow, M.; Flamand, A. Rabies Virus Glycoprotein Is a Trimer. Virology 2004, 187, 627. [Google Scholar] [CrossRef]
- Etessami, R.; Conzelmann, K.K.; Fadai-Ghotbi, B.; Natelson, B.; Tsiang, H.; Ceccaldi, P.E. Spread and Pathogenic Characteristics of a G-Deficient Rabies Virus Recombinant: An in Vitro and in Vivo Study. J. Gen. Virol. 2000, 81, 2147–2153. [Google Scholar] [CrossRef] [PubMed]
- Madavaraju, K.; Koganti, R.; Volety, I.; Yadavalli, T.; Shukla, D. Herpes Simplex Virus Cell Entry Mechanisms: An Update. Front. Cell Infect. Microbiol. 2020, 10, 617578. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gao, J.; Zhang, M.; Zhang, D.; Duan, M.; Guan, Z.; Guo, Y. Dynein- and Kinesin- Mediated Intracellular Transport on Microtubules Facilitates RABV Infection. Vet. Microbiol. 2021, 262, 109241. [Google Scholar] [CrossRef]
- Miranda-Saksena, M.; Denes, C.E.; Diefenbach, R.J.; Cunningham, A.L. Infection and Transport of Herpes Simplex Virus Type 1 in Neurons: Role of the Cytoskeleton. Viruses 2018, 10, 92. [Google Scholar] [CrossRef]
- Piret, J.; Boivin, G. Innate Immune Response during Herpes Simplex Virus Encephalitis and Development of Immunomodulatory Strategies. Rev. Med. Virol. 2015, 25, 300–319. [Google Scholar] [CrossRef] [PubMed]
- Devaux, C.A.; Pontarotti, P.; Levasseur, A.; Colson, P.; Raoult, D. Is It Time to Switch to a Formulation Other than the Live Attenuated Poliovirus Vaccine to Prevent Poliomyelitis? Front. Public Health 2023, 11, 1284337. [Google Scholar] [CrossRef]
- Wolbert, J.G.; Rajnik, M.; Swinkels, H.M.; Higginbotham, K. Poliomyelitis; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Ohka, S.; Nihei, C.I.; Yamazaki, M.; Nomoto, A. Poliovirus Trafficking toward Central Nervous System via Human Poliovirus Receptor-Dependent and -Independent Pathway. Front. Microbiol. 2012, 3, 147. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, N. Chapter 1 The Pathogenesis of Poliomyelitis: What We Don’t Know. Adv. Virus Res. 2008, 71, 1–50. [Google Scholar] [CrossRef]
- Andersen, N.S.B.; Larsen, S.M.; Nissen, S.K.; Jørgensen, S.E.; Mardahl, M.; Christiansen, M.; Kay, L.; Mogensen, T.H. Host Genetics, Innate Immune Responses, and Cellular Death Pathways in Poliomyelitis Patients. Front. Microbiol. 2019, 10, 455664. [Google Scholar] [CrossRef]
- Durrant, D.M.; Ghosh, S.; Klein, R.S. The Olfactory Bulb: An Immunosensory Effector Organ during Neurotropic Viral Infections. ACS Chem. Neurosci. 2016, 7, 464. [Google Scholar] [CrossRef]
- Pacheco-Herrero, M.; Soto-Rojas, L.O.; Harrington, C.R.; Flores-Martinez, Y.M.; Villegas-Rojas, M.M.; León-Aguilar, A.M.; Martínez-Gómez, P.A.; Campa-Córdoba, B.B.; Apátiga-Pérez, R.; Corniel-Taveras, C.N.; et al. Elucidating the Neuropathologic Mechanisms of SARS-CoV-2 Infection. Front. Neurol. 2021, 12, 660087. [Google Scholar] [CrossRef]
- Monu, M.; Ahmad, F.; Olson, R.M.; Balendiran, V.; Singh, P.K. SARS-CoV-2 Infects Cells Lining the Blood-Retinal Barrier and Induces a Hyperinflammatory Immune Response in the Retina via Systemic Exposure. PLoS Pathog. 2024, 20, e1012156. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.F.; Chu, L.W.; Liao, I.C.; Simanjuntak, Y.; Lin, Y.L.; Juan, C.C.; Ping, Y.H. The Mechanism of the Zika Virus Crossing the Placental Barrier and the Blood-Brain Barrier. Front. Microbiol. 2020, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- van Marle, G.; Antony, J.; Ostermann, H.; Dunham, C.; Hunt, T.; Halliday, W.; Maingat, F.; Urbanowski, M.D.; Hobman, T.; Peeling, J.; et al. West Nile Virus-Induced Neuroinflammation: Glial Infection and Capsid Protein-Mediated Neurovirulence. J. Virol. 2007, 81, 10933. [Google Scholar] [CrossRef]
- Marinho, P.E.S.; Kroon, E.G. Flaviviruses as Agents of Childhood Central Nervous System Infections in Brazil. New Microbes New Infect. 2019, 31, 100572. [Google Scholar] [CrossRef]
- Das, S.; Basu, A. Japanese Encephalitis Virus Infects Neural Progenitor Cells and Decreases Their Proliferation. J. Neurochem. 2008, 106, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Potokar, M.; Jorgačevski, J.; Zorec, R. Astrocytes in Flavivirus Infections. Int. J. Mol. Sci. 2019, 20, 691. [Google Scholar] [CrossRef]
- de Vries, L.; Harding, A.T. Mechanisms of Neuroinvasion and Neuropathogenesis by Pathologic Flaviviruses. Viruses 2023, 15, 261. [Google Scholar] [CrossRef] [PubMed]
- Alimonti, J.B.; Ribecco-Lutkiewicz, M.; Sodja, C.; Jezierski, A.; Stanimirovic, D.B.; Liu, Q.; Haqqani, A.S.; Conlan, W.; Bani-Yaghoub, M. Zika Virus Crosses an in Vitro Human Blood Brain Barrier Model. Fluids Barriers CNS 2018, 15, 15. [Google Scholar] [CrossRef] [PubMed]
- Papa, M.P.; Meuren, L.M.; Coelho, S.V.A.; de Oliveira Lucas, C.G.; Mustafá, Y.M.; Matassoli, F.L.; Silveira, P.P.; Frost, P.S.; Pezzuto, P.; Ribeiro, M.R.; et al. Zika Virus Infects, Activates, and Crosses Brain Microvascular Endothelial Cells, without Barrier Disruption. Front. Microbiol. 2017, 8, 2557. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Lo, Y.; Chapagain, M.; Lum, S.; Kumar, M.; Gurjav, U.; Luo, H.; Nakatsuka, A.; Nerurkar, V.R. West Nile Virus Infection Modulates Human Brain Microvascular Endothelial Cells Tight Junction Proteins and Cell Adhesion Molecules: Transmigration across the in Vitro Blood-Brain Barrier. Virology 2009, 385, 425. [Google Scholar] [CrossRef]
- Agrawal, T.; Sharvani, V.; Nair, D.; Medigeshi, G.R. Japanese Encephalitis Virus Disrupts Cell-Cell Junctions and Affects the Epithelial Permeability Barrier Functions. PLoS ONE 2013, 8, 69465. [Google Scholar] [CrossRef]
- Palus, M.; Vancova, M.; Sirmarova, J.; Elsterova, J.; Perner, J.; Ruzek, D. Tick-Borne Encephalitis Virus Infects Human Brain Microvascular Endothelial Cells without Compromising Blood-Brain Barrier Integrity. Virology 2017, 507, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Salimi, H.; Cain, M.D.; Jiang, X.; Roth, R.A.; Beatty, W.L.; Sun, C.; Klimstra, W.B.; Hou, J.; Klein, R.S. Encephalitic Alphaviruses Exploit Caveola-Mediated Transcytosis at the Blood-Brain Barrier for Central Nervous System Entry. mBio 2020, 11, 02731-19. [Google Scholar] [CrossRef]
- Alam, A.M. Nipah Virus, an Emerging Zoonotic Disease Causing Fatal Encephalitis. Clin. Med. 2022, 22, 348. [Google Scholar] [CrossRef] [PubMed]
- Escaffre, O.; Borisevich, V.; Rockx, B. Pathogenesis of Hendra and Nipah Virus Infection in Humans. J. Infect. Dev. Ctries. 2013, 7, 308–311. [Google Scholar] [CrossRef]
- Bruno, L.; Nappo, M.A.; Ferrari, L.; Di Lecce, R.; Guarnieri, C.; Cantoni, A.M.; Corradi, A. Nipah Virus Disease: Epidemiological, Clinical, Diagnostic and Legislative Aspects of This Unpredictable Emerging Zoonosis. Animals 2022, 13, 159. [Google Scholar] [CrossRef]
- Contreras, E.M.; Johnston, G.P.; Buchholz, D.W.; Ortega, V.; Monreal, I.A.; Zamora, J.L.R.; Cheung, T.; Aguilar, H.C. Roles of Cholesterol in Early and Late Steps of the Nipah Virus Membrane Fusion Cascade. J. Virol. 2021, 95, 2323–2343. [Google Scholar] [CrossRef]
- Aditi; Shariff, M. Nipah Virus Infection: A Review. Epidemiol. Infect. 2019, 147, e95. [Google Scholar] [CrossRef] [PubMed]
- Salimi, H.; Klein, R.S.; Salimi, H.; Klein, R.S. Disruption of the Blood-Brain Barrier During Neuroinflammatory and Neuroinfectious Diseases. In Neuroimmune Diseases; Springer: Cham, Switzerland, 2019; Volume 195. [Google Scholar] [CrossRef]
- Mustafá, Y.M.; Meuren, L.M.; Coelho, S.V.A.; De Arruda, L.B. Pathways Exploited by Flaviviruses to Counteract the Blood-Brain Barrier and Invade the Central Nervous System. Front. Microbiol. 2019, 10, 525. [Google Scholar] [CrossRef]
- Lorin, V.; Danckaert, A.; Porrot, F.; Schwartz, O.; Afonso, P.V.; Mouqueta, H. Antibody Neutralization of HIV-1 Crossing the Blood-Brain Barrier. mBio 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Blackhurst, B.M.; Funk, K.E. Molecular and Cellular Mechanisms Underlying Neurologic Manifestations of Mosquito-Borne Flavivirus Infections. Viruses 2023, 15, 2200. [Google Scholar] [CrossRef] [PubMed]
- Mbita, Z.; Hull, R.; Dlamini, Z. Human Immunodeficiency Virus-1 (HIV-1)-Mediated Apoptosis: New Therapeutic Targets. Viruses 2014, 6, 3181. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [PubMed]
- Landi, A.; Iannucci, V.; Van Nuffel, A.; Meuwissen, P.; Verhasselt, B. One Protein to Rule Them All: Modulation of Cell Surface Receptors and Molecules by HIV Nef. Curr. HIV Res. 2011, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.R.; Rachel, G.; Parthasarathy, D. HIV Proteins and Endothelial Dysfunction: Implications in Cardiovascular Disease. Front. Cardiovasc. Med. 2018, 5, 425638. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Ye, A.A.; Shirazinejad, C.; Hurley, J.H.; Drubin, D.G. Kinetic Investigation Reveals an HIV-1 Nef-Dependent Increase in AP-2 Recruitment and Productivity at Endocytic Sites. Mol. Biol. Cell 2024, 35, 24. [Google Scholar] [CrossRef]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue Virus NS1 Protein Activates Cells via Toll-like Receptor 4 and Disrupts Endothelial Cell Monolayer Integrity. Sci. Transl. Med. 2015, 7, 304ra142. [Google Scholar] [CrossRef]
- Puerta-Guardo, H.; Glasner, D.R.; Espinosa, D.A.; Biering, S.B.; Patana, M.; Ratnasiri, K.; Wang, C.; Beatty, P.R.; Harris, E. Flavivirus NS1 Triggers Tissue-Specific Vascular Endothelial Dysfunction Reflecting Disease Tropism. Cell Rep. 2019, 26, 1598–1613.e8. [Google Scholar] [CrossRef] [PubMed]
- Puerta-Guardo, H.; Glasner, D.R.; Harris, E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016, 12, e1005738. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-F.; Lei, H.-Y.; Shiau, A.-L.; Liu, H.-S.; Yeh, T.-M.; Chen, S.-H.; Liu, C.-C.; Chiu, S.-C.; Lin, Y.-S. Endothelial Cell Apoptosis Induced by Antibodies against Dengue Virus Nonstructural Protein 1 via Production of Nitric Oxide. J. Immunol. 2002, 169, 657–664. [Google Scholar] [CrossRef]
- Beatty, P.R.; Puerta-Guardo, H.; Killingbeck, S.S.; Glasner, D.R.; Hopkins, K.; Harris, E. Dengue Virus NS1 Triggers Endothelial Permeability and Vascular Leak That Is Prevented by NS1 Vaccination. Sci. Transl. Med. 2015, 7, 304ra141. [Google Scholar] [CrossRef]
- Zeng, Q.; Liu, J.; Hao, C.; Zhang, B.; Zhang, H. Making Sense of Flavivirus Non-Strctural Protein 1 in Innate Immune Evasion and Inducing Tissue-Specific Damage. Virus Res. 2023, 336, 199222. [Google Scholar] [CrossRef]
- Shrivastava, G.; García-Cordero, J.; León-Juárez, M.; Oza, G.; Tapia-Ramírez, J.; Villegas-Sepulveda, N.; Cedillo-Barrón, L. NS2A Comprises a Putative Viroporin of Dengue Virus 2. Virulence 2017, 8, 1450. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, G.; Visoso-Carvajal, G.; Garcia-Cordero, J.; Leon-Juarez, M.; Chavez-Munguia, B.; Lopez, T.; Nava, P.; Villegas-Sepulveda, N.; Cedillo-Barron, L. Dengue Virus Serotype 2 and Its Non-Structural Proteins 2A and 2B Activate NLRP3 Inflammasome. Front. Immunol. 2020, 11, 469220. [Google Scholar] [CrossRef] [PubMed]
- Latanova, A.; Starodubova, E.; Karpov, V. Flaviviridae Nonstructural Proteins: The Role in Molecular Mechanisms of Triggering Inflammation. Viruses 2022, 14, 1808. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, S.M.; Rotta, I.; Ribeiro, C.E.; Smith, D.; Wang, R.; Judicello, J.; Potter, M.; Vaida, F.; Letendre, S.; Ellis, R.J. Blood-CSF Barrier and Compartmentalization of CNS Cellular Immune Response in HIV Infection. J. Neuroimmunol. 2016, 301, 41. [Google Scholar] [CrossRef]
- Pokorna Formanova, P.; Palus, M.; Salat, J.; Hönig, V.; Stefanik, M.; Svoboda, P.; Ruzek, D. Changes in Cytokine and Chemokine Profiles in Mouse Serum and Brain, and in Human Neural Cells, upon Tick-Borne Encephalitis Virus Infection. J. Neuroinflammation 2019, 16, 205. [Google Scholar] [CrossRef] [PubMed]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 Infection: The Role of Cytokines in COVID-19 Disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Zidovec-Lepej, S.; Vilibic-Cavlek, T.; Barbic, L.; Ilic, M.; Savic, V.; Tabain, I.; Ferenc, T.; Grgic, I.; Gorenec, L.; Bogdanic, M.; et al. Antiviral Cytokine Response in Neuroinvasive and Non-Neuroinvasive West Nile Virus Infection. Viruses 2021, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Berman, J.W. Chemokine-Dependent Mechanisms of Leukocyte Trafficking across a Model of the Blood–Brain Barrier. Methods 2003, 29, 351–361. [Google Scholar] [CrossRef]
- Greene, C.; Connolly, R.; Brennan, D.; Laffan, A.; O’Keeffe, E.; Zaporojan, L.; O’Callaghan, J.; Thomson, B.; Connolly, E.; Argue, R.; et al. Blood–Brain Barrier Disruption and Sustained Systemic Inflammation in Individuals with Long COVID-Associated Cognitive Impairment. Nat. Neurosci. 2024, 27, 421. [Google Scholar] [CrossRef]
- Rhea, E.M.; Logsdon, A.F.; Hansen, K.M.; Williams, L.M.; Reed, M.J.; Baumann, K.K.; Holden, S.J.; Raber, J.; Banks, W.A.; Erickson, M.A. The S1 Protein of SARS-CoV-2 Crosses the Blood–Brain Barrier in Mice. Nat. Neurosci. 2020, 24, 368–378. [Google Scholar] [CrossRef]
- Hernández-Parra, H.; Reyes-Hernández, O.D.; Figueroa-González, G.; González-Del Carmen, M.; González-Torres, M.; Peña-Corona, S.I.; Florán, B.; Cortés, H.; Leyva-Gómez, G. Alteration of the Blood-Brain Barrier by COVID-19 and Its Implication in the Permeation of Drugs into the Brain. Front. Cell Neurosci. 2023, 17, 1125109. [Google Scholar] [CrossRef]
- Chen, J.; Tan, R.; Mo, Y.; Zhang, J. The Blood-Brain Barrier in Health, Neurological Diseases, and COVID-19. Fundam. Res. 2022, 2, 817–826. [Google Scholar] [CrossRef]
- Levast, B.; Barblu, L.; Coutu, M.; Prévost, J.; Brassard, N.; Peres, A.; Stegen, C.; Madrenas, J.; Kaufmann, D.E.; Finzi, A. HIV-1 Gp120 Envelope Glycoprotein Determinants for Cytokine Burst in Human Monocytes. PLoS ONE 2017, 12, e0174550. [Google Scholar] [CrossRef]
- Pandey, N.; Jain, A.; Garg, R.K.; Kumar, R.; Agrawal, O.P.; Lakshmana Rao, P.V. Serum Levels of IL-8, IFNγ, IL-10, and TGF β and Their Gene Expression Levels in Severe and Non-Severe Cases of Dengue Virus Infection. Arch. Virol. 2015, 160, 1463–1475. [Google Scholar] [CrossRef]
- Versele, R.; Sevin, E.; Gosselet, F.; Fenart, L.; Candela, P. TNF-α and IL-1β Modulate Blood-Brain Barrier Permeability and Decrease Amyloid-β Peptide Efflux in a Human Blood-Brain Barrier Model. Int. J. Mol. Sci. 2022, 23, 10235. [Google Scholar] [CrossRef] [PubMed]
- Blecharz-Lang, K.G.; Wagner, J.; Fries, A.; Nieminen-Kelhä, M.; Rösner, J.; Schneider, U.C.; Vajkoczy, P. Interleukin 6-Mediated Endothelial Barrier Disturbances Can Be Attenuated by Blockade of the IL6 Receptor Expressed in Brain Microvascular Endothelial Cells. Transl. Stroke Res. 2018, 9, 631–642. [Google Scholar] [CrossRef]
- Lopez-Ramirez, M.A.; Fischer, R.; Torres-Badillo, C.C.; Davies, H.A.; Logan, K.; Pfizenmaier, K.; Male, D.K.; Sharrack, B.; Romero, I.A. Role of Caspases in Cytokine-Induced Barrier Breakdown in Human Brain Endothelial Cells. J. Immunol. 2012, 189, 3130–3139. [Google Scholar] [CrossRef]
- Veldhuis, W.B.; Floris, S.; Van der Meide, P.H.; Vos, I.M.P.; De Vries, H.E.; Dijkstra, C.D.; Bär, P.R.; Nicolay, K. Interferon-Beta Prevents Cytokine-Induced Neutrophil Infiltration and Attenuates Blood-Brain Barrier Disruption. J. Cereb. Blood Flow Metab. 2003, 23, 1060–1069. [Google Scholar] [CrossRef]
- Haase, A.T.; Stowring, L.; Narayan, O.; Griffin, D.; Price, D. Slow Persistent Infection Caused by Visna Virus: Role of Host Restriction. Science 1977, 195, 175–177. [Google Scholar] [CrossRef]
- Narayan, O.; Wolinsky, J.S.; Clements, J.E.; Strandberg, J.D.; Griffin, D.E.; Cork, L.C. Slow Virus Replication: The Role of Macrophages in the Persistence and Expression of Visna Viruses of Sheep and Goats. J. Gen. Virol. 1982, 59, 345–356. [Google Scholar] [CrossRef]
- Meltzer, M.S.; Skillman, D.R.; Gomatos, P.J.; Kalter, D.C.; Gendelman, H.E. Role of Mononuclear Phagocytes in the Pathogenesis of Human Immunodeficiency Virus Infection. Annu. Rev. Immunol. 1990, 8, 169–194. [Google Scholar] [CrossRef]
- Pulliam, L. Cytomegalovirus Preferentially Infects a Monocyte Derived Macrophage/Microglial Cell in Human Brain Cultures: Neuropathology Differs between Strains. J. Neuropathol. Exp. Neurol. 1991, 50, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Daley-Bauer, L.P.; Roback, L.J.; Wynn, G.M.; Mocarski, E.S. Cytomegalovirus Hijacks CX3CR1hi Patrolling Monocytes as Immune-Privileged Vehicles for Dissemination in Mice. Cell Host Microbe 2014, 15, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Percivalle, E.; Sammartino, J.C.; Cassaniti, I.; Arbustini, E.; Urtis, M.; Smirnova, A.; Concardi, M.; Belgiovine, C.; Ferrari, A.; Lilleri, D.; et al. Macrophages and Monocytes: “Trojan Horses” in COVID-19. Viruses 2021, 13, 2178. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; Hogue, I.B.; Enquist, L.W. Virus Infections in the Nervous System. Cell Host Microbe 2013, 13, 379. [Google Scholar] [CrossRef]
- Ayala-Nunez, N.V.; Gaudin, R. A Viral Journey to the Brain: Current Considerations and Future Developments. PLoS Pathog. 2020, 16, e1008434. [Google Scholar] [CrossRef] [PubMed]
- Rouse, B.T.; Sehrawat, S. Immunity and Immunopathology to Viruses: What Decides the Outcome? Nat. Rev. Immunol. 2010, 10, 514. [Google Scholar] [CrossRef]
- Bearer, E.L. HSV, Axonal Transport and Alzheimer’s Disease: In Vitro and in Vivo Evidence for Causal Relationships. Future Virol. 2012, 7, 885. [Google Scholar] [CrossRef] [PubMed]
- Esiri, M.M. Herpes Simplex Encephalitis. An Immunohistological Study of the Distribution of Viral Antigen within the Brain. J. Neurol. Sci. 1982, 54, 209–226. [Google Scholar] [CrossRef]
- McMahon, A.; Conrick-Martin, I. Commonly Encountered Central Nervous System Infections in the Intensive Care Unit. BJA Educ. 2023, 23, 212. [Google Scholar] [CrossRef]
- Doll, J.R.; Thompson, R.L.; Sawtell, N.M. Infectious Herpes Simplex Virus in the Brain Stem Is Correlated with Reactivation in the Trigeminal Ganglia. J. Virol. 2019, 93, e02209-18. [Google Scholar] [CrossRef] [PubMed]
- Mielcarska, M.B.; Skowrońska, K.; Wyżewski, Z.; Toka, F.N. Disrupting Neurons and Glial Cells Oneness in the Brain—The Possible Causal Role of Herpes Simplex Virus Type 1 (HSV-1) in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 23, 242. [Google Scholar] [CrossRef]
- Pahud, B.A.; Glaser, C.A.; Dekker, C.L.; Arvin, A.M.; Schmid, D.S. Varicella Zoster Disease of the Central Nervous System: Epidemiological, Clinical, and Laboratory Features 10 Years after the Introduction of the Varicella Vaccine. J. Infect. Dis. 2011, 203, 316. [Google Scholar] [CrossRef]
- Yakovenko, V.; Ciobotaro, P.; Bardenstein, R.; Zusev, M.; Zimhony, O. The Role of Varicella Zoster Virus (VZV) in Central Nervous System Infectious Syndromes. Can. J. Infect. Dis. Med. Microbiol. 2024, 2024, 6664417. [Google Scholar] [CrossRef]
- Liedtke, W.; Opalka, B.; Zimmermann, C.W.; Lignitz, E. Age Distribution of Latent Herpes Simplex Virus 1 and Varicella-Zoster Virus Genome in Human Nervous Tissue. J. Neurol. Sci. 1993, 116, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Hemling, N.; Röyttä, M.; Rinne, J.; Pöllänen, P.; Broberg, E.; Tapio, V.; Vahlberg, T.; Hukkanen, V. Herpesviruses in Brains in Alzheimer’s and Parkinson’s Diseases. Ann. Neurol. 2003, 54, 267–271. [Google Scholar] [CrossRef]
- Bubak, A.N.; Como, C.N.; Blackmon, A.M.; Jones, D.; Nagel, M.A. Varicella Zoster Virus Differentially Alters Morphology and Suppresses Proinflammatory Cytokines in Primary Human Spinal Cord and Hippocampal Astrocytes. J. Neuroinflammation 2018, 15, 318. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt-DeMasters, B.K.; Gilden, D.H. Varicella-Zoster Virus Infections of the Nervous SystemClinical and Pathologic Correlates. Arch. Pathol. Lab. Med. 2001, 125, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Chijioke, O.; Azzi, T.; Nadal, D.; Münz, C. Innate Immune Responses against Epstein Barr Virus Infection. J. Leukoc. Biol. 2013, 94, 1185. [Google Scholar] [CrossRef]
- Soldan, S.S.; Lieberman, P.M. Epstein-Barr Virus Infection in the Development of Neurological Disorders. Drug Discov. Today Dis. Models 2020, 32, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zuo, Y.; Jiang, L.; Peng, Y.; Huang, X.; Zuo, L. Epstein-Barr Virus and Neurological Diseases. Front. Mol. Biosci. 2022, 8, 816098. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L. The Enigmatic Links between Epstein-Barr Virus Infection and Multiple Sclerosis. J. Clin. Investig. 2022, 132, e160468. [Google Scholar] [CrossRef]
- Zheng, H.; Savitz, J. Effect of Cytomegalovirus Infection on the Central Nervous System: Implications for Psychiatric Disorders. Curr. Top. Behav. Neurosci. 2023, 61, 215–241. [Google Scholar] [CrossRef]
- Adelman, J.W.; Rosas-Rogers, S.; Schumacher, M.L.; Mokry, R.L.; Terhune, S.S.; Ebert, A.D. Human Cytomegalovirus Induces Significant Structural and Functional Changes in Terminally Differentiated Human Cortical Neurons. mBio 2023, 14, e02251-23. [Google Scholar] [CrossRef]
- Harberts, E.; Yao, K.; Wohler, J.E.; Maric, D.; Ohayon, J.; Henkin, R.; Jacobson, S. Human Herpesvirus-6 Entry into the Central Nervous System through the Olfactory Pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 13734. [Google Scholar] [CrossRef] [PubMed]
- Chapenko, S.; Roga, S.; Skuja, S.; Rasa, S.; Cistjakovs, M.; Svirskis, S.; Zaserska, Z.; Groma, V.; Murovska, M. Detection Frequency of Human Herpesviruses-6A, -6B, and -7 Genomic Sequences in Central Nervous System DNA Samples from Post-Mortem Individuals with Unspecified Encephalopathy. J. Neurovirol. 2016, 22, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Skuja, S.; Svirskis, S.; Murovska, M. Human Herpesvirus-6 and -7 in the Brain Microenvironment of Persons with Neurological Pathology and Healthy People. Int. J. Mol. Sci. 2021, 22, 2364. [Google Scholar] [CrossRef] [PubMed]
- Santpere, G.; Telford, M.; Andrés-Benito, P.; Navarro, A.; Ferrer, I. The Presence of Human Herpesvirus 6 in the Brain in Health and Disease. Biomolecules 2020, 10, 1520. [Google Scholar] [CrossRef]
- Bahramian, E.; Furr, M.; Wu, J.T.; Ceballos, R.M. Differential Impacts of HHV-6A versus HHV-6B Infection in Differentiated Human Neural Stem Cells. Front. Immunol. 2022, 13, 847106. [Google Scholar] [CrossRef]
- Bortolotti, D.; Gentili, V.; Rotola, A.; Caselli, E.; Rizzo, R. HHV-6A Infection Induces Amyloid-Beta Expression and Activation of Microglial Cells. Alzheimers Res. Ther. 2019, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Grut, V.; Biström, M.; Salzer, J.; Stridh, P.; Jons, D.; Gustafsson, R.; Fogdell-Hahn, A.; Huang, J.; Butt, J.; Lindam, A.; et al. Human Herpesvirus 6A and Axonal Injury before the Clinical Onset of Multiple Sclerosis. Brain 2024, 147, 177–185. [Google Scholar] [CrossRef]
- Poliomyelitis. Available online: https://www.who.int/news-room/fact-sheets/detail/poliomyelitis (accessed on 21 December 2024).
- Pavio, N.; Buc-Caron, M.H.; Colbère-Garapin, F. Persistent Poliovirus Infection of Human Fetal Brain Cells. J. Virol. 1996, 70, 6395. [Google Scholar] [CrossRef]
- Wang, W.; Sun, J.; Wang, N.; Sun, Z.; Ma, Q.; Li, J.; Zhang, M.; Xu, J. Enterovirus A71 Capsid Protein VP1 Increases Blood–Brain Barrier Permeability and Virus Receptor Vimentin on the Brain Endothelial Cells. J. Neurovirol. 2019, 26, 84. [Google Scholar] [CrossRef]
- Chang, L.Y.; Lin, H.Y.; Gau, S.S.F.; Lu, C.Y.; Hsia, S.H.; Huang, Y.C.; Huang, L.M.; Lin, T.Y. Enterovirus A71 Neurologic Complications and Long-Term Sequelae. J. Biomed. Sci. 2019, 26, 57. [Google Scholar] [CrossRef]
- Chang, L.Y.; Lin, T.Y.; Hsu, K.H.; Huang, Y.C.; Lin, K.L.; Hsueh, C.; Shih, S.R.; Ning, H.C.; Hwang, M.S.; Wang, H.S.; et al. Clinical Features and Risk Factors of Pulmonary Oedema after Enterovirus-71-Related Hand, Foot, and Mouth Disease. Lancet 1999, 354, 1682–1686. [Google Scholar] [CrossRef] [PubMed]
- Tee, H.K.; Zainol, M.I.; Sam, I.C.; Chan, Y.F. Recent Advances in the Understanding of Enterovirus A71 Infection: A Focus on Neuropathogenesis. Expert. Rev. Anti Infect. Ther. 2021, 19, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Liao, Y.; Gao, Y.; Jiang, G.; Wang, L.; Zhang, Y.; Fan, S.; Xu, X.; Li, Q. Mechanism for the Lethal Effect of Enterovirus A71 Intracerebral Injection in Neonatal Mice. Lab. Investig. 2019, 100, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Ji, W.; Li, D.; Wang, F.; Sun, T.; Yang, H.; Chen, S.; Zhang, W.; Jin, Y.; Duan, G. The Activation of Complement C5a-C5aR1 Axis in Astrocytes Facilitates the Neuropathogenesis Due to EV-A71 Infection by Upregulating CXCL1. J. Virol. 2024, e0151424. [Google Scholar] [CrossRef]
- Majde, J.A.; Bohnet, S.G.; Ellis, G.A.; Churchill, L.; Leyva-Grado, V.; Wu, M.; Szentirmai, E.; Rehman, A.; Krueger, J.M. Detection of Mouse-Adapted Human Influenza Virus in the Olfactory Bulbs of Mice within Hours after Intranasal Infection. J. Neurovirol. 2007, 13, 399–409. [Google Scholar] [CrossRef]
- Shinya, K.; Makino, A.; Hatta, M.; Watanabe, S.; Kim, J.H.; Hatta, Y.; Gao, P.; Ozawa, M.; Le, Q.M.; Kawaoka, Y. Subclinical Brain Injury Caused by H5N1 Influenza Virus Infection. J. Virol. 2011, 85, 5202. [Google Scholar] [CrossRef]
- Van Riel, D.; Leijten, L.M.; Verdijk, R.M.; GeurtsvanKessel, C.; Van Der Vries, E.; Van Rossum, A.M.C.; Osterhaus, A.D.M.E.; Kuiken, T. Evidence for Influenza Virus CNS Invasion along the Olfactory Route in an Immunocompromised Infant. J. Infect. Dis. 2014, 210, 419–423. [Google Scholar] [CrossRef]
- Siegers, J.Y.; van de Bildt, M.W.G.; Lin, Z.; Leijten, L.M.; Lavrijssen, R.A.M.; Bestebroer, T.; Spronken, M.I.J.; De Zeeuw, C.I.; Gao, Z.; Schrauwen, E.J.A.; et al. Viral Factors Important for Efficient Replication of Influenza A Viruses in Cells of the Central Nervous System. J. Virol. 2019, 93, e02273-18. [Google Scholar] [CrossRef]
- Kimura-Ohba, S.; Kitamura, M.; Tsukamoto, Y.; Kogaki, S.; Sakai, S.; Fushimi, H.; Matsuoka, K.; Takeuchi, M.; Itoh, K.; Ueda, K.; et al. Viral Entry and Translation in Brain Endothelia Provoke Influenza-Associated Encephalopathy. Acta Neuropathol. 2024, 147, 77. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly Pathogenic H5N1 Influenza Virus Can Enter the Central Nervous System and Induce Neuroinflammation and Neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.P.; Yip, T.F.; Peiris, J.S.M.; Ip, N.Y.; Lee, S.M.Y. Avian Influenza A H7N9 Virus Infects Human Astrocytes and Neuronal Cells and Induces Inflammatory Immune Responses. J. Neurovirol. 2018, 24, 752. [Google Scholar] [CrossRef] [PubMed]
- Libbey, J.E.; Fujinami, R.S. Adaptive Immune Response to Viral Infections in the Central Nervous System. Handb. Clin. Neurol. 2014, 123, 225–247. [Google Scholar] [CrossRef] [PubMed]
- Mumps. Available online: https://www.ecdc.europa.eu/en/mumps (accessed on 21 December 2024).
- Rubin, S.; Eckhaus, M.; Rennick, L.J.; Bamford, C.G.G.; Duprex, W.P. Molecular Biology, Pathogenesis and Pathology of Mumps Virus. J. Pathol. 2015, 235, 242. [Google Scholar] [CrossRef]
- Julkunen, I.; Lehtokoski-Lehtiniemi, E.; Koskiniemi, M.; Vaheri, A. Elevated Mumps Antibody Titers in the Cerebrospinal Fluid Suggesting Chronic Mumps Virus Infection in the Central Nervous System. Pediatr. Infect. Dis. 1985, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Haginoya, K.; Ike, K.; Linuma, K.; Yagi, T.; Kon, K.; Yokoyama, H.; Numazaki, Y. Chronic Progressive Mumps Virus Encephalitis in a Child. Lancet 1995, 346, 50. [Google Scholar] [CrossRef]
- Rubin, S.A.; Amexis, G.; Pletnikov, M.; Vanderzanden, J.; Mauldin, J.; Sauder, C.; Malik, T.; Chumakov, K.; Carbone, K.M. Changes in Mumps Virus Gene Sequence Associated with Variability in Neurovirulent Phenotype. J. Virol. 2003, 77, 11616. [Google Scholar] [CrossRef]
- Esolen, L.M.; Takahashi, K.; Johnson, R.T.; Vaisberg, A.; Moench, T.R.; Wesselingh, S.L.; Griffin, D.E. Brain Endothelial Cell Infection in Children with Acute Fatal Measles. J. Clin. Investig. 1995, 96, 2478. [Google Scholar] [CrossRef]
- Young, V.A.; Rall, G.F. Making It to the Synapse: Measles Virus Spread in and among Neurons. Curr. Top. Microbiol. Immunol. 2009, 330, 3. [Google Scholar] [CrossRef]
- Bonthius, D.J. Measles Virus and the Central Nervous System: An Update. Semin. Pediatr. Neurol. 2023, 47, 101078. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Schaulies, J.; Niewiesk, S.; Schneider-Schaulies, S.; Ter Meulen, V. Measles Virus in the CNS: The Role of Viral and Host Factors for the Establishment and Maintenance of a Persistent Infection. J. Neurovirol. 1999, 5, 613–622. [Google Scholar] [CrossRef]
- Dando, S.J.; Mackay-Sim, A.; Norton, R.; Currie, B.J.; St. John, J.A.; Ekberg, J.A.K.; Batzloff, M.; Ulett, G.C.; Beacham, I.R. Pathogens Penetrating the Central Nervous System: Infection Pathways and the Cellular and Molecular Mechanisms of Invasion. Clin. Microbiol. Rev. 2014, 27, 691. [Google Scholar] [CrossRef]
- Zacks, M.A.; Paessler, S. Encephalitic alphaviruses. Vet. Microbiol. 2009, 140, 281. [Google Scholar] [CrossRef]
- Wong, K.T.; Robertson, T.; Ong, B.B.; Chong, J.W.; Yaiw, K.C.; Wang, L.F.; Ansford, A.J.; Tannenberg, A. Human Hendra Virus Infection Causes Acute and Relapsing Encephalitis. Neuropathol. Appl. Neurobiol. 2009, 35, 296–305. [Google Scholar] [CrossRef]
- Meier, K.; Olejnik, J.; Hume, A.J.; Mühlberger, E. A Comparative Assessment of the Pathogenic Potential of Newly Discovered Henipaviruses. Pathogens 2024, 13, 587. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Alejandro, B.; Hetman, M.; Hattab, E.M.; Joiner, J.; Schroten, H.; Ishikawa, H.; Chung, D.H. Zika Virus Infects Pericytes in the Choroid Plexus and Enters the Central Nervous System through the Blood-Cerebrospinal Fluid Barrier. PLoS Pathog. 2020, 16, e1008204. [Google Scholar] [CrossRef] [PubMed]
- Danes, L.; Kufner, J.; Hrusková, V.; Rychterova, V. The Role of the Olfactory Route on Infection of the Respiratory Tract with Venezuelan Equine Encephalomyelitis Virus in Normal and Operated Macaca Rhesus Monkeys. I. Results of Virological Examination. Available online: https://pubmed.ncbi.nlm.nih.gov/4405396/ (accessed on 21 December 2024).
- Ma, H.; Albe, J.R.; Gilliland, T.; McMillen, C.M.; Gardner, C.L.; Boyles, D.A.; Cottle, E.L.; Dunn, M.D.; Lundy, J.D.; Salama, N.; et al. Long-Term Persistence of Viral RNA and Inflammation in the CNS of Macaques Exposed to Aerosolized Venezuelan Equine Encephalitis Virus. PLoS Pathog. 2022, 18, e1009946. [Google Scholar] [CrossRef] [PubMed]
- Bantle, C.M.; Phillips, A.T.; Smeyne, R.J.; Rocha, S.M.; Olson, K.E.; Tjalkens, R.B. Infection with Mosquito-Borne Alphavirus Induces Selective Loss of Dopaminergic Neurons, Neuroinflammation and Widespread Protein Aggregation. NPJ Park. Dis. 2019, 5, 20. [Google Scholar] [CrossRef]
- Singer, E.J.; Valdes-Sueiras, M.; Commins, D.; Levine, A. Neurologic Presentations of AIDS. Neurol. Clin. 2010, 28, 253. [Google Scholar] [CrossRef] [PubMed]
- Castellano, P.; Prevedel, L.; Eugenin, E.A. HIV-Infected Macrophages and Microglia That Survive Acute Infection Become Viral Reservoirs by a Mechanism Involving Bim. Sci. Rep. 2017, 7, 12866. [Google Scholar] [CrossRef]
- Pattanaik, A.; Bhandarkar, B.S.; Lodha, L.; Marate, S. SARS-CoV-2 and the Nervous System: Current Perspectives. Arch. Virol. 2023, 168, 171. [Google Scholar] [CrossRef]
- Haverty, R.; McCormack, J.; Evans, C.; Purves, K.; O’Reilly, S.; Gautier, V.; Rochfort, K.; Fabre, A.; Fletcher, N.F. SARS-CoV-2 Infects Neurons, Astrocytes, Choroid Plexus Epithelial Cells and Pericytes of the Human Central Nervous System. J. Gen. Virol. 2024, 105, 002009. [Google Scholar] [CrossRef]
- Steardo, L.; Steardo, L.; Zorec, R.; Verkhratsky, A. Neuroinfection May Contribute to Pathophysiology and Clinical Manifestations of COVID-19. Acta Physiol. 2020, 229, e13473. [Google Scholar] [CrossRef] [PubMed]
- Kist, M.; Vucic, D. Cell Death Pathways: Intricate Connections and Disease Implications. EMBO J. 2021, 40, e106700. [Google Scholar] [CrossRef] [PubMed]
- Said, S.; Kang, M. Viral Encephalitis; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Houston, F.; Andréoletti, O. Animal Prion Diseases: The Risks to Human Health. Brain Pathol. 2019, 29, 248. [Google Scholar] [CrossRef] [PubMed]
- Bogovic, P.; Strle, F. Tick-Borne Encephalitis: A Review of Epidemiology, Clinical Characteristics, and Management. World J. Clin. Cases WJCC 2015, 3, 430. [Google Scholar] [CrossRef]
- Counotte, M.J.; Kim, C.R.; Wang, J.; Bernstein, K.; Deal, C.D.; Broutet, N.J.N.; Low, N. Sexual Transmission of Zika Virus and Other Flaviviruses: A Living Systematic Review. PLoS Med. 2018, 15, e1002611. [Google Scholar] [CrossRef]
- Simon, L.V.; Sandhu, D.S.; Goyal, A.; Kruse, B. Japanese Encephalitis; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Fares, M.; Cochet-Bernoin, M.; Gonzalez, G.; Montero-Menei, C.N.; Blanchet, O.; Benchoua, A.; Boissart, C.; Lecollinet, S.; Richardson, J.; Haddad, N.; et al. Pathological Modeling of TBEV Infection Reveals Differential Innate Immune Responses in Human Neurons and Astrocytes That Correlate with Their Susceptibility to Infection. J. Neuroinflammation 2020, 17, 76. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, A.-B.; Saiz, J.-C. Neurological Manifestations of Zika Virus Infection. World J. Virol. 2016, 5, 135. [Google Scholar] [CrossRef]
- Sips, G.J.; Wilschut, J.; Smit, J.M. Neuroinvasive Flavivirus Infections. Rev. Med. Virol. 2012, 22, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Turtle, L.; Solomon, T. Japanese Encephalitis-the Prospects for New Treatments. Nat. Rev. Neurol. 2018, 14, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R.; Roehrig, J.T.; Hughes, J.M. West Nile Virus Encephalitis. N. Engl. J. Med. 2002, 347, 1225–1226. [Google Scholar] [CrossRef] [PubMed]
- Bogovič, P.; Lusa, L.; Korva, M.; Pavletič, M.; Rus, K.R.; Lotrič-Furlan, S.; Avšič-županc, T.; Strle, K.; Strle, F. Inflammatory Immune Responses in the Pathogenesis of Tick-Borne Encephalitis. J. Clin. Med. 2019, 8, 731. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.M.; Thao, T.T.N.; Duy, N.M.; Nhat, T.M.; Clapham, H.E. Estimates of the Global Burden of Japanese Encephalitis and the Impact of Vaccination from 2000–2015. Elife 2020, 9, e51027. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.B.; Schaefer, T.J. West Nile Virus; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chiffi, G.; Grandgirard, D.; Leib, S.L.; Chrdle, A.; Růžek, D. Tick-Borne Encephalitis: A Comprehensive Review of the Epidemiology, Virology, and Clinical Picture. Rev. Med. Virol. 2023, 33, e2470. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, L.S.; Parra, B.; Pardo, C.A. Neurological Implications of Zika Virus Infection in Adults. J. Infect. Dis. 2017, 216, S897. [Google Scholar] [CrossRef]
- Fischer, M.; Hills, S.; Staples, E.; Johnson, B.; Yaich, M.; Solomon, T. Japanese Encephalitis Prevention and Control: Advances, Challenges, and New Initiatives. In Emerging Infections 8; Wiley Online Library: Minneapolis, MN, USA, 2014; pp. 93–124. [Google Scholar] [CrossRef]
- Clinical Signs and Symptoms of West Nile Virus Disease|West Nile Virus|CDC. Available online: https://www.cdc.gov/west-nile-virus/hcp/clinical-signs/index.html (accessed on 21 December 2024).
- Gyure, K.A. West Nile Virus Infections. J. Neuropathol. Exp. Neurol. 2009, 68, 1053–1060. [Google Scholar] [CrossRef]
- Mandl, C.W. Steps of the Tick-Borne Encephalitis Virus Replication Cycle That Affect Neuropathogenesis. Virus Res. 2005, 111, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, E.; Preusser, M.; Garzuly, F.; Holzmann, H.; Heinz, F.X.; Budka, H. Visualization of Central European Tick-Borne Encephalitis Infection in Fatal Human Cases. J. Neuropathol. Exp. Neurol. 2005, 64, 506–512. [Google Scholar] [CrossRef]
- Kuwayama, M.; Ito, M.; Takao, S.; Shimazu, Y.; Fukuda, S.; Miyazaki, K.; Kurane, I.; Takasaki, T. Japanese Encephalitis Virus in Meningitis Patients, Japan. Emerg. Infect. Dis. 2005, 11, 471. [Google Scholar] [CrossRef]
- Solomon, T.; Kneen, R.; Dung, N.M.; Khanh, V.C.; Thuy, T.T.N.; Ha, D.Q.; Day, N.P.J.; Nisalak, A.; Vaughn, D.W.; White, N.J. Poliomyelitis-like Illness Due to Japanese Encephalitis Virus. Lancet 1998, 351, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Dung, N.M.; Kneen, R.; Gainsborough, M.; Vaughn, D.W.; Khanh, V.T. Japanese Encephalitis. J. Neurol. Neurosurg. Psychiatry 2000, 68, 405. [Google Scholar] [CrossRef]
- Ghosh, D.; Basu, A. Japanese Encephalitis—A Pathological and Clinical Perspective. PLoS Negl. Trop. Dis. 2009, 3, e437. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.Y.; Zhang, Y.H.; Tan, Z.R.; Huang, J.; Zhao, Y.W. Guillain-Barré Syndrome Associated with Japanese Encephalitis Virus Infection in China. Viral Immunol. 2014, 27, 418–420. [Google Scholar] [CrossRef]
- Liu, S.; Wang, J.; Yang, J.; Wen, Y. The Underlying Mechanism of Guillain-Barré Syndrome in a Young Patient Suffered from Japanese Encephalitis Virus Infection: A Case Report. Virol. J. 2022, 19, 139. [Google Scholar] [CrossRef] [PubMed]
- Sejvar, J.J. Clinical Manifestations and Outcomes of West Nile Virus Infection. Viruses 2014, 6, 606. [Google Scholar] [CrossRef] [PubMed]
- Beshai, R.; Bibawy, D.; Bibawy, J. Guillain–Barré Syndrome Secondary to West Nile Virus in New York City. Case Rep. Infect. Dis. 2020, 2020, 6501658. [Google Scholar] [CrossRef]
- Sejvar, J.J.; Bode, A.V.; Marfin, A.A.; Campbell, G.L.; Pape, J.; Biggerstaff, B.J.; Petersen, L.R. West Nile Virus–Associated Flaccid Paralysis Outcome. Emerg. Infect. Dis. 2006, 12, 514. [Google Scholar] [CrossRef]
- Klee, A.L.; Maldin, B.; Edwin, B.; Poshni, I.; Mostashari, F.; Fine, A.; Layton, M.; Nash, D. Long-Term Prognosis for Clinical West Nile Virus Infection. Emerg. Infect. Dis. 2004, 10, 1405. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.O.; Resnick, M.; Miller, V. Depression after Infection with West Nile Virus. Emerg. Infect. Dis. 2007, 13, 479. [Google Scholar] [CrossRef]
- Carson, P.J.; Konewko, P.; Wold, K.S.; Mariani, P.; Goli, S.; Bergloff, P.; Crosby, R.D. Long-Term Clinical and Neuropsychological Outcomes of West Nile Virus Infection. Clin. Infect. Dis. 2006, 43, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, J.G.; Vachalová, I.; Großkopf, J. Tick-Borne Encephalitis Complicated by Guillain-Barré Syndrome. Acta Neurol. Belg. 2019, 119, 649–651. [Google Scholar] [CrossRef]
- Gritsun, T.S.; Lashkevich, V.A.; Gould, E.A. Tick-Borne Encephalitis. Antivir. Res. 2003, 57, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Ampudia, Y.; Monsalve, D.M.; Castillo-Medina, L.F.; Rodríguez, Y.; Pacheco, Y.; Halstead, S.; Willison, H.J.; Anaya, J.M.; Ramírez-Santana, C. Autoimmune Neurological Conditions Associated with Zika Virus Infection. Front. Mol. Neurosci. 2018, 11, 116. [Google Scholar] [CrossRef]
- Kumar, S.; Verma, A.; Yadav, P.; Dubey, S.K.; Azhar, E.I.; Maitra, S.S.; Dwivedi, V.D. Molecular Pathogenesis of Japanese Encephalitis and Possible Therapeutic Strategies. Arch. Virol. 2022, 167, 1739–1762. [Google Scholar] [CrossRef]
- Yadav, P.; Chakraborty, P.; Jha, N.K.; Dewanjee, S.; Jha, A.K.; Panda, S.P.; Mishra, P.C.; Dey, A.; Jha, S.K. Molecular Mechanism and Role of Japanese Encephalitis Virus Infection in Central Nervous System-Mediated Diseases. Viruses 2022, 14, 2686. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, S.; Chakravarty, A. Neurological Complications of Dengue Fever. Curr. Neurol. Neurosci. Rep. 2022, 22, 515–529. [Google Scholar] [CrossRef]
- Pustijanac, E.; Buršić, M.; Talapko, J.; Škrlec, I.; Meštrović, T.; Lišnjić, D. Tick-Borne Encephalitis Virus: A Comprehensive Review of Transmission, Pathogenesis, Epidemiology, Clinical Manifestations, Diagnosis, and Prevention. Microorganisms 2023, 11, 1634. [Google Scholar] [CrossRef]
- Sharma, S.; Mathur, A.; Prakash, V.; Kulshreshtha, R.; Kumar, R.; Chaturvedi, U.C. Japanese Encephalitis Virus Latency in Peripheral Blood Lymphocytes and Recurrence of Infection in Children. Clin. Exp. Immunol. 1991, 85, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Patabendige, A.; Michael, B.D.; Craig, A.G.; Solomon, T. Brain Microvascular Endothelial-Astrocyte Cell Responses Following Japanese Encephalitis Virus Infection in an in Vitro Human Blood-Brain Barrier Model. Mol. Cell Neurosci. 2018, 89, 60–70. [Google Scholar] [CrossRef]
- Filgueira, L.; Lannes, N. Review of Emerging Japanese Encephalitis Virus: New Aspects and Concepts about Entry into the Brain and Inter-Cellular Spreading. Pathogens 2019, 8, 111. [Google Scholar] [CrossRef]
- Cheeran, M.C.J.; Hu, S.; Sheng, W.S.; Rashid, A.; Peterson, P.K.; Lokensgard, J.R. Differential Responses of Human Brain Cells to West Nile Virus Infection. J. Neurovirol. 2005, 11, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Gottlieb, D.; Diamond, M.S. Infection and Injury of Neurons by West Nile Encephalitis Virus. J. Virol. 2003, 77, 13203. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kumar, M.; Gurjav, U.; Lum, S.; Nerurkar, V.R. Reversal of West Nile Virus-Induced Blood-Brain Barrier Disruption and Tight Junction Proteins Degradation by Matrix Metalloproteinases Inhibitor. Virology 2010, 397, 130. [Google Scholar] [CrossRef] [PubMed]
- Constant, O.; Maarifi, G.; Barthelemy, J.; Martin, M.F.; Tinto, B.; Savini, G.; Van de Perre, P.; Nisole, S.; Simonin, Y.; Salinas, S. Differential Effects of Usutu and West Nile Viruses on Neuroinflammation, Immune Cell Recruitment and Blood–Brain Barrier Integrity. Emerg. Microbes Infect. 2023, 12, 2156815. [Google Scholar] [CrossRef]
- Ghita, L.; Breitkopf, V.; Mulenge, F.; Pavlou, A.; Gern, O.L.; Durán, V.; Prajeeth, C.K.; Kohls, M.; Jung, K.; Stangel, M.; et al. Sequential MAVS and MyD88/TRIF Signaling Triggers Anti-Viral Responses of Tick-Borne Encephalitis Virus-Infected Murine Astrocytes. J. Neurosci. Res. 2021, 99, 2478–2492. [Google Scholar] [CrossRef]
- Pranclova, V.; Nedvedova, L.; Kotounova, E.; Vaclav, H.; Dvorakova, M.; Davidkova, M.; Bily, T.; Vancova, M.; Ruzek, D.; Palus, M. Unraveling the Role of Human Microglia in Tick-Borne Encephalitis Virus Infection: Insights into Neuroinflammation and Viral Pathogenesis. Microbes Infect. 2024, 26, 105383. [Google Scholar] [CrossRef]
- Schultz, V.; Barrie, J.A.; Donald, C.L.; Crawford, C.L.; Mullin, M.; Anderson, T.J.; Solomon, T.; Barnett, S.C.; Linington, C.; Kohl, A.; et al. Oligodendrocytes Are Susceptible to Zika Virus Infection in a Mouse Model of Perinatal Exposure: Implications for CNS Complications. Glia 2021, 69, 2023–2036. [Google Scholar] [CrossRef]
- Mutso, M.; John, J.A.S.; Ling, Z.L.; Burt, F.J.; Poo, Y.S.; Liu, X.; Žusinaite, E.; Grau, G.E.; Hueston, L.; Merits, A.; et al. Basic Insights into Zika Virus Infection of Neuroglial and Brain Endothelial Cells. J. Gen. Virol. 2020, 101, 622–634. [Google Scholar] [CrossRef]
- Ferraris, P.; Cochet, M.; Hamel, R.; Gladwyn-Ng, I.; Alfano, C.; Diop, F.; Garcia, D.; Talignani, L.; Montero-Menei, C.N.; Nougairède, A.; et al. Zika Virus Differentially Infects Human Neural Progenitor Cells According to Their State of Differentiation and Dysregulates Neurogenesis through the Notch Pathway. Emerg. Microbes Infect. 2019, 8, 1003. [Google Scholar] [CrossRef]
- Garcez, P.P.; Nascimento, J.M.; De Vasconcelos, J.M.; Madeiro Da Costa, R.; Delvecchio, R.; Trindade, P.; Loiola, E.C.; Higa, L.M.; Cassoli, J.S.; Vitória, G.; et al. Zika Virus Disrupts Molecular Fingerprinting of Human Neurospheres. Sci. Rep. 2017, 7, srep40780. [Google Scholar] [CrossRef] [PubMed]
- Thongtan, T.; Thepparit, C.; Smith, D.R. The Involvement of Microglial Cells in Japanese Encephalitis Infections. J. Immunol. Res. 2012, 2012, 890586. [Google Scholar] [CrossRef] [PubMed]
- Thongtan, T.; Cheepsunthorn, P.; Chaiworakul, V.; Rattanarungsan, C.; Wikan, N.; Smith, D.R. Highly Permissive Infection of Microglial Cells by Japanese Encephalitis Virus: A Possible Role as a Viral Reservoir. Microbes Infect. 2010, 12, 37–45. [Google Scholar] [CrossRef]
- Bai, F.; Kong, K.F.; Dai, J.; Qian, F.; Zhang, L.; Brown, C.R.; Fikrig, E.; Montgomery, R.R. A Paradoxical Role for Neutrophils in the Pathogenesis of West Nile Virus. J. Infect. Dis. 2010, 202, 1804. [Google Scholar] [CrossRef]
- Mladinich, M.C.; Schwedes, J.; Mackow, E.R. Zika Virus Persistently Infects and Is Basolaterally Released from Primary Human Brain Microvascular Endothelial Cells. mBio 2017, 8, e00952-17. [Google Scholar] [CrossRef]
- Ravi, V.; Desai, A.S.; Shenoy, P.K.; Satishchandra, P.; Chandramuki, A.; Gourie-Devi, M. Persistence of Japanese Encephalitis Virus in the Human Nervous System. J. Med. Virol. 1993, 40, 326–329. [Google Scholar] [CrossRef]
- Murray, K.; Walker, C.; Herrington, E.; Lewis, J.A.; McCormick, J.; Beasley, D.W.C.; Tesh, R.B.; Fisher-Hoch, S. Persistent Infection with West Nile Virus Years after Initial Infection. J. Infect. Dis. 2010, 201, 2. [Google Scholar] [CrossRef]
- Nicholls, C.M.R.; Sevvana, M.; Kuhn, R.J. Structure-Guided Paradigm Shifts in Flavivirus Assembly and Maturation Mechanisms. Adv. Virus Res. 2020, 108, 33. [Google Scholar] [CrossRef]
- Chowdhury, P.; Khan, S.A. Global Emergence of West Nile Virus: Threat & Preparedness in Special Perspective to India. Indian J. Med. Res. 2021, 154, 36. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Dai, X. The Global Incidence and Trends of Three Common Flavivirus Infections (Dengue, Yellow Fever, and Zika) from 2011 to 2021. Front. Microbiol. 2024, 15, 1458166. [Google Scholar] [CrossRef]
- Wang, H.; Liang, G. Epidemiology of Japanese Encephalitis: Past, Present, and Future Prospects. Ther. Clin. Risk Manag. 2015, 11, 435–448. [Google Scholar] [CrossRef]
- Gole, S.; Anand, A. Autoimmune Encephalitis; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Lancaster, E. Autoantibody Encephalitis: Presentation, Diagnosis, and Management. J. Clin. Neurol. 2022, 18, 373. [Google Scholar] [CrossRef]
- Zang, Q.; Wang, Y.; Guo, J.; Long, L.; Zhang, S.; Cui, C.; Song, D.; Yu, B.; Tang, F.; Teng, J.; et al. Treatment of Severe Japanese Encephalitis Complicated with Hashimoto’s Thyroiditis and Guillain-Barré Syndrome with Protein A Immunoadsorption: A Case Report. Front. Immunol. 2022, 12, 807937. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, P.; Alexopoulos, H.; Sourdi, A.; Papadimitriou, D.; Dimitrakopoulos, A.N.; Moutsopoulos, H.M. West Nile Virus Infection Triggering Autoimmune Encephalitis: Pathophysiological and Therapeutic Implications. Clin. Immunol. 2019, 207, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, A.C.; Foris, L.A.; Tadi, P. Acute Disseminated Encephalomyelitis; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Kamel, M.G.; Nam, N.T.; Han, N.H.B.; El-Shabouny, A.E.; Makram, A.E.R.M.; Abd-Elhay, F.A.E.; Dang, T.N.; Hieu, N.L.T.; Huong, V.T.Q.; Tung, T.H.; et al. Post-Dengue Acute Disseminated Encephalomyelitis: A Case Report and Meta-Analysis. PLoS Negl. Trop. Dis. 2017, 11, e0005715. [Google Scholar] [CrossRef]
- Karoli, R.; Siddiqi, Z.; Fatima, J.; Maini, S. Was It a Case of Acute Disseminated Encephalomyelitis? A Rare Association Following Dengue Fever. J. Neurosci. Rural. Pract. 2013, 4, 318–321. [Google Scholar] [CrossRef]
- Alves-Leon, S.V.; Lima, M.D.R.; Nunes, P.C.G.; Chimelli, L.M.C.; Rabelo, K.; Nogueira, R.M.R.; De Bruycker-Nogueira, F.; De Azeredo, E.L.; Bahia, P.R.; Rueda Lopes, F.C.; et al. Zika Virus Found in Brain Tissue of a Multiple Sclerosis Patient Undergoing an Acute Disseminated Encephalomyelitis-like Episode. Mult. Scler. 2019, 25, 427–430. [Google Scholar] [CrossRef]
- Ohtaki, E.; Matsuishi, T.; Hirano, Y.; Maekawa, K.; Ohtaki, J.E.; Matsuishi, T.; Hirano, Y. Acute Disseminated Encephalomyelitis after Treatment with Japanese B Encephalitis Vaccine (Nakayama-Yoken and Beijing Strains). J. Neurol. Neurosurg. Psychiatry 1995, 59, 316. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.P.; Taylor, R.S. Guillain-Barre Syndrome; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Lim, C.S.; Kaisbain, N.; Lim, W.J. A Rare Combination: Dengue Fever Complicated with Guillain-Barre Syndrome. Cureus 2023, 15, e40957. [Google Scholar] [CrossRef]
- Barbi, L.; Coelho, A.V.C.; de Alencar, L.C.A.; Crovella, S. Prevalence of Guillain-Barré Syndrome among Zika Virus Infected Cases: A Systematic Review and Meta-Analysis. Braz. J. Infect. Dis. 2018, 22, 137–141. [Google Scholar] [CrossRef]
- Basumatary, L.J.; Raja, D.; Bhuyan, D.; Das, M.; Goswami, M.; Kayal, A.K. Clinical and Radiological Spectrum of Japanese Encephalitis. J. Neurol. Sci. 2013, 325, 15–21. [Google Scholar] [CrossRef]
- Shrimanker, I.; Tadi, P.; Schoo, C.; Sánchez-Manso, J.C. Parkinsonism. In Encyclopedia of the Neurological Sciences; Academic Press: Cambridge, MA, USA, 2024; pp. 820–823. [Google Scholar] [CrossRef]
- Poponnikova, T.V. The Clinical Picture of Chronic Tick-Borne Encephalitis in Children. Int. J. Med. Microbiol. 2008, 298, 351–355. [Google Scholar] [CrossRef]
- Fulton, C.D.M.; Beasley, D.W.C.; Bente, D.A.; Dineley, K.T. Long-Term, West Nile Virus-Induced Neurological Changes: A Comparison of Patients and Rodent Models. Brain Behav. Immun. Health 2020, 7, 100105. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.C.; Chou, H.P.; Chiang, Y.C.; Chang, R.; Chen, Y.S.; Juan, Y.C. Neurological or Psychiatric Disorders After Dengue Fever. JAMA Netw. Open 2024, 7, E2410075. [Google Scholar] [CrossRef] [PubMed]
- Rico-Hesse, R. Microevolution and Virulence of Dengue Viruses. Adv. Virus Res. 2003, 59, 315–341. [Google Scholar] [CrossRef]
- Kakde, U.; Khatib, M.N. Neurological Complications in Dengue Among Males of the Adult Age Group. Cureus 2024, 16, e51586. [Google Scholar] [CrossRef]
- Ireland, D.D.C.; Manangeeswaran, M.; Lewkowicz, A.P.; Engel, K.; Clark, S.M.; Laniyan, A.; Sykes, J.; Lee, H.N.; McWilliams, I.L.; Kelley-Baker, L.; et al. Long-Term Persistence of Infectious Zika Virus: Inflammation and Behavioral Sequela in Mice. PLoS Pathog. 2020, 16, e1008689. [Google Scholar] [CrossRef]
- Swinkels, H.M.; Vaillant, A.A.J.; Nguyen, A.D.; Gulick, P.G. HIV and AIDS. In Geriatric Gastroenterology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2024; pp. 659–666. [Google Scholar] [CrossRef]
- Campbell, J.H.; Hearps, A.C.; Martin, G.E.; Williams, K.C.; Crowe, S.M. The Importance of Monocytes and Macrophages in HIV Pathogenesis, Treatment, and Cure. AIDS 2014, 28, 2175. [Google Scholar] [CrossRef]
- Subra, C.; Trautmann, L. Role of T Lymphocytes in HIV Neuropathogenesis. Curr. HIV/AIDS Rep. 2019, 16, 236. [Google Scholar] [CrossRef]
- Kincer, L.P.; Schnell, G.; Swanstrom, R.; Miller, M.B.; Spudich, S.; Eron, J.J.; Price, R.W.; Joseph, S.B. HIV-1 Is Transported into the Central Nervous System by Trafficking Infected Cells. Pathog. Immun. 2023, 7, 131. [Google Scholar] [CrossRef]
- Gras, G.; Kaul, M. Molecular Mechanisms of Neuroinvasion by Monocytes-Macrophages in HIV-1 Infection. Retrovirology 2010, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Burdo, T.H.; Lackner, A.; Williams, K.C. Monocyte/Macrophages and Their Role in HIV Neuropathogenesis. Immunol. Rev. 2013, 254, 102. [Google Scholar] [CrossRef] [PubMed]
- Cenker, J.J.; Stultz, R.D.; McDonald, D. Brain Microglial Cells Are Highly Susceptible to HIV-1 Infection and Spread. AIDS Res. Hum. Retroviruses 2017, 33, 1155. [Google Scholar] [CrossRef]
- Schnell, G.; Joseph, S.; Spudich, S.; Price, R.W.; Swanstrom, R. HIV-1 Replication in the Central Nervous System Occurs in Two Distinct Cell Types. PLoS Pathog. 2011, 7, e1002286. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Wesselingh, S.L.; Cowley, D.; Pardo, C.A.; McArthur, J.C.; Brew, B.J.; Gorry, P.R. Extensive Astrocyte Infection Is Prominent in Human Immunodeficiency Virus-Associated Dementia. Ann. Neurol. 2009, 66, 253–258. [Google Scholar] [CrossRef]
- Kaalund, S.S.; Johansen, A.; Fabricius, K.; Pakkenberg, B. Untreated Patients Dying with AIDS Have Loss of Neocortical Neurons and Glia Cells. Front. Neurosci. 2020, 13, 507164. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Berman, J.W. Cytochrome c Dysregulation Induced by HIV Infection of Astrocytes Results in Bystander Apoptosis of Uninfected Astrocytes by an IP3 and Calcium-Dependent Mechanism. J. Neurochem. 2013, 127, 644. [Google Scholar] [CrossRef]
- Ash, M.K.; Al-Harthi, L.; Schneider, J.R. HIV in the Brain: Identifying Viral Reservoirs and Addressing the Challenges of an HIV Cure. Vaccines 2021, 9, 867. [Google Scholar] [CrossRef]
- Malik, S.; Eugenin, E.A. Mechanisms of HIV Neuropathogenesis: Role of Cellular Communication Systems. Curr. HIV Res. 2016, 14, 400. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, N.; Chomont, N.; Estaquier, J.; Cohen, E.A.; Power, C. Is the Central Nervous System Reservoir a Hurdle for an HIV Cure? Viruses 2023, 15, 2385. [Google Scholar] [CrossRef]
- Elendu, C.; Aguocha, C.M.; Okeke, C.V.; Okoro, C.B.; Peterson, J.C. HIV-Related Neurocognitive Disorders: Diagnosis, Treatment, and Mental Health Implications: A Review. Medicine 2023, 102, E35652. [Google Scholar] [CrossRef]
- Chan, P.; Valcour, V. Neurocognition and the Aging Brain in People with HIV: Implications for Screening. Top. Antivir. Med. 2021, 29, 423. [Google Scholar]
- Marquine, M.J.; Umlauf, A.; Rooney, A.S.; Fazeli, P.L.; Gouaux, B.D.; Woods, S.P.; Letendre, S.L.; Ellis, R.J.; Grant, I.; Moore, D.J. The Veterans Aging Cohort Study (VACS) Index Is Associated with Concurrent Risk for Neurocognitive Impairment. J. Acquir. Immune Defic. Syndr. 2014, 65, 190. [Google Scholar] [CrossRef]
- Daliparty, V.M.; Balasubramanya, R. HIV Encephalitis. In Neuroinfections, 1st ed.; Gilden, D., Ed.; Oxford University Press: Oxford, UK, 2022; pp. 23–26. [Google Scholar] [CrossRef]
- Kaul, M.; Lipton, S.A. Chemokines and Activated Macrophages in HIV Gp120-Induced Neuronal Apoptosis. Proc. Natl. Acad. Sci. USA 1999, 96, 8212. [Google Scholar] [CrossRef] [PubMed]
- Kovalevich, J.; Langford, D. Neuronal Toxicity in HIV CNS Disease. Future Virol. 2012, 7, 687. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xie, J.; Dutta, D.; Xiong, H. HIV-1 Envelope Protein Gp120 Modulation of Glutamate Effects on Cortical Neuronal Synapses: Implications for HIV-1-Associated Neuropathogenesis. Int. J. Physiol. Pathophysiol. Pharmacol. 2023, 15, 75. [Google Scholar]
- Zhou, L.; Saksena, N.K. HIV Associated Neurocognitive Disorders. Infect. Dis. Rep. 2013, 5, 38–50. [Google Scholar] [CrossRef]
- Ben Haij, N.; Planès, R.; Leghmari, K.; Serrero, M.; Delobel, P.; Izopet, J.; BenMohamed, L.; Bahraoui, E. HIV-1 Tat Protein Induces Production of Proinflammatory Cytokines by Human Dendritic Cells and Monocytes/Macrophages through Engagement of TLR4-MD2-CD14 Complex and Activation of NF-ΚB Pathway. PLoS ONE 2015, 10, 129425. [Google Scholar] [CrossRef]
- Periyasamy, P.; Thangaraj, A.; Bendi, V.S.; Buch, S. HIV-1 Tat-Mediated Microglial Inflammation Involves a Novel MiRNA-34a-NLRC5-NFκB Signaling Axis. Brain Behav. Immun. 2019, 80, 227. [Google Scholar] [CrossRef]
- Ru, W.; Liu, X.; Bae, C.; Shi, Y.; Walikonis, R.; Chung, J.M.; Tang, S.J. Microglia Mediate HIV-1 Gp120-Induced Synaptic Degeneration in Spinal Pain Neural Circuits. J. Neurosci. 2019, 39, 8408. [Google Scholar] [CrossRef]
- Mangino, G.; Percario, Z.A.; Fiorucci, G.; Vaccari, G.; Acconcia, F.; Chiarabelli, C.; Leone, S.; Noto, A.; Horenkamp, F.A.; Manrique, S.; et al. HIV-1 Nef Induces Proinflammatory State in Macrophages through Its Acidic Cluster Domain: Involvement of TNF Alpha Receptor Associated Factor 2. PLoS ONE 2011, 6, e22982. [Google Scholar] [CrossRef]
- Wilson, K.M.; He, J.J. HIV Nef Expression Down-Modulated GFAP Expression and Altered Glutamate Uptake and Release and Proliferation in Astrocytes. Aging Dis. 2023, 14, 152. [Google Scholar] [CrossRef]
- Li, G.; Makar, T.; Gerzanich, V.; Kalakonda, S.; Ivanova, S.; Pereira, E.F.R.; Andharvarapu, S.; Zhang, J.; Marc Simard, J.; Zhao, R.Y. HIV-1 Vpr-Induced Proinflammatory Response and Apoptosis Are Mediated through the Sur1-Trpm4 Channel in Astrocytes. mBio 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Thangaraj, A.; Periyasamy, P.; Liao, K.; Bendi, V.S.; Callen, S.; Pendyala, G.; Buch, S. HIV-1 TAT-Mediated Microglial Activation: Role of Mitochondrial Dysfunction and Defective Mitophagy. Autophagy 2018, 14, 1596. [Google Scholar] [CrossRef]
- Dubrovsky, L.; Brichacek, B.; Prashant, N.M.; Pushkarsky, T.; Mukhamedova, N.; Fleetwood, A.J.; Xu, Y.; Dragoljevic, D.; Fitzgerald, M.; Horvath, A.; et al. Extracellular Vesicles Carrying HIV-1 Nef Induce Long-Term Hyperreactivity of Myeloid Cells. Cell Rep. 2022, 41, 111674. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Wang, Q.; Xu, Y.; Tsao, L.C.; Nakagawa, T.; Guo, H.; Su, L.; Xiong, Y. Vpr Targets TET2 for Degradation by CRL4VprBP E3 Ligase to Sustain IL-6 Expression and Enhance HIV-1 Replication. Mol. Cell 2018, 70, 961. [Google Scholar] [CrossRef] [PubMed]
- Langer, S.; Hammer, C.; Hopfensperger, K.; Klein, L.; Hotter, D.; Jesus, P.D.D.; Herbert, K.M.; Pache, L.; Smith, N.; van der Merwe, J.A.; et al. HIV-1 Vpu Is a Potent Transcriptional Suppressor of NF-ΚB-Elicited Antiviral Immune Responses. Elife 2019, 8, e41930. [Google Scholar] [CrossRef]
- Zhang, R.Z.; Kane, M. Insights into the Role of HIV-1 Vpu in Modulation of NF-ĸB Signaling Pathways. mBio 2023, 14, e0092023. [Google Scholar] [CrossRef]
- Shrikant, P.; Benos, D.J.; Tang, L.P.; Benveniste, E.N. HIV Glycoprotein 120 Enhances Intercellular Adhesion Molecule-1 Gene Expression in Glial Cells. Involvement of Janus Kinase/Signal Transducer and Activator of Transcription and Protein Kinase C Signaling Pathways. J. Immunol. 1950, 156, 1307–1314. Available online: https://pubmed.ncbi.nlm.nih.gov/8558011/ (accessed on 23 December 2024). [CrossRef]
- Pu, H.; Tian, J.; Flora, G.; Lee, Y.W.; Nath, A.; Hennig, B.; Toborek, M. HIV-1 Tat Protein Upregulates Inflammatory Mediators and Induces Monocyte Invasion into the Brain. Mol. Cell. Neurosci. 2003, 24, 224–237. [Google Scholar] [CrossRef]
- Duan, M.; Yao, H.; Hu, G.; Chen, X.M.; Lund, A.K.; Buch, S. HIV Tat Induces Expression of ICAM-1 in HUVECs: Implications for MiR-221/-222 in HIV-Associated Cardiomyopathy. PLoS ONE 2013, 8, e60170. [Google Scholar] [CrossRef]
- Swingler, S.; Brichacek, B.; Jacque, J.M.; Ulich, C.; Zhou, J.; Stevenson, M. HIV-1 Nef Intersects the Macrophage CD40L Signalling Pathway to Promote Resting-Cell Infection. Nature 2003, 424, 213. [Google Scholar] [CrossRef] [PubMed]
- Fabryova, H.; Strebel, K. Vpr and Its Cellular Interaction Partners: R We There Yet? Cells 2019, 8, 1310. [Google Scholar] [CrossRef]
- Ghaly, M.; Proulx, J.; Borgmann, K.; Park, I.W. Novel Role of HIV-1 Nef in Regulating the Ubiquitination of Cellular Proteins. Front. Cell Infect. Microbiol. 2023, 13, 1106591. [Google Scholar] [CrossRef]
- Badley, A.D.; Pilon, A.A.; Landay, A.; Lynch, D.H. Mechanisms of HIV-Associated Lymphocyte Apoptosis. Blood 2000, 96, 2951–2964. [Google Scholar] [CrossRef]
- Jabea Ekabe, C.; Asaba Clinton, N.; Agyei, E.K.; Kehbila, J. Role of Apoptosis in HIV Pathogenesis. Adv. Virol. 2022, 2022, 8148119. [Google Scholar] [CrossRef]
- Cummins, N.W.; Badley, A.D. Anti-Apoptotic Mechanisms of HIV: Lessons and Novel Approaches to Curing HIV. Cell Mol. Life Sci. 2012, 70, 3355. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shackelford, J.M.; Selliah, N.; Shivers, D.K.; O’Neill, E.; Garcia, J.V.; Muthumani, K.; Weiner, D.; Yu, X.F.; Gabuzda, D.; et al. The HIV-1 Vif Protein Mediates Degradation of Vpr and Reduces Vpr-Induced Cell Cycle Arrest. DNA Cell Biol. 2008, 27, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.H.; Williams, D.A.; McManus, C.; Baribaud, F.; Doms, R.W.; Schols, D.; De Clercq, E.; Kotlikoff, M.I.; Collman, R.G.; Freedman, B.D. HIV-1 Gp120 and Chemokines Activate Ion Channels in Primary Macrophages through CCR5 and CXCR4 Stimulation. Proc. Natl. Acad. Sci. USA 2000, 97, 4832. [Google Scholar] [CrossRef]
- Hu, X.-T. HIV-1 Tat-Mediated Calcium Dysregulation and Neuronal Dysfunction in Vulnerable Brain Regions. Curr. Drug Targets 2016, 17, 4–14. [Google Scholar] [CrossRef]
- Shrestha, J.; Santerre, M.; Allen, C.N.; Arjona, S.P.; Hooper, R.; Mukerjee, R.; Kaul, M.; Shcherbik, N.; Soboloff, J.; Sawaya, B.E. HIV-1 Gp120 Protein Promotes HAND through the Calcineurin Pathway Activation. Mitochondrion 2023, 70, 31. [Google Scholar] [CrossRef]
- Dubey, R.C.; Mishra, N.; Gaur, R. G Protein-Coupled and ATP-Sensitive Inwardly Rectifying Potassium Ion Channels Are Essential for HIV Entry. Sci. Rep. 2019, 9, 4113. [Google Scholar] [CrossRef]
- Ferrucci, A.; Nonnemacher, M.R.; Wigdahl, B. Human Immunodeficiency Virus Viral Protein R as an Extracellular Protein in Neuropathogenesis. Adv. Virus Res. 2011, 81, 165. [Google Scholar] [CrossRef]
- Ewart, G.D.; Sutherland, T.; Gage, P.W.; Cox, G.B. The Vpu Protein of Human Immunodeficiency Virus Type 1 Forms Cation-Selective Ion Channels. J. Virol. 1996, 70, 7108. [Google Scholar] [CrossRef] [PubMed]
- Ebenbichler, C.F.; Stoiber, H.; Schneider, R.; Patsch, J.R.; Dierich, M.P. The Human Immunodeficiency Virus Type 1 Transmembrane Gp41 Protein Is a Calcium-Binding Protein and Interacts with the Putative Second-Receptor Molecules in a Calcium-Dependent Manner. J. Virol. 1996, 70, 1723–1728. [Google Scholar] [CrossRef]
- Majeed, S.; Dang, L.; Islam, M.M.; Ishola, O.; Borbat, P.P.; Ludtke, S.J.; Georgieva, E.R. HIV-1 Vpu Protein Forms Stable Oligomers in Aqueous Solution via Its Transmembrane Domain Self-Association. Sci. Rep. 2023, 13, 14691. [Google Scholar] [CrossRef]
- Clark, E.; Nava, B.; Caputi, M. Tat Is a Multifunctional Viral Protein That Modulates Cellular Gene Expression and Functions. Oncotarget 2017, 8, 27569–27581. [Google Scholar] [CrossRef]
- Liu, X.; Bae, C.; Gelman, B.B.; Chung, J.M.; Tang, S.J. A Neuron-to-Astrocyte Wnt5a Signal Governs Astrogliosis during HIV-Associated Pain Pathogenesis. Brain 2022, 145, 4108. [Google Scholar] [CrossRef] [PubMed]
- Mukerji, J.; Olivieri, K.C.; Misra, V.; Agopian, K.A.; Gabuzda, D. Proteomic Analysis of HIV-1 Nef Cellular Binding Partners Reveals a Role for Exocyst Complex Proteins in Mediating Enhancement of Intercellular Nanotube Formation. Retrovirology 2012, 9, 33. [Google Scholar] [CrossRef]
- Buffalo, C.Z.; Iwamoto, Y.; Hurley, J.H.; Ren, X. How HIV Nef Proteins Hijack Membrane Traffic To Promote Infection. J. Virol. 2019, 93, e01322-19. [Google Scholar] [CrossRef]
- Goes, L.R.; Sajani, A.; Sivro, A.; Olowojesiku, R.; Ray, J.C.; Perrone, I.; Yolitz, J.; Girard, A.; Leyre, L.; Wibmer, C.K.; et al. The V2 Loop of HIV Gp120 Delivers Costimulatory Signals to CD4+ T Cells through Integrin A4β7 and Promotes Cellular Activation and Infection. Proc. Natl. Acad. Sci. USA 2020, 117, 32566–32573. [Google Scholar] [CrossRef] [PubMed]
- Valentín-Guillama, G.; López, S.; Kucheryavykh, Y.V.; Chorna, N.E.; Pérez, J.; Ortiz-Rivera, J.; Inyushin, M.; Makarov, V.; Valentín-Acevedo, A.; Quinones-Hinojosa, A.; et al. HIV-1 Envelope Protein Gp120 Promotes Proliferation and the Activation of Glycolysis in Glioma Cell. Cancers 2018, 10, 301. [Google Scholar] [CrossRef] [PubMed]
- Vignoli, A.L.; Martini, I.; Haglid, K.H.; Silvestroni, L.; Augusti-Tocco, G.; Biagioni, S. Neuronal Glycolytic Pathway Impairment Induced by HIV Envelope Glycoprotein Gp120. Mol. Cell Biochem. 2000, 215, 73–80. [Google Scholar] [CrossRef]
- Kang, S.; Tang, H. HIV-1 Infection and Glucose Metabolism Reprogramming of T Cells: Another Approach Toward Functional Cure and Reservoir Eradication. Front. Immunol. 2020, 11, 572677. [Google Scholar] [CrossRef] [PubMed]
- Barrero, C.A.; Datta, P.K.; Sen, S.; Deshmane, S.; Amini, S.; Khalili, K.; Merali, S. HIV-1 Vpr Modulates Macrophage Metabolic Pathways: A SILAC-Based Quantitative Analysis. PLoS ONE 2013, 8, e68376. [Google Scholar] [CrossRef] [PubMed]
- Sivalingam, K.; Cirino, T.J.; McLaughlin, J.P.; Samikkannu, T. HIV-Tat and Cocaine Impact Brain Energy Metabolism: Redox Modification and Mitochondrial Biogenesis Influence NRF Transcription-Mediated Neurodegeneration. Mol. Neurobiol. 2021, 58, 490. [Google Scholar] [CrossRef]
- Yan, J.; Shun, M.C.; Hao, C.; Zhang, Y.; Qian, J.; Hrecka, K.; Delucia, M.; Monnie, C.; Ahn, J.; Skowronski, J. HIV-1 Vpr Reprograms CLR4DCAF1 E3 Ubiquitin Ligase to Antagonize Exonuclease 1-Mediated Restriction of HIV-1 Infection. mBio 2018, 9, 10-1128. [Google Scholar] [CrossRef]
- Wen, X.; Klockow, L.; Nekorchuk, M.; Sharifi, H.J.; de Noronha, C.M.C. The HIV1 Protein Vpr Acts to Enhance Constitutive DCAF1-Dependent UNG2 Turnover. PLoS ONE 2012, 7, e30939. [Google Scholar] [CrossRef]
- Zhou, X.; Delucia, M.; Hao, C.; Hrecka, K.; Monnie, C.; Skowronski, J.; Ahn, J. HIV-1 Vpr Protein Directly Loads Helicase-like Transcription Factor (HLTF) onto the CRL4-DCAF1 E3 Ubiquitin Ligase. J. Biol. Chem. 2017, 292, 21117–21127. [Google Scholar] [CrossRef]
- Erdmann, N.B.; Whitney, N.P.; Zheng, J. Potentiation of Excitotoxicity in HIV-1 Associated Dementia and the Significance of Glutaminase. Clin. Neurosci. Res. 2006, 6, 315. [Google Scholar] [CrossRef]
- El-Amine, R.; Germini, D.; Zakharova, V.V.; Tsfasman, T.; Sheval, E.V.; Louzada, R.A.N.; Dupuy, C.; Bilhou-Nabera, C.; Hamade, A.; Najjar, F.; et al. HIV-1 Tat Protein Induces DNA Damage in Human Peripheral Blood B-Lymphocytes via Mitochondrial ROS Production. Redox Biol. 2018, 15, 97. [Google Scholar] [CrossRef]
- Hu, S.; Sheng, W.S.; Lokensgard, J.R.; Peterson, P.K.; Rock, R.B. Preferential Sensitivity of Human Dopaminergic Neurons to Gp120-Induced Oxidative Damage. J. Neurovirol. 2009, 15, 401. [Google Scholar] [CrossRef]
- Mangino, G.; Famiglietti, M.; Capone, C.; Veroni, C.; Percario, Z.A.; Leone, S.; Fiorucci, G.; Lülf, S.; Romeo, G.; Agresti, C.; et al. HIV-1 Myristoylated Nef Treatment of Murine Microglial Cells Activates Inducible Nitric Oxide Synthase, NO2 Production and Neurotoxic Activity. PLoS ONE 2015, 10, e0130189. [Google Scholar] [CrossRef]
- Borrajo, A.; Spuch, C.; Penedo, M.A.; Olivares, J.M.; Agís-Balboa, R.C. Important Role of Microglia in HIV-1 Associated Neurocognitive Disorders and the Molecular Pathways Implicated in Its Pathogenesis. Ann. Med. 2021, 53, 43. [Google Scholar] [CrossRef] [PubMed]
- Hategan, A.; Bianchet, M.A.; Steiner, J.; Karnaukhova, E.; Masliah, E.; Fields, A.; Lee, M.H.; Dickens, A.M.; Haughey, N.; Dimitriadis, E.K.; et al. HIV-Tat Protein and Amyloid β Peptide Form Multifibrillar Structures That Cause Neurotoxicity. Nat. Struct. Mol. Biol. 2017, 24, 379. [Google Scholar] [CrossRef]
- Naghavi, M.H. “APP”reciating the Complexity of HIV-Induced Neurodegenerative Diseases. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef]
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s Disease: Pathophysiology and Therapeutic Strategies. Mol. Neurodegener. 2022, 17, e1007309. [Google Scholar] [CrossRef]
- Kwon, H.S.; Brent, M.M.; Getachew, R.; Jayakumar, P.; Chen, L.F.; Schnolzer, M.; McBurney, M.W.; Marmorstein, R.; Greene, W.C.; Ott, M. Human Immunodeficiency Virus Type 1 Tat Protein Inhibits the SIRT1 Deacetylase and Induces T-Cell Hyperactivation. Cell Host Microbe 2008, 3, 158. [Google Scholar] [CrossRef] [PubMed]
- Abraham, L.; Fackler, O.T. HIV-1 Nef: A Multifaceted Modulator of T Cell Receptor Signaling. Cell Commun. Signal 2012, 10, 39. [Google Scholar] [CrossRef]
- Abbas, W.; Herbein, G. T-Cell Signaling in HIV-1 Infection. Open Virol. J. 2013, 7, 57. [Google Scholar] [CrossRef]
- Reuschl, A.K.; Mesner, D.; Shivkumar, M.; Whelan, M.V.X.; Pallett, L.J.; Guerra-Assunção, J.A.; Madansein, R.; Dullabh, K.J.; Sigal, A.; Thornhill, J.P.; et al. HIV-1 Vpr Drives a Tissue Residency-like Phenotype during Selective Infection of Resting Memory T Cells. Cell Rep. 2022, 39, 110650. [Google Scholar] [CrossRef]
- Bauby, H.; Ward, C.C.; Hugh-White, R.; Swanson, C.M.; Schulz, R.; Goujon, C.; Malima, M.H. HIV-1 Vpr Induces Widespread Transcriptomic Changes in CD4+ T Cells Early Postinfection. mBio 2021, 12, e0136921. [Google Scholar] [CrossRef]
- Berman, J.W.; Carvallo, L.; Buckner, C.M.; Luers, A.; Prevedel, L.; Bennett, M.V.; Eugenin, E.A. HIV-Tat Alters Connexin43 Expression and Trafficking in Human Astrocytes: Role in NeuroAIDS. J. Neuroinflammation 2016, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Zheng, J.; Okamoto, S.; Gendelman, H.E.; Lipton, S.A. HIV-1 Infection and AIDS: Consequences for the Central Nervous System. Cell Death Differ. 2005, 12 (Suppl. 1), 878–892. [Google Scholar] [CrossRef]
- Guha, D.; Wagner, M.C.E.; Ayyavoo, V. Human Immunodeficiency Virus Type 1 (HIV-1)-Mediated Neuroinflammation Dysregulates Neurogranin and Induces Synaptodendritic Injury. J. Neuroinflammation 2018, 15, 126. [Google Scholar] [CrossRef]
- Dahm, T.; Rudolph, H.; Schwerk, C.; Schroten, H.; Tenenbaum, T. Neuroinvasion and Inflammation in Viral Central Nervous System Infections. Mediat. Inflamm. 2016, 2016, 8562805. [Google Scholar] [CrossRef]
- Bryant, A.K.; Ellis, R.J.; Umlauf, A.; Gouaux, B.; Soontornniyomkij, V.; Letendre, S.L.; Achim, C.L.; Masliah, E.; Grant, I.; Moore, D.J. Antiretroviral Therapy Reduces Neurodegeneration in Human Immunodeficiency Virus Infection. AIDS 2015, 29, 323. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-Associated Neurocognitive Disorders Persist in the Era of Potent Antiretroviral Therapy: CHARTER Study. Neurology 2010, 75, 2087. [Google Scholar] [CrossRef] [PubMed]
- Krut, J.J.; Mellberg, T.; Price, R.W.; Hagberg, L.; Fuchs, D.; Rosengren, L.; Nilsson, S.; Zetterberg, H.; Gisslén, M. Biomarker Evidence of Axonal Injury in Neuroasymptomatic HIV-1 Patients. PLoS ONE 2014, 9, e88591. [Google Scholar] [CrossRef]
- Sacktor, N. Changing Clinical Phenotypes of HIV-Associated Neurocognitive Disorders. J. Neurovirol. 2018, 24, 141. [Google Scholar] [CrossRef] [PubMed]
- Clifford, D.B.; Fagan, A.M.; Holtzman, D.M.; Morris, J.C.; Teshome, M.; Shah, A.R.; Kauwe, J.S.K. CSF Biomarkers of Alzheimer Disease in HIV-Associated Neurologic Disease. Neurology 2009, 73, 1982. [Google Scholar] [CrossRef]
- Rubin, L.H.; Sundermann, E.E.; Moore, D.J. The Current Understanding of Overlap between Characteristics of HIV-Associated Neurocognitive Disorders and Alzheimer’s Disease. J. Neurovirol. 2019, 25, 661. [Google Scholar] [CrossRef]
- Ortega, M.; Ances, B.M. Role of HIV in Amyloid Metabolism. J. Neuroimmune Pharmacol. 2014, 9, 483–491. [Google Scholar] [CrossRef]
- Howdle, G.C.; Quidé, Y.; Kassem, M.S.; Johnson, K.; Rae, C.D.; Brew, B.J.; Cysique, L.A. Brain Amyloid in Virally Suppressed HIV-Associated Neurocognitive Disorder. Neurol. Neuroimmunol. Neuroinflamm 2020, 7. [Google Scholar] [CrossRef]
- Sundermann, E.E.; Bondi, M.W.; Campbell, L.M.; Gouaux, B.; Moore, R.C.; Soontornniyomkij, V.; Moore, D.J. Distinguishing Amnestic Mild Cognitive Impairment From HIV-Associated Neurocognitive Disorders. J. Infect. Dis. 2021, 224, 435. [Google Scholar] [CrossRef]
- Mirsattari, S.M.; Power, C.; Nath, A. Parkinsonism with HIV Infection. Mov. Disord. 1998, 13, 684–689. [Google Scholar] [CrossRef]
- Koutsilieri, E.; Sopper, S.; Scheller, C.; Ter Meulen, V.; Riederer, P. Parkinsonism in HIV Dementia. J. Neural Transm. 2002, 109, 767–775. [Google Scholar] [CrossRef]
- DeVaughn, S.; Müller-Oehring, E.M.; Markey, B.; Brontë-Stewart, H.M.; Schulte, T. Aging with HIV-1 Infection: Motor Functions, Cognition, and Attention—A Comparison with Parkinson’s Disease. Neuropsychol. Rev. 2015, 25, 424. [Google Scholar] [CrossRef]
- Dehner, L.F.; Spitz, M.; Pereira, J.S. Parkinsonism in HIV Infected Patients during Antiretroviral Therapy—Data from a Brazilian Tertiary Hospital. Braz. J. Infect. Dis. 2016, 20, 499. [Google Scholar] [CrossRef]
- Amod, F.; Holla, V.V.; Ojha, R.; Pandey, S.; Yadav, R.; Pal, P.K. A Review of Movement Disorders in Persons Living with HIV. Park. Relat. Disord. 2023, 114, 105774. [Google Scholar] [CrossRef]
- Cysique, L.A.; Brew, B.J. Vascular Cognitive Impairment and HIV-Associated Neurocognitive Disorder: A New Paradigm. J. Neurovirol. 2019, 25, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Moulignier, A.; Moulonguet, A.; Pialoux, G.; Rozenbaum, W. Reversible ALS-like Disorder in HIV Infection. Neurology 2001, 57, 995–1001. [Google Scholar] [CrossRef]
- Bowen, L.N.; Tyagi, R.; Li, W.; Alfahad, T.; Smith, B.; Wright, M.; Singer, E.J.; Nath, A. HIV-Associated Motor Neuron Disease: HERV-K Activation and Response to Antiretroviral Therapy. Neurology 2016, 87, 1756. [Google Scholar] [CrossRef]
- Spudich, S.; González-Scarano, F. HIV-1-Related Central Nervous System Disease: Current Issues in Pathogenesis, Diagnosis, and Treatment. Cold Spring Harb. Perspect. Med. 2012, 2, a007120. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-Associated Neurocognitive Disorder—Pathogenesis and Prospects for Treatment. Nat. Rev. Neurol. 2016, 12, 234. [Google Scholar] [CrossRef]
- Clifford, D.B. HIV Associated Neurocognitive Disorder. Curr. Opin. Infect. Dis. 2017, 30, 117. [Google Scholar] [CrossRef]
- Thakur, K.T.; Boubour, A.; Saylor, D.; Das, M.; Bearden, D.R.; Birbeck, G.L. Global HIV Neurology: A Comprehensive Review. AIDS 2019, 33, 163. [Google Scholar] [CrossRef]
- Kaul, M. HIV-1 Associated Dementia: Update on Pathological Mechanisms and Therapeutic Approaches. Curr. Opin. Neurol. 2009, 22, 315–320. [Google Scholar] [CrossRef]
- Lingor, P.; Demleitner, A.F.; Wolff, A.W.; Feneberg, E. SARS-CoV-2 and Neurodegenerative Diseases: What We Know and What We Don’t. J. Neural Transm. 2022, 129, 1155. [Google Scholar] [CrossRef] [PubMed]
- Siow, I.; Lee, K.S.; Zhang, J.J.Y.; Saffari, S.E.; Ng, A. Encephalitis as a Neurological Complication of COVID-19: A Systematic Review and Meta-analysis of Incidence, Outcomes, and Predictors. Eur. J. Neurol. 2021, 28, 3491. [Google Scholar] [CrossRef]
- Krahel, W.D.; Bartak, M.; Cymerys, J. Acute and Long-Term SARS-CoV-2 Infection and Neurodegeneration Processes—Circulus Vitiosus. Acta Virol. 2024, 68, 12765. [Google Scholar] [CrossRef]
- Pandya, D.; Johnson, T.P. Chronic and Delayed Neurological Manifestations of Persistent Infections. Curr. Opin. Neurol. 2023, 36, 198. [Google Scholar] [CrossRef]
- Choutka, J.; Jansari, V.; Hornig, M.; Iwasaki, A. Unexplained Post-Acute Infection Syndromes. Nat. Med. 2022, 28, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brünink, S.; Greuel, S.; et al. Olfactory Transmucosal SARS-CoV-2 Invasion as a Port of Central Nervous System Entry in Individuals with COVID-19. Nat. Neurosci. 2020, 24, 168–175. [Google Scholar] [CrossRef]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in Human and Mouse Brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P.; et al. Dysregulation of Brain and Choroid Plexus Cell Types in Severe COVID-19. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef]
- Lee, M.H.; Perl, D.P.; Steiner, J.; Pasternack, N.; Li, W.; Maric, D.; Safavi, F.; Horkayne-Szakaly, I.; Jones, R.; Stram, M.N.; et al. Neurovascular Injury with Complement Activation and Inflammation in COVID-19. Brain 2022, 145, 2555–2568. [Google Scholar] [CrossRef]
- Bayat, A.H.; Azimi, H.; Hassani Moghaddam, M.; Ebrahimi, V.; Fathi, M.; Vakili, K.; Mahmoudiasl, G.R.; Forouzesh, M.; Boroujeni, M.E.; Nariman, Z.; et al. COVID-19 Causes Neuronal Degeneration and Reduces Neurogenesis in Human Hippocampus. Apoptosis 2022, 27, 852. [Google Scholar] [CrossRef]
- Kryńska, K.; Kuliś, K.; Mazurek, W.; Gudowska-Sawczuk, M.; Zajkowska, M.; Mroczko, B. The Influence of SARS-CoV-2 Infection on the Development of Selected Neurological Diseases. Int. J. Mol. Sci. 2024, 25, 8715. [Google Scholar] [CrossRef]
- Jarczak, D.; Nierhaus, A. Cytokine Storm—Definition, Causes, and Implications. Int. J. Mol. Sci. 2022, 23, 11740. [Google Scholar] [CrossRef]
- Ningrum, D.N.A.; Kung, W.M. Challenges and Perspectives of Neurological Disorders. Brain Sci. 2023, 13, 676. [Google Scholar] [CrossRef]
- Feigin, V.L.; Vos, T.; Nichols, E.; Owolabi, M.O.; Carroll, W.M.; Dichgans, M.; Deuschl, G.; Parmar, P.; Brainin, M.; Murray, C. The Global Burden of Neurological Disorders: Translating Evidence into Policy. Lancet Neurol. 2020, 19, 255. [Google Scholar] [CrossRef]
- Van Schependom, J.; D’haeseleer, M. Advances in Neurodegenerative Diseases. J. Clin. Med. 2023, 12, 12. [Google Scholar] [CrossRef]
- Dehhaghi, M.; Kazemi Shariat Panahi, H.; Guillemin, G.J. Microorganisms’ Footprint in Neurodegenerative Diseases. Front. Cell Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef]
- Flint, B.; Tadi, P. Physiology, Aging; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ramanoël, S.; Hoyau, E.; Kauffmann, L.; Renard, F.; Pichat, C.; Boudiaf, N.; Krainik, A.; Jaillard, A.; Baciu, M. Gray Matter Volume and Cognitive Performance during Normal Aging. A Voxel-Based Morphometry Study. Front. Aging Neurosci. 2018, 10, 350276. [Google Scholar] [CrossRef]
- Li, Y.; Choi, W.J.; Wei, W.; Song, S.; Zhang, Q.; Liu, J.; Wang, R.K. Aging-Associated Changes in Cerebral Vasculature and Blood Flow as Determined by Quantitative Optical Coherence Tomography Angiography. Neurobiol. Aging 2018, 70, 148. [Google Scholar] [CrossRef]
- Costantini, E.; D’Angelo, C.; Reale, M. The Role of Immunosenescence in Neurodegenerative Diseases. Mediat. Inflamm. 2018, 2018, 6039171. [Google Scholar] [CrossRef]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative Disease: Models, Mechanisms, and a New Hope. Dis. Model. Mech. 2017, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef]
- Maiti, P.; Manna, J.; Dunbar, G.L.; Maiti, P.; Dunbar, G.L. Current Understanding of the Molecular Mechanisms in Parkinson’s Disease: Targets for Potential Treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef]
- Abdelmoaty, M.M.; Lu, E.; Kadry, R.; Foster, E.G.; Bhattarai, S.; Mosley, R.L.; Gendelman, H.E. Clinical Biomarkers for Lewy Body Diseases. Cell Biosci. 2023, 13, 209. [Google Scholar] [CrossRef] [PubMed]
- Baizabal-Carvallo, J.; Alonso-Juarez, M. The Role of Viruses in the Pathogenesis of Parkinson’s Disease. Neural Regen. Res. 2021, 16, 1200. [Google Scholar] [CrossRef]
- Olsen, L.K.; Dowd, E.; McKernan, D.P. A Role for Viral Infections in Parkinson’s Etiology? Neuronal Signal 2018, 2, 20170166. [Google Scholar] [CrossRef]
- German, D.C.; Manaye, K.; Smith, W.K.; Woodward, D.J.; Saper, C.B. Midbrain Dopaminergic Cell Loss in Parkinson’s Disease: Computer Visualization. Ann. Neurol. 1989, 26, 507–514. [Google Scholar] [CrossRef]
- Arai, H.; Furuya, T.; Mizuno, Y.; Mochizuki, H. Inflammation and Infection in Parkinson’s Disease. Histol. Histopathol. 2006, 21, 673–678. [Google Scholar] [CrossRef]
- Henry, J.; Smeyne, R.J.; Jang, H.; Miller, B.; Okun, M.S. Parkinsonism and Neurological Manifestations of Influenza Throughout the 20th and 21st Centuries. Park. Relat. Disord. 2010, 16, 566. [Google Scholar] [CrossRef]
- Takahashi, M.; Yamada, T.; Nakajima, S.; Nakajima, K.; Yamamoto, T.; Okada, H. The Substantia Nigra Is a Major Target for Neurovirulent Influenza A Virus. J. Exp. Med. 1995, 181, 2161. [Google Scholar] [CrossRef]
- Wang, H.; Liu, X.; Tan, C.; Zhou, W.; Jiang, J.; Peng, W.; Zhou, X.; Mo, L.; Chen, L. Bacterial, Viral, and Fungal Infection-related Risk of Parkinson’s Disease: Meta-analysis of Cohort and Case–Control Studies. Brain Behav. 2020, 10, e01549. [Google Scholar] [CrossRef] [PubMed]
- Vlajinac, H.; Dzoljic, E.; Maksimovic, J.; Marinkovic, J.; Sipetic, S.; Kostic, V. Infections as a Risk Factor for Parkinson’s Disease: A Case-Control Study. Int. J. Neurosci. 2013, 123, 329–332. [Google Scholar] [CrossRef]
- Lin, W.Y.; Lin, M.S.; Weng, Y.H.; Yeh, T.H.; Lin, Y.S.; Fong, P.Y.; Wu, Y.R.; Lu, C.S.; Chen, R.S.; Huang, Y.Z. Association of Antiviral Therapy with Risk of Parkinson Disease in Patients with Chronic Hepatitis C Virus Infection. JAMA Neurol. 2019, 76, 1019. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Boltz, D.A.; Webster, R.G.; Smeyne, R.J. Viral Parkinsonism. Biochim. Biophys. Acta 2009, 1792, 714. [Google Scholar] [CrossRef]
- Sasco, A.J.; Paffenbarger, R.S. Measles Infection and Parkinson’s Disease. Am. J. Epidemiol. 1985, 122, 1017–1031. [Google Scholar] [CrossRef]
- The Lancet: Global Burden of Disease. Available online: https://www.thelancet.com/gbd (accessed on 22 December 2024).
- Qiu, C.; Kivipelto, M.; Von Strauss, E. Epidemiology of Alzheimer’s Disease: Occurrence, Determinants, and Strategies toward Intervention. Dialogues Clin. Neurosci. 2009, 11, 111. [Google Scholar] [CrossRef]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.D.C.; Diaz-Cintra, S.; et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef]
- O’Brien, J.T.; Thomas, A. Vascular Dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C. Vascular Dementia Prevention: A Risk Factor Analysis. Cerebrovasc. Dis. 2005, 20 (Suppl. 2), 91–100. [Google Scholar] [CrossRef]
- Emmady, P.D.; Schoo, C.; Tadi, P. Major Neurocognitive Disorder (Dementia); StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s Disease-Associated Amyloid β-Protein Is an Antimicrobial Peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef]
- Morris, J.C.; Chang Wong, E.; Chang Chui, H. Vascular Cognitive Impairment and Dementia. CONTINUUM Lifelong Learn. Neurol. 2022, 28, 750. [Google Scholar] [CrossRef]
- Custodio, N.; Montesinos, R.; Lira, D.; Herrera-Pérez, E.; Bardales, Y.; Valeriano-Lorenzo, L. Mixed Dementia: A Review of the Evidence. Dement. Neuropsychol. 2017, 11, 364–370. [Google Scholar] [CrossRef]
- Morales, I.; Guzmán-Martínez, L.; Cerda-Troncoso, C.; Farías, G.A.; Maccioni, R.B. Neuroinflammation in the Pathogenesis of Alzheimer’s Disease. A Rational Framework for the Search of Novel Therapeutic Approaches. Front. Cell Neurosci. 2014, 8, 112. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and Neuroinflammation: A Pathological Perspective. J. Neuroinflammation 2004, 1, 14. [Google Scholar] [CrossRef]
- Lima, M.N.; Barbosa-Silva, M.C.; Maron-Gutierrez, T. Microglial Priming in Infections and Its Risk to Neurodegenerative Diseases. Front. Cell Neurosci. 2022, 16, 878987. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yuan, M.; Yang, E.; Chen, D.; Su, A.; Wu, Z. Enterovirus 71 Infection Induced Aquaporin-4 Depolarization by Increasing Matrix Metalloproteinase-9 Activity. Neurosci. Lett. 2021, 759, 136049. [Google Scholar] [CrossRef]
- Sousa Junior, I.P.; Vieira, T.C.R.G. Enterovirus Infection and Its Relationship with Neurodegenerative Diseases. Mem. Inst. Oswaldo Cruz 2023, 118, e220252. [Google Scholar] [CrossRef]
- Rippee-Brooks, M.D.; Wu, W.; Dong, J.; Pappolla, M.; Fang, X.; Bao, X. Viral Infections, Are They a Trigger and Risk Factor of Alzheimer’s Disease? Pathogens 2024, 13, 240. [Google Scholar] [CrossRef]
- Sait, A.; Angeli, C.; Doig, A.J.; Day, P.J.R. Viral Involvement in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 1049. [Google Scholar] [CrossRef] [PubMed]
- Sipilä, P.N.; Heikkilä, N.; Lindbohm, J.V.; Hakulinen, C.; Vahtera, J.; Elovainio, M.; Suominen, S.; Väänänen, A.; Koskinen, A.; Nyberg, S.T.; et al. Hospital-Treated Infectious Diseases and the Risk of Dementia: A Large, Multicohort, Observational Study with a Replication Cohort. Lancet Infect. Dis. 2021, 21, 1557–1567. [Google Scholar] [CrossRef]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus Exposure and Neurodegenerative Disease Risk across National Biobanks. Neuron 2023, 111, 1086–1093.e2. [Google Scholar] [CrossRef]
- Schnier, C.; Janbek, J.; Lathe, R.; Haas, J. Reduced Dementia Incidence after Varicella Zoster Vaccination in Wales 2013–2020. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2022, 8, e12293. [Google Scholar] [CrossRef]
- Verreault, R.; Laurin, D.; Lindsay, J.; De Serres, G. Past Exposure to Vaccines and Subsequent Risk of Alzheimer’s Disease. Can. Med. Assoc. J. 2001, 165, 1495. [Google Scholar]
- Wollebo, H.S.; White, M.K.; Gordon, J.; Berger, J.R.; Khalili, K. Persistence and Pathogenesis of the Neurotropic Polyomavirus JC. Ann. Neurol. 2015, 77, 560. [Google Scholar] [CrossRef] [PubMed]
- Pietropaolo, V.; Prezioso, C.; Bagnato, F.; Antonelli, G. John Cunningham Virus: An Overview on Biology and Disease of the Etiological Agent of the Progressive Multifocal Leukoencephalopathy. Rev. New Microbiol. 2018, 41, 1121–7138. [Google Scholar]
- Garg, M.; Arora, A.; Kulkarni, S.D.; Hegde, A.U.; Shah, K.N. Subacute Sclerosing Panencephalitis (SSPE): Experience from a Tertiary-Care Pediatric Center. J. Neurosci. Rural Pract. 2022, 13, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Jafri, S.K.; Kumar, R.; Ibrahim, S. Subacute Sclerosing Panencephalitis–Current Perspectives. Pediatr. Health Med. Ther. 2018, 9, 67. [Google Scholar] [CrossRef]
- Poli, S.; Donisi, V.; Mazzi, M.A.; Gobbin, F.; Giusto, G.; Orlandi, R.; Schena, F.; Del Piccolo, L.; das Nair, R.; Gajofatto, A.; et al. Fostering Quality of Life in Young Adults Living with Multiple Sclerosis: A Pilot Study of a Co-Created Integrated Intervention. Front. Psychol. 2024, 15, 1342166. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated with Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef]
- Miljković, D.; Spasojević, I. Multiple Sclerosis: Molecular Mechanisms and Therapeutic Opportunities. Antioxid. Redox Signal 2013, 19, 2286. [Google Scholar] [CrossRef]
- Virtanen, J.O.; Jacobson, S. Viruses and Multiple Sclerosis. CNS Neurol. Disord. Drug Targets 2012, 11, 528. [Google Scholar] [CrossRef]
- Koprowski, H.; Defreitas, E.C.; Harper, M.E.; Sandberg-Wollheim, M.; Sheremata, W.A.; Robert-Guroff, M.; Saxinger, C.W.; Feinberg, M.B.; Wong-Staal, F.; Gallo, R.C. Multiple Sclerosis and Human T-Cell Lymphotropic Retroviruses. Nature 1985, 318, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Koprowski, H. Search for Viruses in Multiple Sclerosis. Neurology 1976, 26, 80–82. [Google Scholar] [CrossRef]
- Tsunoda, I.; Fujinami, R.S. Neuropathogenesis of Theiler’s Murine Encephalomyelitis Virus Infection, An Animal Model for Multiple Sclerosis. J. Neuroimmune Pharmacol. 2009, 5, 355. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated with Multiple Sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Thacker, E.L.; Mirzaei, F.; Ascherio, A. Infectious Mononucleosis and Risk for Multiple Sclerosis: A Meta-Analysis. Ann. Neurol. 2006, 59, 499–503. [Google Scholar] [CrossRef]
- DeLorenze, G.N.; Munger, K.L.; Lennette, E.T.; Orentreich, N.; Vogelman, J.H.; Ascherio, A. Epstein-Barr Virus and Multiple Sclerosis: Evidence of Association from a Prospective Study with Long-Term Follow-Up. Arch. Neurol. 2006, 63, 839–844. [Google Scholar] [CrossRef]
- Brotman, R.G.; Moreno-Escobar, M.C.; Joseph, J.; Munakomi, S.; Pawar, G. Amyotrophic Lateral Sclerosis; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef]
- Yu, B.; Pamphlett, R. Environmental Insults: Critical Triggers for Amyotrophic Lateral Sclerosis. Transl. Neurodegener. 2017, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.C.; Feuer, R.; Cashman, N.; Luo, H. Enteroviral Infection: The Forgotten Link to Amyotrophic Lateral Sclerosis? Front. Mol. Neurosci. 2018, 11, 63. [Google Scholar] [CrossRef]
- Wang, X.; Hu, Y.; Xu, R. The Pathogenic Mechanism of TAR DNA-Binding Protein 43 (TDP-43) in Amyotrophic Lateral Sclerosis. Neural Regen. Res. 2023, 19, 800. [Google Scholar] [CrossRef]
- Tyagi, R.; Li, W.; Parades, D.; Bianchet, M.A.; Nath, A. Inhibition of Human Endogenous Retrovirus-K by Antiretroviral Drugs. Retrovirology 2017, 14, 21. [Google Scholar] [CrossRef]
- Belshaw, R.; Katzourakis, A.; Pačes, J.; Burt, A.; Tristem, M. High Copy Number in Human Endogenous Retrovirus Families Is Associated with Copying Mechanisms in Addition to Reinfection. Mol. Biol. Evol. 2005, 22, 814–817. [Google Scholar] [CrossRef]
- Shin, W.; Lee, J.; Son, S.Y.; Ahn, K.; Kim, H.S.; Han, K. Human-Specific HERV-K Insertion Causes Genomic Variations in the Human Genome. PLoS ONE 2013, 8, e60605. [Google Scholar] [CrossRef]
- Wildschutte, J.H.; Williams, Z.H.; Montesion, M.; Subramanian, R.P.; Kidd, J.M.; Coffin, J.M. Discovery of Unfixed Endogenous Retrovirus Insertions in Diverse Human Populations. Proc. Natl. Acad. Sci. USA 2016, 113, E2326–E2334. [Google Scholar] [CrossRef]
- Villesen, P.; Aagaard, L.; Wiuf, C.; Pedersen, F.S. Identification of Endogenous Retroviral Reading Frames in the Human Genome. Retrovirology 2004, 1, 32. [Google Scholar] [CrossRef] [PubMed]
- Gröger, V.; Emmer, A.; Staege, M.S.; Cynis, H. Endogenous Retroviruses in Nervous System Disorders. Pharmaceuticals 2021, 14, 70. [Google Scholar] [CrossRef]
- Mao, J.; Zhang, Q.; Cong, Y.S. Human Endogenous Retroviruses in Development and Disease. Comput. Struct. Biotechnol. J. 2021, 19, 5978. [Google Scholar] [CrossRef]
- Pal, S.; Tyler, J.K. Epigenetics and Aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef]
- Latifi, T.; Zebardast, A.; Marashi, S.M. The Role of Human Endogenous Retroviruses (HERVs) in Multiple Sclerosis and the Plausible Interplay between HERVs, Epstein–Barr Virus Infection, and Vitamin D. Mult. Scler. Relat. Disord. 2022, 57, 103318. [Google Scholar] [CrossRef]
- Bhetariya, P.; Kriesel, J.; Fischer, K. Analysis of Human Endogenous Retrovirus Expression in Multiple Sclerosis Plaques. J. Emerg. Dis. Virol. 2017, 3. [Google Scholar] [CrossRef]
- Arru, G.; Mameli, G.; Deiana, G.A.; Rassu, A.L.; Piredda, R.; Sechi, E.; Caggiu, E.; Bo, M.; Nako, E.; Urso, D.; et al. Humoral Immunity Response to Human Endogenous Retroviruses K/W Differentiates between Amyotrophic Lateral Sclerosis and Other Neurological Diseases. Eur. J. Neurol. 2018, 25, 1076-e84. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Samimi, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic Tau-Induced PiRNA Depletion Promotes Neuronal Death through Transposable Element Dysregulation in Neurodegenerative Tauopathies. Nat. Neurosci. 2018, 21, 1038. [Google Scholar] [CrossRef]
- Phan, K.; He, Y.; Fu, Y.H.; Dzamko, N.; Bhatia, S.; Gold, J.; Rowe, D.; Ke, Y.D.; Ittner, L.M.; Hodges, J.R.; et al. Pathological Manifestation of Human Endogenous Retrovirus K in Frontotemporal Dementia. Commun. Med. 2021, 1, 60. [Google Scholar] [CrossRef]
- Wallace, A.D.; Wendt, G.A.; Barcellos, L.F.; de Smith, A.J.; Walsh, K.M.; Metayer, C.; Costello, J.F.; Wiemels, J.L.; Francis, S.S. To ERV Is Human: A Phenotype-Wide Scan Linking Polymorphic Human Endogenous Retrovirus-K Insertions to Complex Phenotypes. Front. Genet. 2018, 9, 298. [Google Scholar] [CrossRef]
- Dolei, A.; Ibba, G.; Piu, C.; Serra, C. Expression of HERV Genes as Possible Biomarker and Target in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3706. [Google Scholar] [CrossRef] [PubMed]
- Christensen, T. Human Endogenous Retroviruses in Neurologic Disease. APMIS 2016, 124, 116–126. [Google Scholar] [CrossRef]
- Küry, P.; Nath, A.; Créange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379. [Google Scholar] [CrossRef]
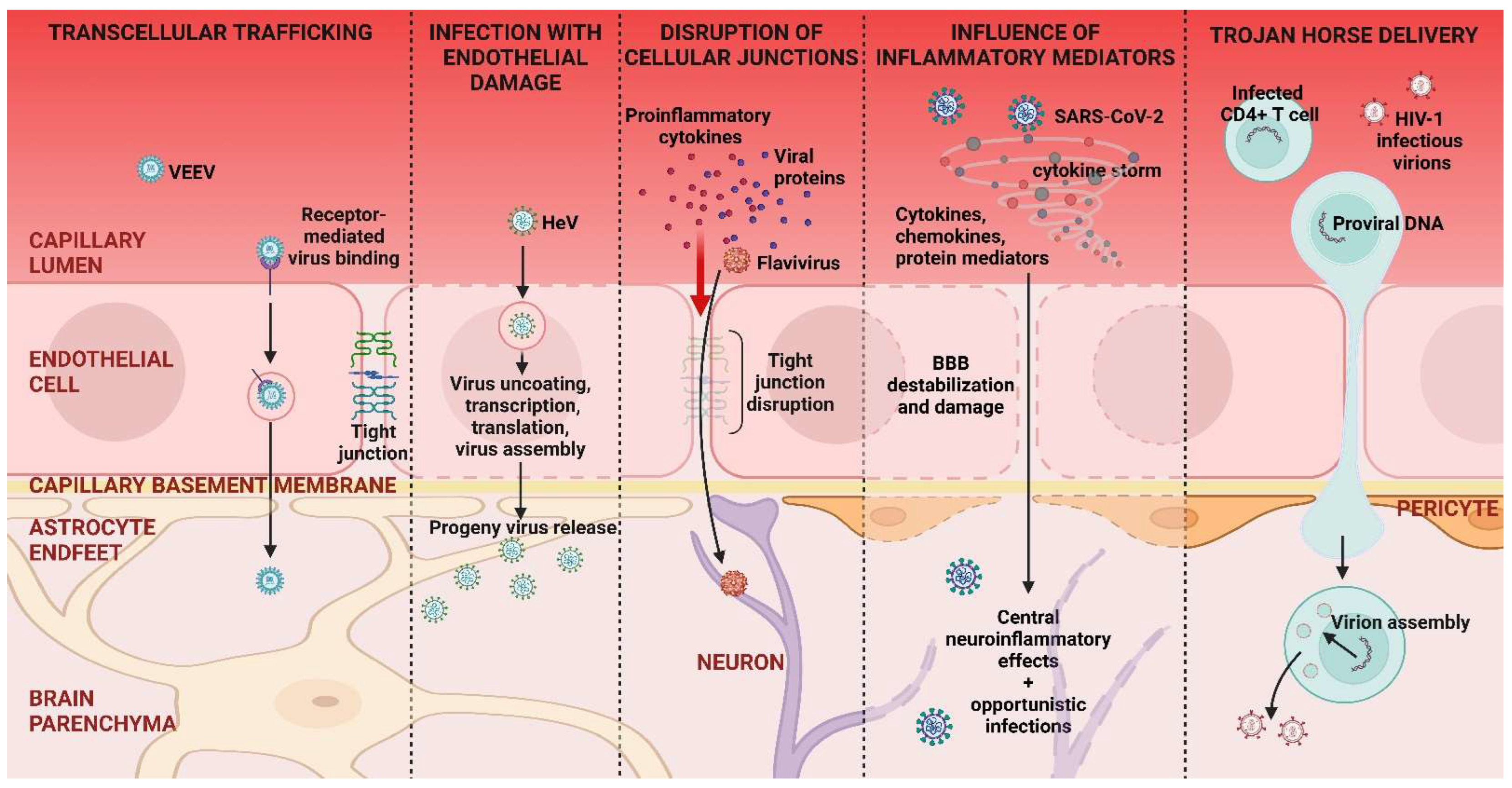
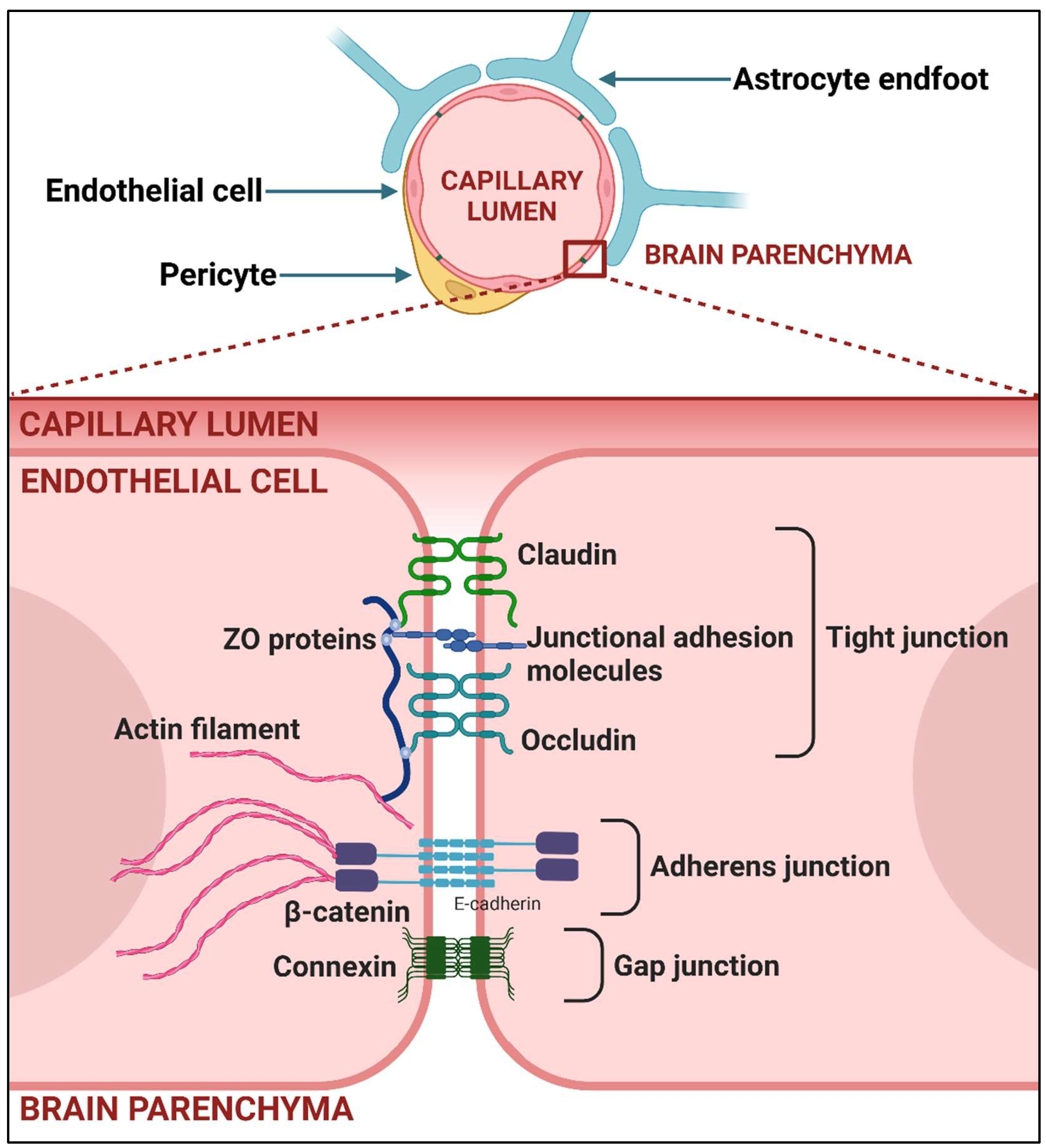

{kind=link}
{kind=link}
| RABV | HSV-1 | |
|---|---|---|
| Cells through which the virus enters the host’s body | Muscle cells | Epithelial cells |
| Method of entry into the peripheral neuron | Receptor-dependent clathrin-mediated endocytosis [39,62] | Fusion of virus lipid envelope with host cell membrane [63] |
| Viral proteins responsible for neuron entry | RABV G—the only protein exposed on the surface of the viral envelope that can bind to various receptors [64,65] | At least 12 different surface glycoproteins bind to more than one receptor [66] |
| Type of neuron used on the way to the CNS | Motor neuron | Sensory neuron |
| Mechanism of virus transport in a neuron | Fast dynein- and kinesin-mediated transport on microtubules in endosomes [67] | Dynein, dynactin, and kinesin-1-mediated transport on microtubules of the non-enveloped capsid [68] |
| Reaching the brain and the fate of the host | The virus always reaches the brain cells, ultimately causing the host’s death | If the virus reaches the brain cells, it may cause severe acute, potentially fatal encephalomyelitis, but this rarely happens |
| Ability to persist in the brain without causing the host’s death | No | Yes, in a latent form in the brainstem, as well as in other brain regions until the host’s natural death (not explained how it reaches them) |
| Main factor(s) determining tissue and cell tropism | Multiple—high level of receptors on the host cell surface, lower protective immune response of neurons, low immune surveillance over cells, others | The occurrence of specific receptors on host cells’ surface |
| Inflammatory response in the brain during acute infection | Specific suppression of the host immunity through diverse mechanisms [46] | Brain parenchyma inflammation is accompanied by an inflammatory response that may become exaggerated and further increases brain injury [69] |
| Possible Hematogenous Neuroinvasion Through Brain Barriers | Viruses |
|---|---|
| 1. Transcellular pathway with none or minimal barrier damage | Flaviviridae: WNV, JEV, TBEV, ZIKV; Togaviridae: VEEV, WEEV; Retroviridae: HIV-1 |
| 2.1. Infection with general damage to barrier cells | Flaviviridae, Paramyxoviridae: HeV, NiV |
| 2.2. Paracellular pathway with direct impairment of intercellular junctions of barrier cells | Flaviviridae: WNV, JEV, DENV Picornaviridae: Enterovirus A71 |
| 2.3. Barrier damage by inflammatory mediators | Coronaviridae: SARS-CoV-2 |
| 3. Trojan horse mechanism | Flaviviridae: JEV, WNV, ZIKV Retroviridae: HIV-1, Visna virus |
| Virus | Common Way of Entry to the CNS | Frequent Clinical Presentation | Outcome and Viral Persistence | Infected CNS Cells | |
|---|---|---|---|---|---|
| Family: Herpesviridae | HSV-1, HSV-2 | Anterograde transport from TG (HSV-1) or sacral dorsal ganglion (HSV-2) by sensory neuron projections [137], via olfactory pathway [138]. | Severe acute necrotizing encephalitis (HSV-1), meningitis (HSV-2) [139], or asymptomatic presence—accumulating data from the autopsy of people deceased without neurological illnesses (2–100%) [140]. | Encephalitis often leads to lifelong brain damage or death. Virus undergoes latency and persists permanently within neural tissue. | Neurons, astrocytes, microglia, oligodendrocytes [141]. |
| VZV | Anterograde transport by neuron projections from dorsal root ganglia and cranial nerve ganglia. | Meningitis or encephalitis often without concurrent zoster rash [142,143] or asymptomatic presence—data from the autopsy of people with no evidence of neurological disease (1–27.5%) [144,145]. | Resolution of symptoms with or without neurologic sequelae, encephalitis can result in fatal outcome. Virus is suspected to persist in the CNS, as it establishes latency in neurons. | Neurons, astrocytes [146], oligodendrocytes [147]. | |
| EBV | Via infected lymphocytes. | Meningitis, encephalitis, and CNS-lymphoma. | Resolution of symptoms without or with long-term consequences (depending on the host immune status and other factors [148]), may end with death. Establishes lifelong infection of the PNS and/or CNS, EBV-infected cells, free virus, and gene products can be found in the CNS [149]. | Neurons [150], astrocytes, microglia [151]. | |
| CMV | Presumably via infected monocytes [132]. | Meningitis, encephalitis (particularly in immunosuppressed individuals). | In immune-naïve and immunocompromised persons can cause CNS complications or result in fatal outcome [152]. Routinely establishes lifelong infection of the PNS and/or CNS [149]. | Neurons (debated), astrocytes, microglia, oligodendrocytes, neural progenitor cells [153]. | |
| HHV-6A, HHV-6B | Via olfactory pathway [154], infected lymphocytes, or optic tract [155]. | Encephalitis or asymptomatic presence—data from autopsies of individuals who passed away without neurological disorders (19–87.5%) [145,155,156]. | Encephalitis often has devastating sequelae or may cause death [157]. Viruses establish the lifelong infection of the PNS and/or CNS [149]. | Neurons, astrocytes [158], microglia [159], oligodendrocytes [160]. | |
| Family: Picornaviridae | Poliovirus | Via the BBB and motor neurons [72]. | Paralytic poliomyelitis. | Severe and irreversible damage of motor neurons, among those paralyzed, 5–10% die [161]. It is suggested that the virus may persist in surviving patients [162]. | Neurons [72]. |
| Enterovirus-A71 | Hematogenous way—transcellular pathway and/or infection of endothelial cells [163]. | Meningitis, encephalitis, polio-like syndrome, encephalomyelitis (the risk may be associated with younger age) [164]. | CNS involvement may entail severe sequelae, death is rarely reported [165]. The mechanisms and extent of EV-A71 persistence in the human CNS is unknown [166]. | Neurons, astrocytes [167], microglia [168]. | |
| Family: Orthomyxoviridae | Subtypes of influenza A virus | Presumably via olfactory nerve pathway [169,170,171], cranial nerves [172], via BBB endothelial cells [173]. | Acute encephalitis or influenza-associated encephalopathy. | Severe onset, often resulting in significant morbidity and high mortality within a matter of days [173]. | Neurons [174], astrocytes [175]. |
| Family: Paramyxoviridae | Mumps virus (MuV) | Through the choroid plexus—virus infects choroidal and ependymal epithelial cells [176], on mononuclear cells transiting the BBB during viremia. | Meningitis. | Usually mild or asymptomatic disease with often complete recovery [177]. Symptomatic CNS infection is less common and rarely fatal [178]. In rare instances, the virus is believed to persist in the CNS [179,180]. | Neurons [181]. |
| Measles virus | Hematogenous way—infection of brain endothelial cells [182], in infected CNS infiltrating macrophages [183]. | Primary encephalitis, postinfectious encephalomyelitis, SSPE. | All diseases can lead to severe and permanent brain injury [184], SSPE is invariably fatal. Virus may persist in neurons [185]. | Neurons, astrocytes, oligodendrocytes [183]. | |
| Henipaviruses (HeV, NiV) | Directly from the nasal mucosa and in a hematogenous way [186]. | Febrile illness or encephalitis. | The disease may be fatal, survivors may experience severe neurological sequelae [187]. | Endothelial cells of the CNS microvasculature, neurons [188], astrocytes, oligodendrocytes [189]. | |
| Family: Rhabdoviridae | RABV | Retrograde axonal transport. | Progressive rabies encephalitis. | Invariably fatal outcome. | Neurons, astrocytes (field strains) [53]. |
| Family: Flaviviridae | Encephalitic flaviviruses | Along the peripheral nervous system and axonal transport, hematogenous route—the infection of choroid plexus cells [190], the infection of BBB cells without cytopathic effect, with the downregulation of junction proteins, or crossing the BBB in leukocytes [83]. | Severe encephalitides and other neurologic syndromes. | Encephalitides are often fatal or may lead to transient or permanent neurological deficits [96]. Viruses may persist in the CNS. | Neurons, astrocytes, microglia, oligodendrocytes (please ref. to Table 5). |
| Family: Togaviridae | VEEV, WEEV, EEEV | Anterograde transport along peripheral nerves, olfactory route [191], hematogenous route—transcytosis across intact BBB [89]. | Mild, febrile illness to severe encephalitis. | Encephalitides have 1–75% fatality rate, depending on the virus strain [89]. Virus replication and/or genomic RNA may persist in the CNS [192]. | Neurons, astrocytes, microglia [193]. |
| Family: Retroviridae | HIV | In HIV-infected monocytes. | HAND. | Cognitive, motor and behavioral abnormalities [194]. Virus persists in the CNS. | Microglia, astrocytes [195]. |
| Family: Coronaviridae | SARS-CoV-2 | Via olfactory epithelium, via optic nerve, hematogenous pathway—by infection of brain endothelial cells [196]. | Encephalopathy (in most critically ill COVID-19 patients), PASC. | With serious or long-term neurological dysfunctions and deficits. | Neurons, astrocytes, choroid plexus epithelial cells [197]. |
| JEV | WNV | TBEV | ZIKV | |
|---|---|---|---|---|
| Disease transmission | Mosquito bite | Mosquito bite | Tick bite, consumption of unpasteurized milk from infected ruminants (1% of infections) [202] | Mosquito bite, placental transmission, sexual transmission [203] |
| Geographical regions of most encephalitis cases | Asia, Australia, Western Pacific [204] | Europe and North America [82] | Central/Northern Europe and North-Eastern Asia [205] | Africa, South-East Asia, Micronesia, French Polynesia, both Americas [206] |
| Most frequently encephalitis-bearing patients | Children | Elderly or inmunocompromised individuals [207] | Adults | Newborns |
| Vaccines available | Yes | No | Yes | No |
|
% of people infected with the virus who develop encephalitis | 0.1–1% [208] | 0.67% [209] | 2–25% [210] | Unknown |
| Number of encephalitis cases annually | ~100,000 [211] | Few thousand [212] | 10,000–15,000 [213] | Unknown in newborns, very few cases of adult people [214] |
| Encephalitis mortality rate | 20–30% [215] | Over 10% [216], 10–20% for WNV meningitis [217] | 1–40% [218]— in severe infections, TBE manifests as meningoencephalitis with significant mortality rate [219] | Unknown |
| Other neurological syndromes associated with infection | Meningitis [220], poliomyelitis-like flaccid paralysis [221], Parkinsonian syndrome [222], neuropsychiatric sequelae, cognitive and language impairment [223], GBS [224,225] | Meningitis, poliomyelitis [226], GBS [227], acute flaccid paralysis [228], movement disorders, cognitive difficulties [229], neuropsychiatric sequelae [230,231] | Meningitis, GBS [232], permanent paresis, postencephalitic syndrome [210], epilepsy, lateral sclerosis, dispersed sclerosis, Parkinson-like disease, mental deterioration leading to severe dementia and/or death [233] | Microcephaly and other congenital malformations in newborns, GBS—predominate presentaion in adults [234], encephalopathy, meningitis, myelitis, seizures [214] |
| Flavivirus | Non-neuronal CNS cells that can be infected |
| JEV | astrocytes [82], microglia, BMECs [240], pericytes [241], neural progenitor stem cells [81] |
| WNV | astrocytes [242], oligodendrocytes [243], BMECs [244], pericytes [245] |
| TBEV | astrocytes, oligodendrocytes [205], microglia [246], BMECs, pericytes [247] |
| ZIKV | astrocytes, oligodendrocytes [248], microglia, BMECs [249], pericytes [190], neural progenitor cells [250], glial progenitor cells [251] |
| Autoimmune Condition | Definition | Onset Time | Flaviviral Infections Associated with Autoimmune Condition |
|---|---|---|---|
| Autoimmune encephalitis, AIE | Acute to subacute progressive inflammation of the brain, associated with the presence of antibodies against self-antigens expressed in the CNS, often neuronal proteins [262]. | Can develop early or during onset of encephalitis, usually during recovery from viral infection or several weeks later [263]. | JEV, WNV [264,265] |
| Acute disseminated encephalomy-elitis, ADEM | Acute post-infectious progressive inflammatory disorder characterized by brain and spinal cord demyelination and other types of neural damage [266] can also occur post-vaccination. | Develops during or within days to weeks after CNS infections or after illnesses with CNS involvement. | DENV, ZIKV [237,267,268,269]. Immunization against JEV [270]. |
| Guillain–Barre Syndrome, GBS | Post-infectious neuropathy resulting from the autoimmune destruction of nerves in the PNS [271]. | Typically begins within a few days to several weeks after a viral infection. | DENV, WNV, JEV, and ZIKV [224,227,272,273]. |
| Direct injury of neurons | gp120, gp41, Tat, Vpr [300,301,302,303] |
| Induction of pro-inflammatory cytokine expression and release; glial and myeloid cell activation | Tat, gp120, Nef, Vpr [122,304,305,306,307,308,309,310,311,312] |
| Suppression of NF-κB-elicited antiviral immune responses | Vpu [313,314] |
| Induction of adhesion molecules expression and mediation of adhesion molecules’ release | Tat, gp120, Nef [315,316,317,318] |
| Regulation of protein stability through modulation of ubiquitination | Vif, Vpu, Vpx, Vpr, Nef [319,320] |
| Promotion of apoptosis | gp120, Tat, Vpu, Nef [319,321,322] |
| Inhibition of apoptosis | Tat, Vpr, Nef, Vif [323,324] |
| Alteration of intracellular ionic homeostasis | Tat, gp120, Nef, Vpr, Vpu, gp41 [325,326,327,328,329,330,331,332] |
| Disruption of cell–cell communication | Tat, Nef, gp120 [333,334,335,336,337] |
| Disruption of metabolic pathways | gp120, Tat, Vpr [338,339,340,341,342] |
| Disruption of DNA repair | Vpr [343,344,345] |
| Disruption of neurotransmitter signaling | [346] |
| Induction of ROS and RNS production | Tat, Vpr, Vpu, gp120, gp41, Nef [347,348,349,350] |
| Interaction with Aβ and/or Tau resulting in their accumulation (disruption of processing, localization, and phagocytosis by microglial cells) | Tat, gp120, Vpr, Nef [351,352,353] |
| Modulation of infected T cell activation status | gp120, Env, Nef, Tat, Vpr [354,355,356,357,358] |
| Neurodegenerative Disorder | HERVs Implicated in Disease |
|---|---|
| MS | HERV-W, HERV-K, HERV-H, HERV-E, HERV-15 [467,468]. |
| ALS | HERV-K, HERV-W [469] |
| AD | HERV-H, HERV-K, HERV-L, HERV-W [5,470] |
| progressive supranuclear palsy | HERV-H, HERV-K, HERV-L [470] |
| FTD | HERV-K [471] |
| PD | HERV-K [5,472] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mielcarska, M.B.; Rouse, B.T. Viruses and the Brain—A Relationship Prone to Trouble. Viruses 2025, 17, 203. https://doi.org/10.3390/v17020203
Mielcarska MB, Rouse BT. Viruses and the Brain—A Relationship Prone to Trouble. Viruses. 2025; 17(2):203. https://doi.org/10.3390/v17020203
Chicago/Turabian StyleMielcarska, Matylda Barbara, and Barry T. Rouse. 2025. "Viruses and the Brain—A Relationship Prone to Trouble" Viruses 17, no. 2: 203. https://doi.org/10.3390/v17020203
APA StyleMielcarska, M. B., & Rouse, B. T. (2025). Viruses and the Brain—A Relationship Prone to Trouble. Viruses, 17(2), 203. https://doi.org/10.3390/v17020203