
Short-Read and Long-Read Whole Genome Sequencing for SARS-CoV-2 Variants Identification
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Total Nucleic Acids (TNA) Extraction
2.2. Genomic Sequencing by Illumina NovaSeq Platform
2.3. Genomic Sequencing by ONT MinION Platform
2.4. Genomic Sequencing by PacBio Sequel II Platform
2.5. Quality Control and Trimming
2.6. Genome Mapping and Variant Calling
2.7. Phylogenetic Placement and Clade/Lineage Assignment
2.8. Statistical Analyses and Visualization
3. Results
3.1. Sequencing Quality Statistics
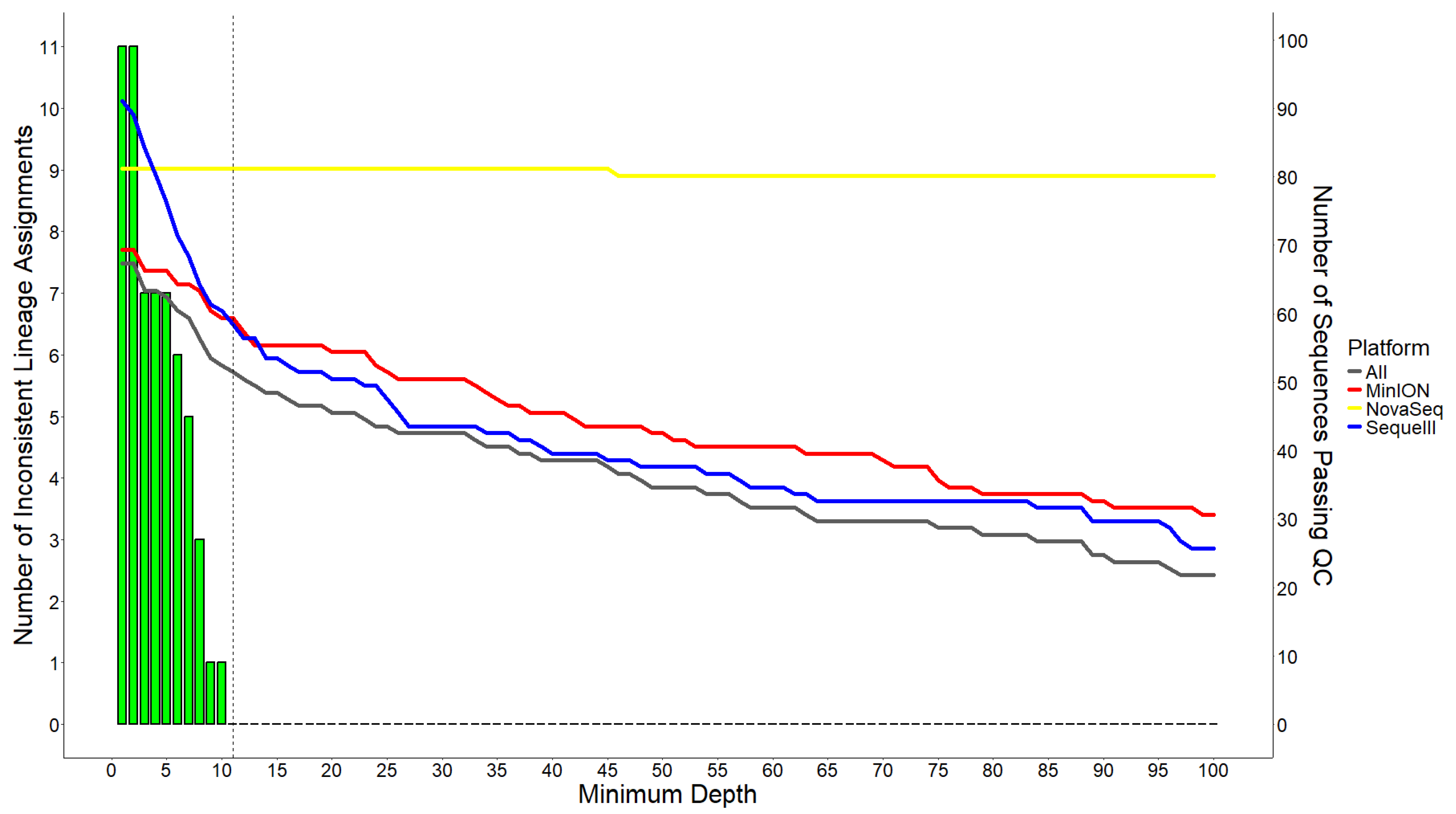
3.2. Sequence Pass Rate and Lineage Assignment Inconsistency
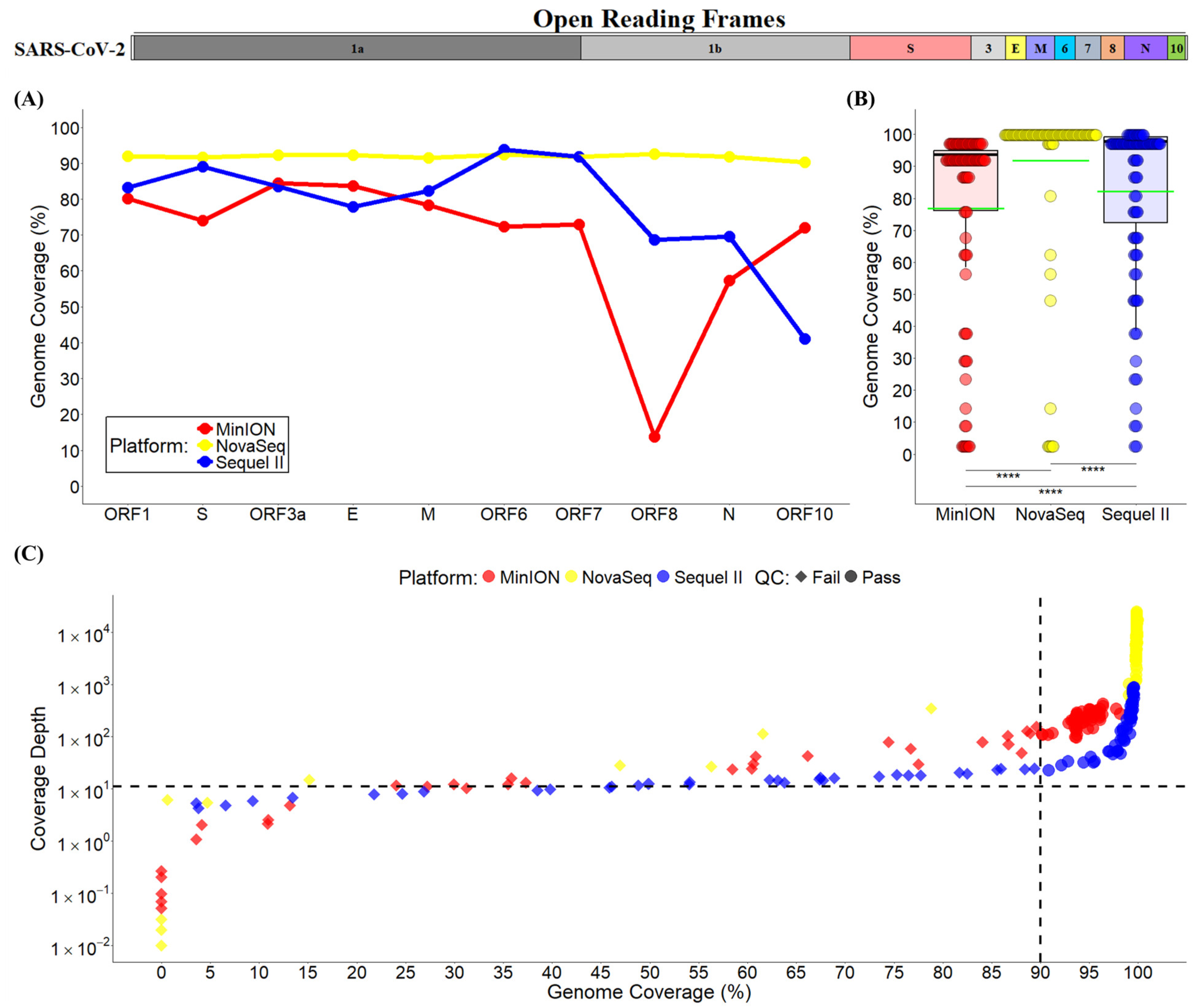
3.3. Mapping Quality and Genome Coverage
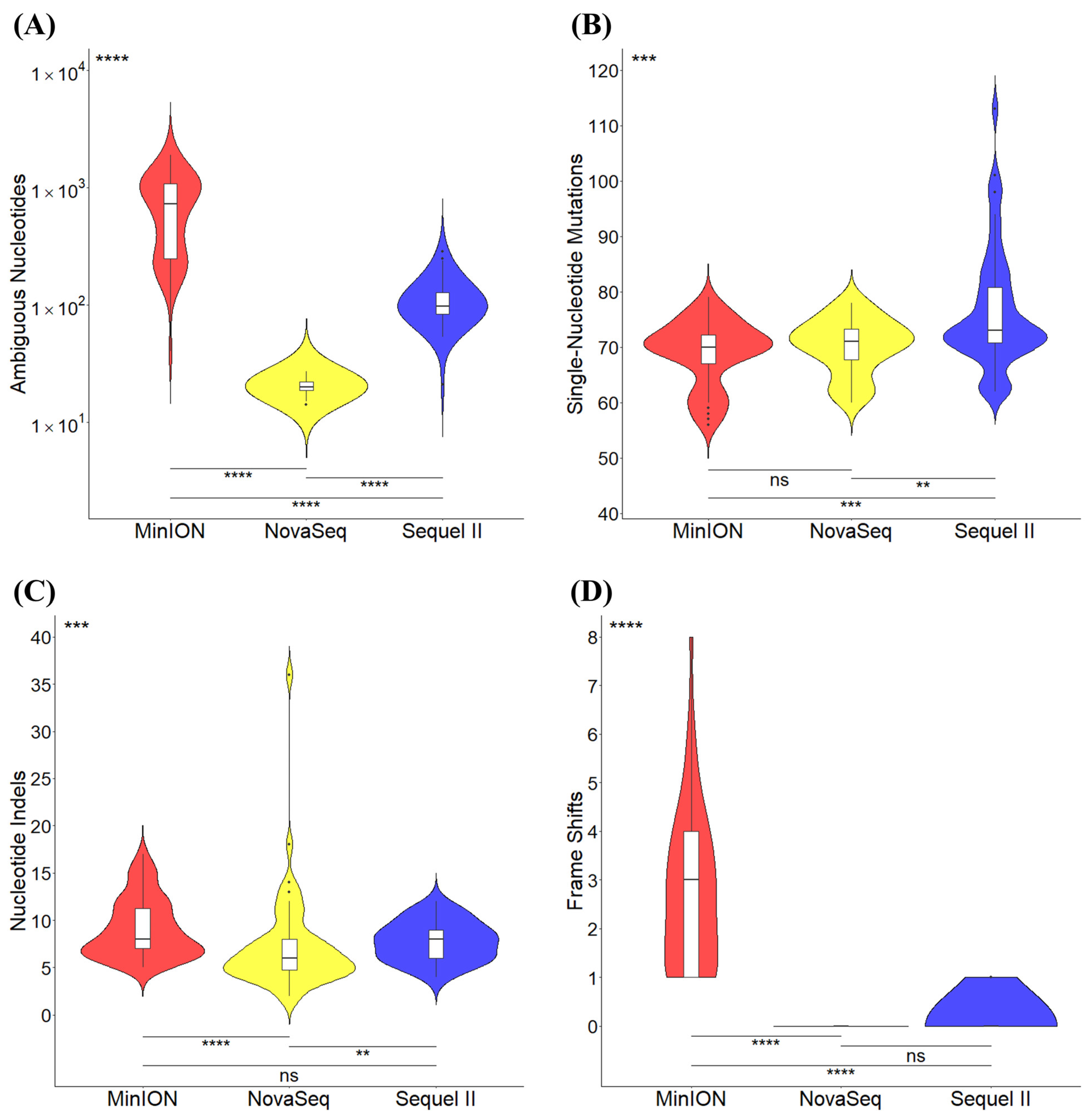
3.4. Consensus Genome Ambiguity and Variant Calling
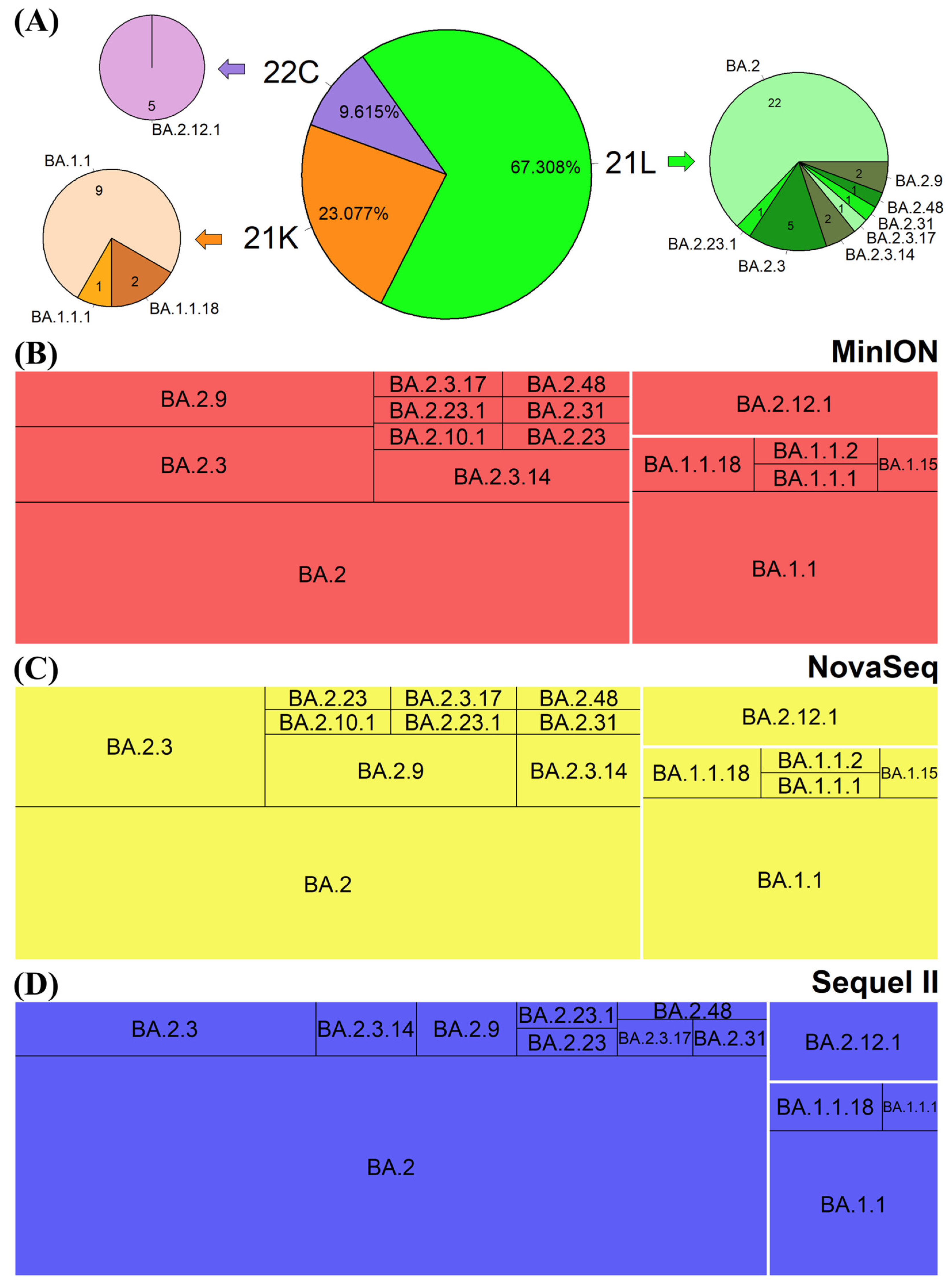
3.5. Phylogenetic Placement of the SARS-CoV-2 Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Msemburi, W.; Karlinsky, A.; Knutson, V.; Aleshin-Guendel, S.; Chatterji, S.; Wakefield, J. The WHO estimates of excess mortality associated with the COVID-19 pandemic. Nature 2023, 613, 130–137. [Google Scholar] [CrossRef]
- Yao, Z.; Zhang, L.; Duan, Y.; Tang, X.; Lu, J. Molecular insights into the adaptive evolution of SARS-CoV-2 spike protein. J. Infect. 2024, 88, 106121. [Google Scholar] [CrossRef] [PubMed]
- Reuschl, A.K.; Thorne, L.G.; Whelan, M.V.X.; Ragazzini, R.; Furnon, W.; Cowton, V.M.; De Lorenzo, G.; Mesner, D.; Turner, J.L.E.; Dowgier, G.; et al. Evolution of enhanced innate immune suppression by SARS-CoV-2 Omicron subvariants. Nat. Microbiol. 2024, 9, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Tosta, S.; Moreno, K.; Schuab, G.; Fonseca, V.; Segovia, F.M.C.; Kashima, S.; Elias, M.C.; Sampaio, S.C.; Ciccozzi, M.; Alcantara, L.C.J.; et al. Global SARS-CoV-2 genomic surveillance: What we have learned (so far). Infect. Genet. Evol. 2023, 108, 105405. [Google Scholar] [CrossRef] [PubMed]
- Sookaromdee, P.; Wiwanitkit, V. Next-Generation Sequencing for Epidemiological Study of COVID and False Negative. Clin. Lab. 2024, 70. [Google Scholar] [CrossRef]
- John, G.; Sahajpal, N.S.; Mondal, A.K.; Ananth, S.; Williams, C.; Chaubey, A.; Rojiani, A.M.; Kolhe, R. Next-Generation Sequencing (NGS) in COVID-19: A Tool for SARS-CoV-2 Diagnosis, Monitoring New Strains and Phylodynamic Modeling in Molecular Epidemiology. Curr. Issues Mol. Biol. 2021, 43, 845–867. [Google Scholar] [CrossRef]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next generation sequencing of SARS-CoV-2 genomes: Challenges, applications and opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef]
- Cook, R.; Brown, N.; Rihtman, B.; Michniewski, S.; Redgwell, T.; Clokie, M.; Stekel, D.J.; Chen, Y.; Scanlan, D.J.; Hobman, J.L.; et al. The long and short of it: Benchmarking viromics using Illumina, Nanopore and PacBio sequencing technologies. Microb. Genom. 2024, 10, 001198. [Google Scholar] [CrossRef]
- Meslier, V.; Quinquis, B.; Da Silva, K.; Plaza Onate, F.; Pons, N.; Roume, H.; Podar, M.; Almeida, M. Benchmarking second and third-generation sequencing platforms for microbial metagenomics. Sci. Data 2022, 9, 694. [Google Scholar] [CrossRef]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Foox, J.; Tighe, S.W.; Nicolet, C.M.; Zook, J.M.; Byrska-Bishop, M.; Clarke, W.E.; Khayat, M.M.; Mahmoud, M.; Laaguiby, P.K.; Herbert, Z.T.; et al. Performance assessment of DNA sequencing platforms in the ABRF Next-Generation Sequencing Study. Nat. Biotechnol. 2021, 39, 1129–1140. [Google Scholar] [CrossRef]
- Carbo, E.C.; Mourik, K.; Boers, S.A.; Munnink, B.O.; Nieuwenhuijse, D.; Jonges, M.; Welkers, M.R.A.; Matamoros, S.; Slooten, J.v.H.T.; Kraakman, M.E.M.; et al. A comparison of five Illumina, Ion Torrent, and nanopore sequencing technology-based approaches for whole genome sequencing of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 2023, 42, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, D.; Jayadas, T.T.P.; Jayathilaka, D.; Jeewandara, C.; Dissanayake, O.; Guruge, D.; Ariyaratne, D.; Gunasinghe, D.; Gomes, L.; Wijesinghe, A.; et al. Comparison of different sequencing techniques for identification of SARS-CoV-2 variants of concern with multiplex real-time PCR. PLoS ONE 2022, 17, e0265220. [Google Scholar] [CrossRef] [PubMed]
- Papa Mze, N.; Beye, M.; Kacel, I.; Tola, R.; Basco, L.; Bogreau, H.; Colson, P.; Fournier, P.-E. Simultaneous SARS-CoV-2 Genome Sequencing of 384 Samples on an Illumina MiSeq Instrument through Protocol Optimization. Genes 2022, 13, 1648. [Google Scholar] [CrossRef]
- Liu, T.; Chen, Z.; Chen, W.; Chen, X.; Hosseini, M.; Yang, Z.; Li, J.; Ho, D.; Turay, D.; Gheorghe, C.P.; et al. A benchmarking study of SARS-CoV-2 whole-genome sequencing protocols using COVID-19 patient samples. iScience 2021, 24, 102892. [Google Scholar] [CrossRef]
- Freed, N.E.; Vlkova, M.; Faisal, M.B.; Silander, O.K. Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and Oxford Nanopore Rapid Barcoding. Biol. Methods Protoc. 2020, 5, bpaa014. [Google Scholar] [CrossRef]
- Shepard, S.S.; Meno, S.; Bahl, J.; Wilson, M.M.; Barnes, J.; Neuhaus, E. Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genom. 2016, 17, 708. [Google Scholar]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakisa, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Brito, A.F.; Semenova, E.; Dudas, G.; Hassler, G.W.; Kalinich, C.C.; Kraemer, M.U.G.; Ho, J.; Tegally, H.; Githinji, G.; Agoti, C.N.; et al. Global disparities in SARS-CoV-2 genomic surveillance. Nat. Commun. 2022, 13, 7003. [Google Scholar] [CrossRef]
- Pillay, S.; Giandhari, J.; Tegally, H.; Wilkinson, E.; Chimukangara, B.; Lessells, R.; Moosa, Y.; Mattison, S.; Gazy, I.; Fish, M.; et al. Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic. Genes 2020, 11, 949. [Google Scholar] [CrossRef]
- Seth-Smith, H.M.B.; Bonfiglio, F.; Cuenod, A.; Reist, J.; Egli, A.; Wuthrich, D. Evaluation of Rapid Library Preparation Protocols for Whole Genome Sequencing Based Outbreak Investigation. Front. Public Health 2019, 7, 241. [Google Scholar] [CrossRef] [PubMed]
- Gerber, Z.; Daviaud, C.; Delafoy, D.; Sandron, F.; Alidjinou, E.K.; Mercier, J.; Gerber, S.; Meyer, V.; Boland, A.; Bocket, L.; et al. A comparison of high-throughput SARS-CoV-2 sequencing methods from nasopharyngeal samples. Sci. Rep. 2022, 12, 12561. [Google Scholar] [CrossRef] [PubMed]
- Bhoyar, R.C.; Jain, A.; Sehgal, P.; Divakar, M.K.; Sharma, D.; Imran, M.; Jolly, B.; Ranjan, G.; Rophina, M.; Sharma, S.; et al. High throughput detection and genetic epidemiology of SARS-CoV-2 using COVIDSeq next-generation sequencing. PLoS ONE 2021, 16, e0247115. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, S.; Cao, S.; Liu, Y.; Cui, Z.; Wang, Y.; Guo, H. Long-read sequencing settings for efficient structural variation detection based on comprehensive evaluation. BMC Bioinform. 2021, 22, 552. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tong, Y.; Wang, K. Genome-wide detection of short tandem repeat expansions by long-read sequencing. BMC Bioinform. 2020, 21 (Suppl. 21), 542. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bhattacharya, M.; Nag, S.; Dhama, K.; Chakraborty, C. A Detailed Overview of SARS-CoV-2 Omicron: Its Sub-Variants, Mutations and Pathophysiology, Clinical Characteristics, Immunological Landscape, Immune Escape, and Therapies. Viruses 2023, 15, 167. [Google Scholar] [CrossRef]
- O’Toole, A.; Pybus, O.G.; Abram, M.E.; Kelly, E.J.; Rambaut, A. Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. BMC Genom. 2022, 23, 121. [Google Scholar] [CrossRef]
- de Bernardi Schneider, A.; Su, M.; Hinrichs, A.S.; Wang, J.; Amin, H.; Bell, J.; Wadford, D.A.; O’Toole, Á.; Scher, E.; Perry, M.D.; et al. SARS-CoV-2 lineage assignments using phylogenetic placement/UShER are superior to pangoLEARN machine-learning method. Virus Evol. 2024, 10, vead085. [Google Scholar] [CrossRef]
- Gangavarapu, K.; Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C.M.; Zaiets, K.; et al. Outbreak.info genomic reports: Scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nat. Methods 2023, 20, 512–522. [Google Scholar] [CrossRef]
- Zou, J.; Kurhade, C.; Xia, H.; Liu, M.; Xie, X.; Ren, P.; Shi, P.-Y. Cross-neutralization of Omicron BA.1 against BA.2 and BA.3 SARS-CoV-2. Nat. Commun. 2022, 13, 2956. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Ajay, S.S.; Parker, S.C.; Abaan, H.O.; Fajardo, K.V.; Margulies, E.H. Accurate and comprehensive sequencing of personal genomes. Genome Res. 2011, 21, 1498–1505. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Connor, R.; Shakya, M.; Yarmosh, D.A.; Maier, W.; Martin, R.; Bradford, R.; Brister, J.R.; Chain, P.S.G.; Copeland, C.A.; di Iulio, J.; et al. Recommendations for Uniform Variant Calling of SARS-CoV-2 Genome Sequence across Bioinformatic Workflows. Viruses 2024, 16, 430. [Google Scholar] [CrossRef]
- Jayme, G.; Liu, J.L.; Galvez, J.H.; Reiling, S.J.; Celikkol, S.; N’Guessan, A.; Lee, S.; Chen, S.-H.; Tsitouras, A.; Sanchez-Quete, F.; et al. Combining Short- and Long-Read Sequencing Technologies to Identify SARS-CoV-2 Variants in Wastewater. Viruses 2024, 16, 1495. [Google Scholar] [CrossRef]
- Ashraf, H.; Ebler, J.; Marschall, T. Allele detection using k-mer-based sequencing error profiles. Bioinform. Adv. 2023, 3, vbad149. [Google Scholar] [CrossRef] [PubMed]
- Laehnemann, D.; Borkhardt, A.; McHardy, A.C. Denoising DNA deep sequencing data-high-throughput sequencing errors and their correction. Brief. Bioinform. 2016, 17, 154–179. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Zhou, P.; Mudge, J.; Gurtowski, J.; Lee, H.; Ramaraj, T.; Walenz, B.P.; Liu, J.; Stupar, R.M.; Denny, R.; et al. Hybrid assembly with long and short reads improves discovery of gene family expansions. BMC Genom. 2017, 18, 541. [Google Scholar] [CrossRef]
- Pirooznia, M.; Kramer, M.; Parla, J.; Goes, F.S.; Potash, J.B.; McCombie, W.R.; Zandi, P.P. Validation and assessment of variant calling pipelines for next-generation sequencing. Hum. Genom. 2014, 8, 14. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequencing Platform | TOTAL | AVERAGE | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yield (Mbp) | Reads | %Q20 | %Q30 | %Passing QC Rate 1 | Read Length (bp) | %Genome Coverage | Depth | %N | SNPs | Indels | FSs | |
| NovaSeq | 16,889.40 | 261,428,474 | 100.00 | 100.00 | 89.13% (82/92) | 117.42 | 91.83 | 9642.84 | 6.98 | 66.82 | 6.75 | 0.10 |
| MinION | 576.16 | 932,604 | 97.83 | 0.00 | 76.09% (70/92) | 584.02 | 76.91 | 157.10 | 15.70 | 58.79 | 9.01 | 4.68 |
| Sequel II | 505.91 | 630,330 | 100.00 | 100.00 | 100.00% (92/92) | 1129.45 | 82.22 | 176.85 | 0.95 | 90.55 | 8.26 | 0.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, M.; Davis, M.L.; Bentz, M.L.; Burgin, A.; Burroughs, M.; Padilla, J.; Nobles, S.; Unoarumhi, Y.; Tang, K. Short-Read and Long-Read Whole Genome Sequencing for SARS-CoV-2 Variants Identification. Viruses 2025, 17, 584. https://doi.org/10.3390/v17040584
Peng M, Davis ML, Bentz ML, Burgin A, Burroughs M, Padilla J, Nobles S, Unoarumhi Y, Tang K. Short-Read and Long-Read Whole Genome Sequencing for SARS-CoV-2 Variants Identification. Viruses. 2025; 17(4):584. https://doi.org/10.3390/v17040584
Chicago/Turabian StylePeng, Mengfei, Morgan L. Davis, Meghan L. Bentz, Alex Burgin, Mark Burroughs, Jasmine Padilla, Sarah Nobles, Yvette Unoarumhi, and Kevin Tang. 2025. "Short-Read and Long-Read Whole Genome Sequencing for SARS-CoV-2 Variants Identification" Viruses 17, no. 4: 584. https://doi.org/10.3390/v17040584
APA StylePeng, M., Davis, M. L., Bentz, M. L., Burgin, A., Burroughs, M., Padilla, J., Nobles, S., Unoarumhi, Y., & Tang, K. (2025). Short-Read and Long-Read Whole Genome Sequencing for SARS-CoV-2 Variants Identification. Viruses, 17(4), 584. https://doi.org/10.3390/v17040584