
Transmission and Characterization of Creutzfeldt–Jakob Disease and Chronic Wasting Disease in the North American Deer Mouse
 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Brain Inocula
2.2. Animal Procedures
2.3. Genotyping of Deer Mice
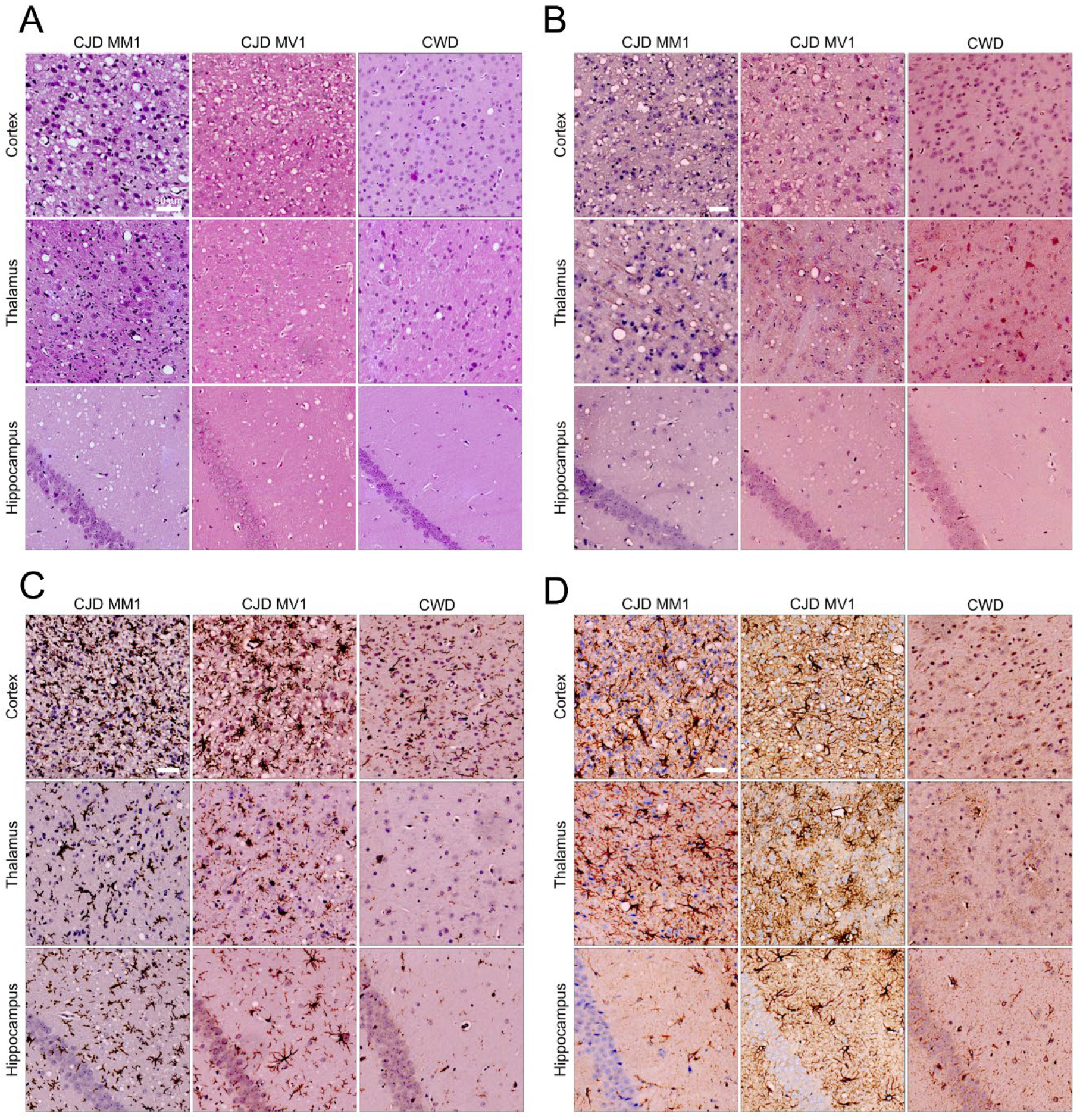
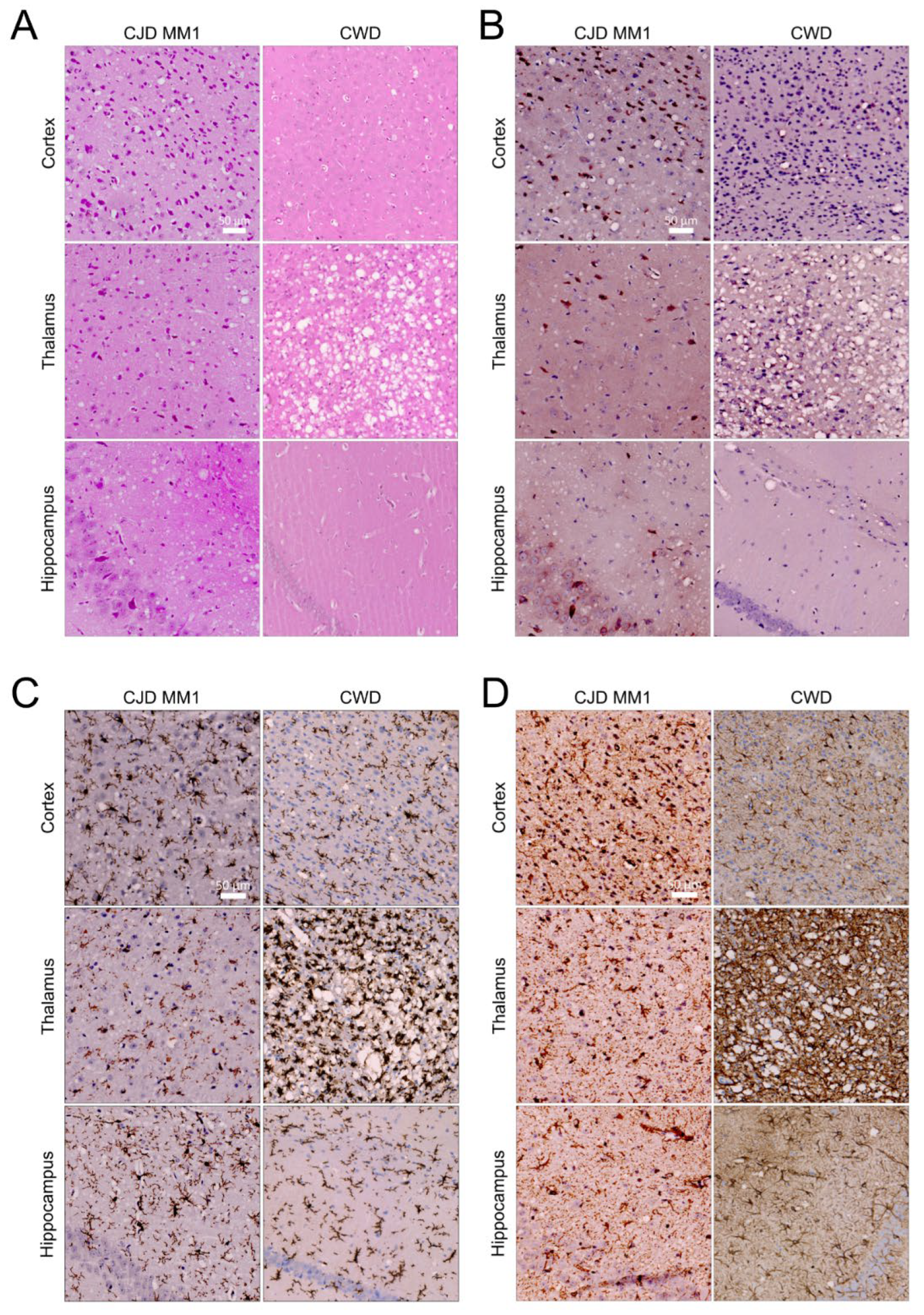
2.4. Profiling and Immunochemistry of Histopathological Lesions
2.5. Western Blotting
2.6. PTA Precipitation and Capillary Electrophoresis
2.7. RT-QuIC Seeding
3. Results
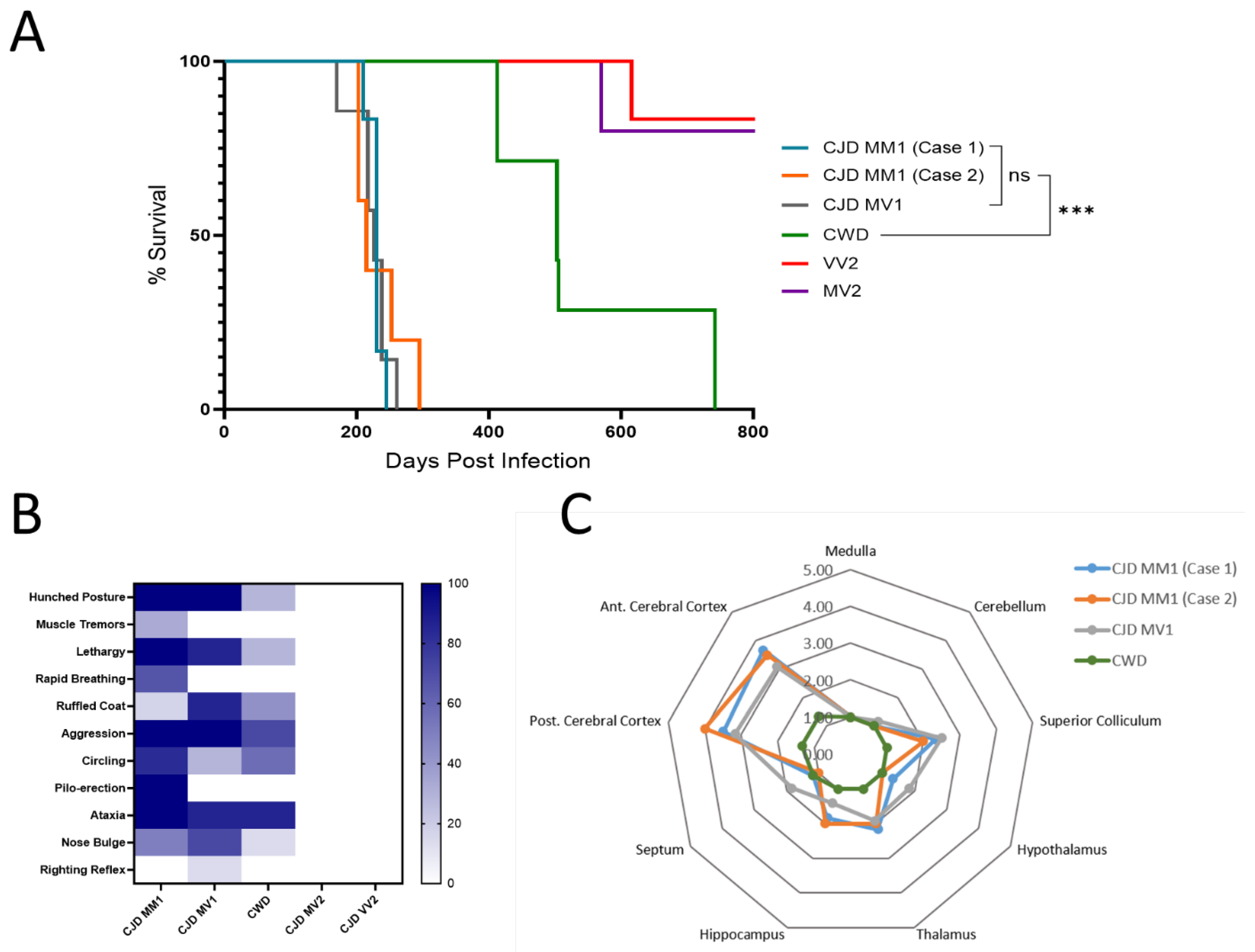
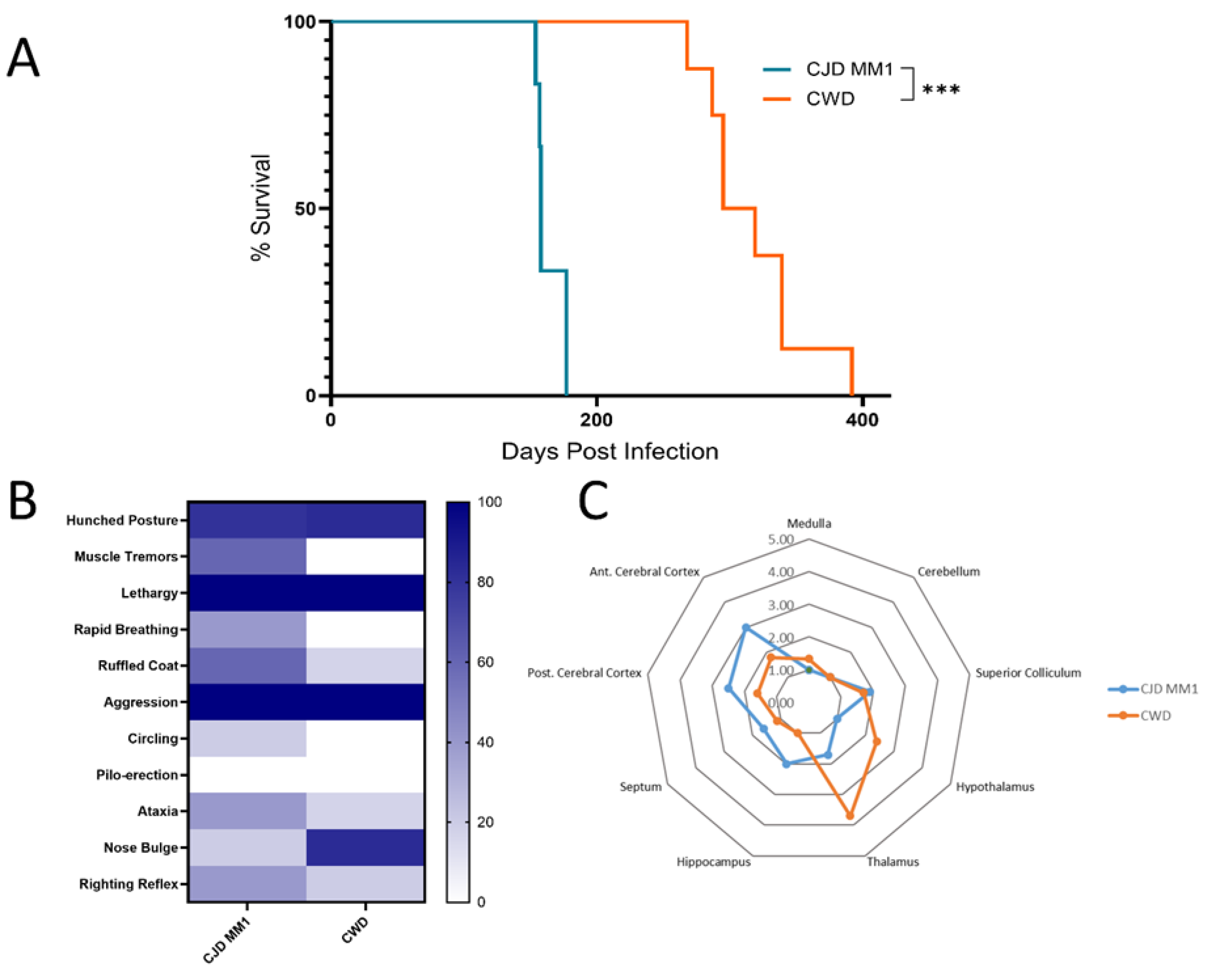
3.1. The North American Deer Mouse, Peromyscus maniculatus, Is Susceptible to sCJD and CWD
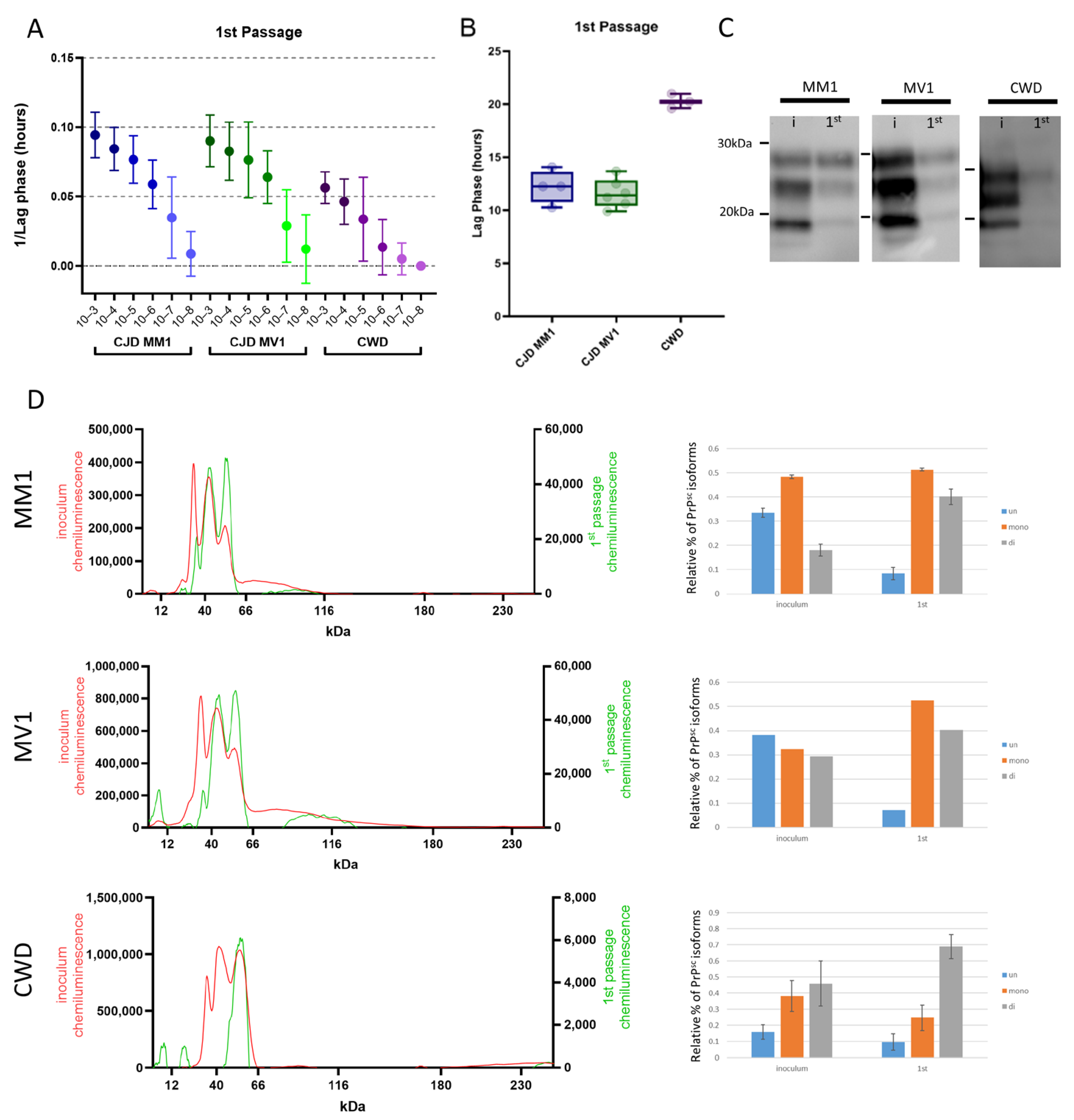
3.2. Protease-Resistant PrPSc and Prion Seeding Activity Is Abundant in CJD- and CWD-Infected Deer Mice
3.3. Adaptation of sCJD MM1 and CWD on Second Passage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mok, T.H.; Mead, S. Preclinical biomarkers of prion infection and neurodegeneration. Curr. Opin. Neurobiol. 2020, 61, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Baiardi, S.; Parchi, P. Understanding Prion Strains: Evidence from Studies of the Disease Forms Affecting Humans. Viruses 2019, 11, 309. [Google Scholar] [CrossRef] [PubMed]
- Baskakov, I.V. The many shades of prion strain adaptation. Prion 2014, 8, 169–172. [Google Scholar] [CrossRef]
- Heisey, D.M.; Mickelsen, N.A.; Schneider, J.R.; Johnson, C.J.; Langenberg, J.A.; Bochsler, P.N.; Keane, D.P.; Barr, D.J. Chronic Wasting Disease (CWD) Susceptibility of Several North American Rodents That Are Sympatric with Cervid CWD Epidemics. J. Virol. 2010, 84, 210–215. [Google Scholar] [CrossRef]
- Hall, E.R. The Mammals of North America; Wiley: New York, NY, USA, 1981; 722p, Available online: http://archive.org/details/mammalsofnortham0000hall (accessed on 23 August 2024).
- Burke, C.M.; Mark, K.M.K.; Walsh, D.J.; Noble, G.P.; Steele, A.D.; Diack, A.B.; Manson, J.C.; Watts, J.C.; Supattapone, S. Identification of a homology-independent linchpin domain controlling mouse and bank vole prion protein conversion. PLoS Pathog. 2020, 16, e1008875. [Google Scholar] [CrossRef]
- Arshad, H.; Patel, Z.; Al-Azzawi, Z.A.M.; Amano, G.; Li, L.; Mehra, S.; Eid, S.; Schmitt-Ulms, G.; Watts, J.C. The molecular determinants of a universal prion acceptor. PLoS Pathog. 2024, 20, e1012538. [Google Scholar] [CrossRef]
- Rivera, N.A.; Brandt, A.L.; Novakofski, J.E.; Mateus-Pinilla, N.E. Chronic Wasting Disease In Cervids: Prevalence, Impact And Management Strategies. Vet. Med. 2019, 10, 123–139. [Google Scholar] [CrossRef]
- Marsh, R.F.; Kincaid, A.E.; Bessen, R.A.; Bartz, J.C. Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus). J. Virol. 2005, 79, 13794–13796. [Google Scholar] [CrossRef]
- Race, B.; Williams, K.; Orrú, C.D.; Hughson, A.G.; Lubke, L.; Chesebro, B. Lack of Transmission of Chronic Wasting Disease to Cynomolgus Macaques. J. Virol. 2018, 92, e00550-18. [Google Scholar] [CrossRef]
- Watts, J.C.; Prusiner, S.B. Mouse Models for Studying the Formation and Propagation of Prions. J. Biol. Chem. 2014, 289, 19841–19849. [Google Scholar] [CrossRef]
- Wadsworth, J.D.F.; Joiner, S.; Linehan, J.M.; Jack, K.; Al-Doujaily, H.; Costa, H.; Ingold, T.; Taema, M.; Zhang, F.; Sandberg, M.K.; et al. Humanized Transgenic Mice Are Resistant to Chronic Wasting Disease Prions From Norwegian Reindeer and Moose. J. Infect. Dis. 2021, 226, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.L.R.; Peterson, A.; Phillipson, C.; Walker, J.M.; Richmond, M.; Jansen, G.H.; Knox, J.D. Prospective Study Demonstrates Utility of EP-QuIC in Creutzfeldt–Jakob Disease Diagnoses. Can. J. Neurol. Sci. 2021, 48, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Vendramelli, R.; Sloan, A.; Waitt, B.; Podhorodecki, L.; Godal, D.; Knox, J.D. Endpoint Quaking-Induced Conversion: A Sensitive, Specific, and High-Throughput Method for Antemortem Diagnosis of Creutzfeldt-Jacob Disease. J. Clin. Microbiol. 2016, 54, 1751–1754. [Google Scholar] [CrossRef] [PubMed]
- Argue, C.K.; Ribble, C.; Lees, V.W.; McLane, J.; Balachandran, A. Epidemiology of an outbreak of chronic wasting disease on elk farms in Saskatchewan. Can. Vet. J. 2007, 48, 1241–1248. [Google Scholar]
- Spraker, T.R.; Balachandran, A.; Zhuang, D.; O’Rourke, K.I. Variable patterns of distribution of PrPCWD in the obex and cranial lymphoid tissues of Rocky Mountain elk (Cervus elaphus nelsoni) with subclinical chronic wasting disease. Vet. Rec. 2004, 155, 295–302. [Google Scholar] [CrossRef]
- Fraser, H.; Dickinson, A.G. The sequential development of the brain lesions of scrapie in three strains of mice. J. Comp. Pathol. 1968, 78, 301–311. [Google Scholar] [CrossRef]
- Levine, D.J.; Stöhr, J.; Falese, L.E.; Ollesch, J.; Wille, H.; Prusiner, S.B.; Long, J.R. Mechanism of Scrapie Prion Precipitation with Phosphotungstate Anions. ACS Chem. Biol. 2015, 10, 1269–1277. [Google Scholar] [CrossRef]
- Woerman, A.L.; Stöhr, J.; Aoyagi, A.; Rampersaud, R.; Krejciova, Z.; Watts, J.C.; Ohyama, T.; Patel, S.; Widjaja, K.; Oehler, A.; et al. Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4949–E4958. [Google Scholar] [CrossRef]
- Myskiw, J.; Lamoureux, L.; Peterson, A.; Knox, D.; Jansen, G.H.; Coulthart, M.B.; Booth, S.A. Development of an Automated Capillary Immunoassay to Detect Prion Glycotypes in Creutzfeldt-Jakob Disease. Lab. Investig. 2023, 103, 100029. [Google Scholar] [CrossRef]
- Orrù, C.D.; Groveman, B.R.; Hughson, A.G.; Manca, M.; Raymond, L.D.; Raymond, G.J.; Campbell, K.J.; Anson, K.J.; Kraus, A.; Caughey, B. RT-QuIC Assays for Prion Disease Detection and Diagnostics. Methods Mol. Biol. 2017, 1658, 185–203. [Google Scholar]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Nonno, R.; Di Bari, M.A.; Pirisinu, L.; D’Agostino, C.; Vanni, I.; Chiappini, B.; Marcon, S.; Riccardi, G.; Tran, L.; Vikøren, T.; et al. Studies in bank voles reveal strain differences between chronic wasting disease prions from Norway and North America. Proc. Natl. Acad. Sci. USA 2020, 117, 31417–31426. [Google Scholar] [CrossRef] [PubMed]
- Bocharova, O.; Makarava, N.; Pandit, N.P.; Molesworth, K.; Baskakov, I.V. Multiple steps of prion strain adaptation to a new host. Front. Neurosci 2024, 18, 1329010. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Giles, K.; Patel, S.; Oehler, A.; De Armond, S.J.; Prusiner, S.B. Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog. 2014, 10, e1003990. [Google Scholar] [CrossRef]
- Di Bari, M.A.; Chianini, F.; Vaccari, G.; Esposito, E.; Conte, M.; Eaton, S.L.; Hamilton, S.; Finlayson, J.; Steele, P.J.; Dagleish, M.P.; et al. The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J. Gen. Virol. 2008, 89 Pt 12, 2975–2985. [Google Scholar] [CrossRef]
- Agrimi, U.; Nonno, R.; Dell’Omo, G.; Di Bari, M.A.; Conte, M.; Chiappini, B.; Esposito, E.; Di Guardo, G.; Windl, O.; Vaccari, G.; et al. Prion protein amino acid determinants of differential susceptibility and molecular feature of prion strains in mice and voles. PLoS Pathog. 2008, 4, e1000113. [Google Scholar] [CrossRef]
- Di Bari, M.A.; Nonno, R.; Castilla, J.; D’Agostino, C.; Pirisinu, L.; Riccardi, G.; Conte, M.; Richt, J.; Kunkle, R.; Langeveld, J.; et al. Chronic wasting disease in bank voles: Characterisation of the shortest incubation time model for prion diseases. PLoS Pathog. 2013, 9, e1003219. [Google Scholar] [CrossRef]
- Orrú, C.D.; Groveman, B.R.; Raymond, L.D.; Hughson, A.G.; Nonno, R.; Zou, W.; Ghetti, B.; Gambetti, P.; Caughey, B. Bank Vole Prion Protein As an Apparently Universal Substrate for RT-QuIC-Based Detection and Discrimination of Prion Strains. PLoS Pathog. 2015, 11, e1004983. [Google Scholar] [CrossRef] [PubMed Central]
- Angers, R.C.; Kang, H.E.; Napier, D.; Browning, S.; Seward, T.; Mathiason, C.; Balachandran, A.; McKenzie, D.; Castilla, J.; Soto, C.; et al. Prion Strain Mutation Determined by Prion Protein Conformational Compatibility and Primary Structure. Science 2010, 328, 1154–1158. [Google Scholar] [CrossRef]
- Nonno, R.; Bari, M.A.D.; Cardone, F.; Vaccari, G.; Fazzi, P.; Dell’Omo, G.; Cartoni, C.; Ingrosso, L.; Boyle, A.; Galeno, R.; et al. Efficient Transmission and Characterization of Creutzfeldt–Jakob Disease Strains in Bank Voles. PLOS Pathog. 2006, 2, e12. [Google Scholar] [CrossRef]
- Parchi, P.; Cescatti, M.; Notari, S.; Schulz-Schaeffer, W.J.; Capellari, S.; Giese, A.; Zou, W.-Q.; Kretzschmar, H.; Ghetti, B.; Brown, P. Agent strain variation in human prion disease: Insights from a molecular and pathological review of the National Institutes of Health series of experimentally transmitted disease. Brain 2010, 133, 3030–3042. [Google Scholar] [CrossRef] [PubMed]
- Korth, C.; Kaneko, K.; Groth, D.; Heye, N.; Telling, G.; Mastrianni, J.; Parchi, P.; Gambetti, P.; Will, R.; Ironside, J.; et al. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc. Natl. Acad. Sci. USA 2003, 100, 4784–4789. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.T.; Will, R.G.; Manson, J.C. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc. Natl. Acad. Sci. USA 2010, 107, 12005–12010. [Google Scholar] [CrossRef] [PubMed]
- Kurt, T.D.; Aguilar-Calvo, P.; Jiang, L.; Rodriguez, J.A.; Alderson, N.; Eisenberg, D.S.; Sigurdson, C.J. Asparagine and glutamine ladders promote cross-species prion conversion. J. Biol. Chem. 2017, 292, 19076–19086. [Google Scholar] [CrossRef]
- Kobayashi, A.; Matsuura, Y.; Takeuchi, A.; Yamada, M.; Miyoshi, I.; Mohri, S.; Kitamoto, T. A domain responsible for spontaneous conversion of bank vole prion protein. Brain Pathol. 2018, 29, 155–163. [Google Scholar] [CrossRef]
- Arshad, H.; Eid, S.; Mehra, S.; Williams, D.; Kaczmarczyk, L.; Stuart, E.; Jackson, W.S.; Schmitt-Ulms, G.; Watts, J.C. The Brain Interactome of a Permissive Prion Replication Substrate. bioRxiv 2024. bioRxiv:2024.08.19.608704. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Passage | Second Passage | % Decrease in Survival Time Upon Passaging | |||
|---|---|---|---|---|---|
| Inoculum | Survival Time (days ± SD) | Clinical Disease | Survival Time (Days ± SD) | Clinical Disease | |
| MM1 (Group 1) | 229 ± 10 | 6/6 | 164 ± 10 | 6/6 | −28.4% |
| MM1 (Group 2) | 234 ± 36 | 5/5 | |||
| MV1 | 224 ± 26 | 7/7 | |||
| MV2 | >600 * | 0/7 | |||
| VV2 | >1000 * | 0/7 | |||
| CWD | 546 ± 130 | 7/7 | 317 ± 34 | 8/8 | −41.9% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myskiw, J.; Lamoureux, L.; Frost, K.; Fox, R.; Slota, J.A.; Mitchell, G.; Bailey-Elkin, B.A.; Booth, S.A. Transmission and Characterization of Creutzfeldt–Jakob Disease and Chronic Wasting Disease in the North American Deer Mouse. Viruses 2025, 17, 576. https://doi.org/10.3390/v17040576
Myskiw J, Lamoureux L, Frost K, Fox R, Slota JA, Mitchell G, Bailey-Elkin BA, Booth SA. Transmission and Characterization of Creutzfeldt–Jakob Disease and Chronic Wasting Disease in the North American Deer Mouse. Viruses. 2025; 17(4):576. https://doi.org/10.3390/v17040576
Chicago/Turabian StyleMyskiw, Jennifer, Lise Lamoureux, Kathy Frost, Rebecca Fox, Jessy A. Slota, Gordon Mitchell, Ben A. Bailey-Elkin, and Stephanie A. Booth. 2025. "Transmission and Characterization of Creutzfeldt–Jakob Disease and Chronic Wasting Disease in the North American Deer Mouse" Viruses 17, no. 4: 576. https://doi.org/10.3390/v17040576
APA StyleMyskiw, J., Lamoureux, L., Frost, K., Fox, R., Slota, J. A., Mitchell, G., Bailey-Elkin, B. A., & Booth, S. A. (2025). Transmission and Characterization of Creutzfeldt–Jakob Disease and Chronic Wasting Disease in the North American Deer Mouse. Viruses, 17(4), 576. https://doi.org/10.3390/v17040576