
Immunological Control of Herpes Simplex Virus Type 1 Infection: A Non-Thermal Plasma-Based Approach

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HSV-1 as the Etiological Agent of Herpes Labialis
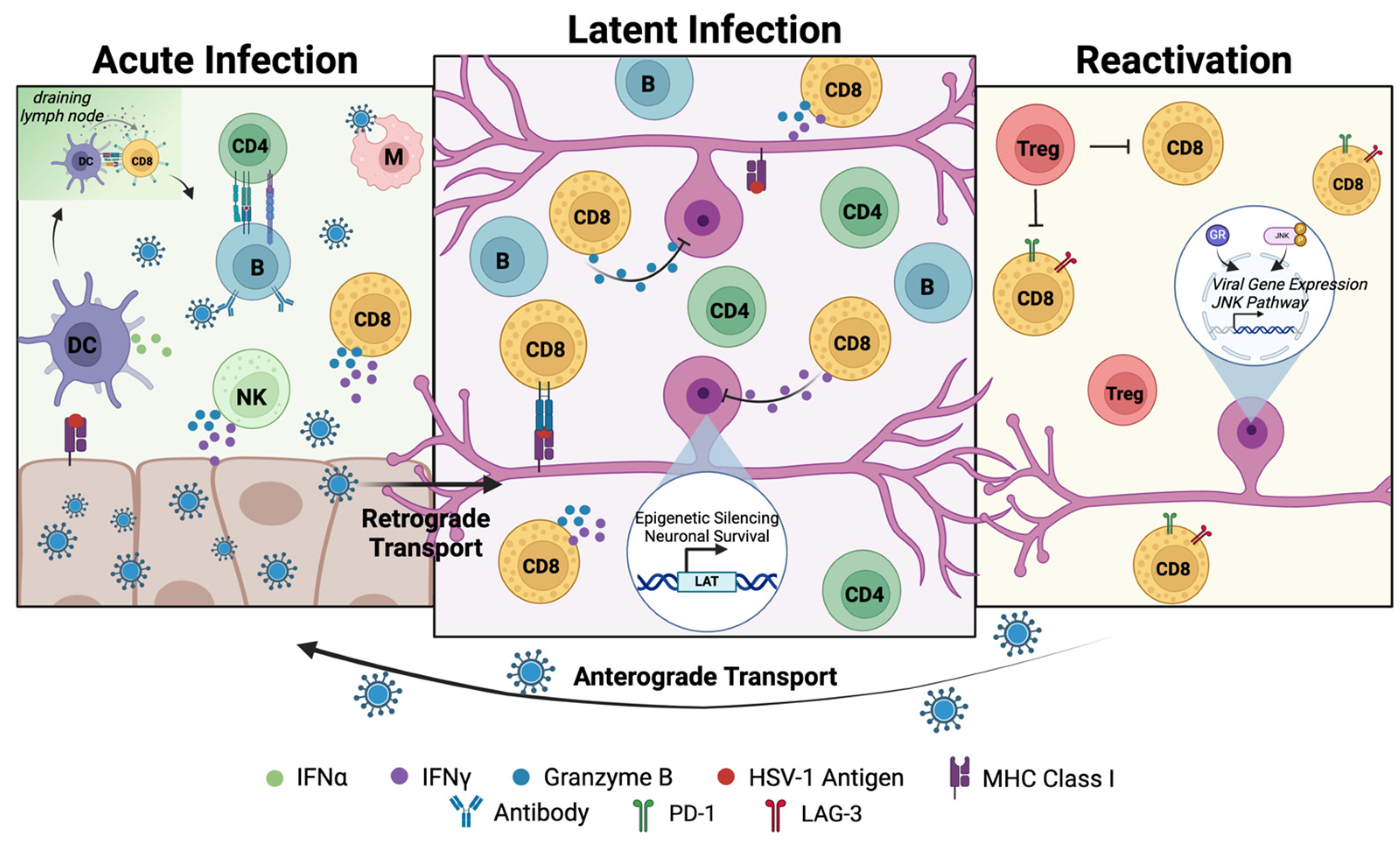
2.1. Acute Epithelial Infection in Herpes Labialis
2.2. Establishment and Maintenance of Latent Virus Reservoirs in Neurons
2.3. Reactivation from Latent Infection
3. Host Responses and Immune Control of Herpes Labialis
3.1. Innate Immune Responses to HSV-1 Transmission and Acute Infection
3.1.1. HSV-1 Is Detected by Cellular Sensors During Acute Infection
3.1.2. Innate Immune Cells Mediate Antiviral Immune Responses Against HSV-1 and Promote Adaptive Immunity
3.2. Adaptive Immune Responses to Productive Infection
3.2.1. CD8+ T Cells
3.2.2. CD4+ T Cells
3.3. Adaptive Immune Responses to Latent Infection and Reactivation
3.3.1. Effects of Immune Responses on Established Reservoirs in Innervating Neurons
3.3.2. Adaptive Immune Responses Do Not Result in the Clearance of Latently Infected Neurons
3.3.3. Effects of Adaptive Immune Responses on Virus Reactivation
In the Nervous System
In the Recurrent Lesion Caused by Reactivated Productive Infection
4. Current Immunological Interventions for Treating Herpes Labialis
4.1. Standard of Care Pharmaceutical Options May Be Detrimental to Host Immune Responses to HSV-1 Infection
4.2. Lessons Learned from Vaccines Under Development for HSV-1 Infection
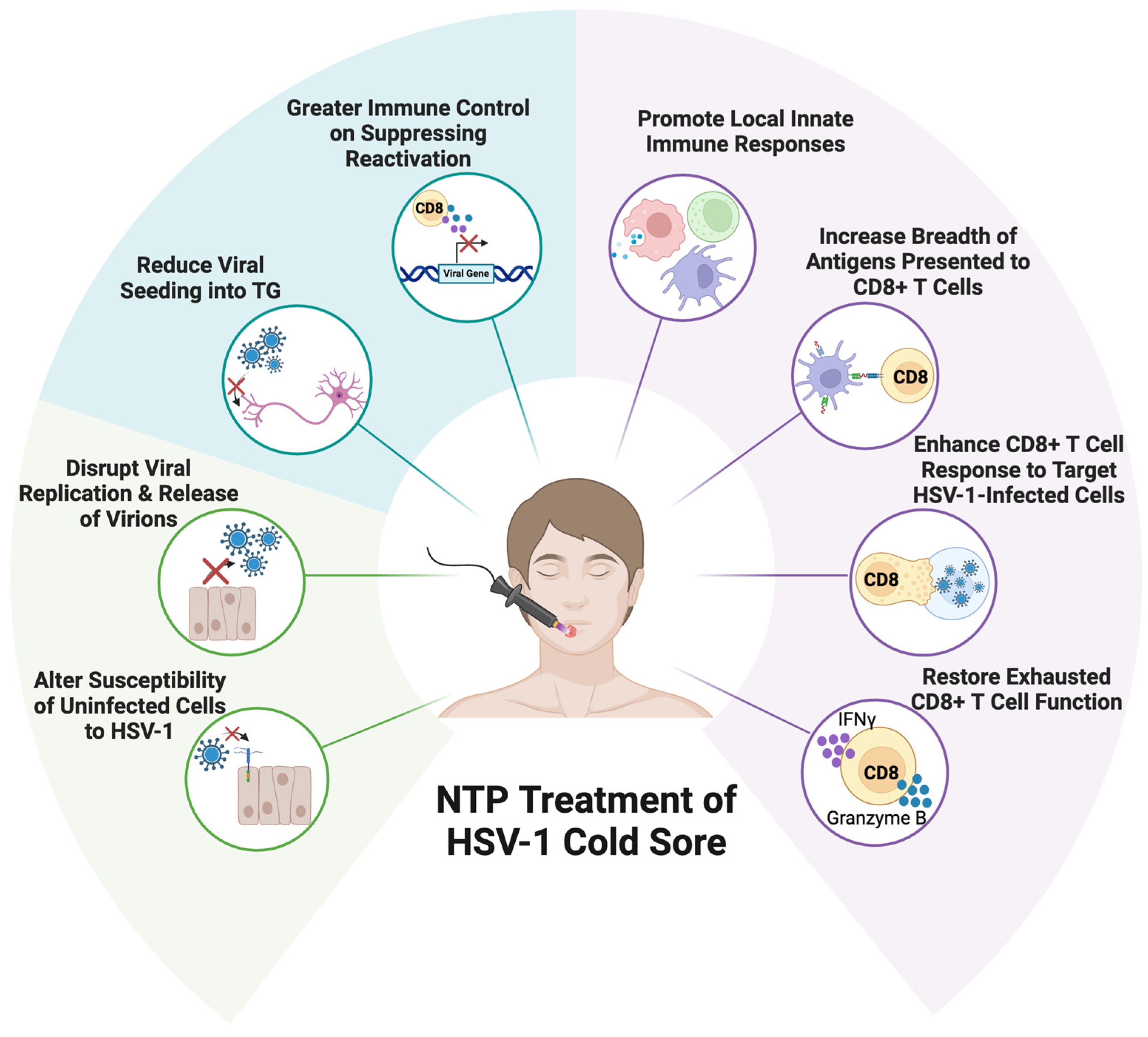
5. Non-Thermal Plasma as the Basis for a Novel Antiviral and Immunological Therapy for Herpes Labialis
5.1. NTP Has Multiple Effects on HSV-1 Infection and Replication
5.1.1. NTP Disrupts HSV-1 Replication in Infected Cells
5.1.2. NTP Reduces Keratinocyte Susceptibility to HSV-1 Infection
5.2. NTP as a Multi-Functional Therapy for HSV-1 Infection
5.2.1. NTP Antiviral Activities Will Limit Virus Spread and Pathogenesis in the Lesion
5.2.2. Potential Effects of NTP on the HSV-1 Latent Reservoir in Neurons
5.2.3. NTP-Associated Immunomodulation During HSV-1 Infection
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HSV-1 | Herpes simplex virus type 1 |
| NTP | Non-thermal plasma |
| TG | Trigeminal ganglia |
| CNS | Central nervous system |
| SOC | Standard of care |
| DC | Dendritic cell |
| LAT | Latency-associated transcript |
| JNK | Jun-N-terminal kinase |
| GR | Glucocorticoid receptor |
| PRR | Pattern recognition receptor |
| LC | Langerhan cell |
| APC | Antigen-presenting cell |
| PAMP | Pathogen-associated molecular pattern |
| DAMP | Damage-associated molecular pattern |
| IFN | Interferon |
| NK | Natural killer cell |
| TLR | Toll-like receptor |
| NK-κB | Nuclear factor-κB |
| Myd88 | Myeloid differentiation factor 88 |
| TRAF6 | Tumor necrosis factor receptor-associated factor 6 |
| cGas | Cyclic GMP-AMP synthase |
| STING | Stimulator of interferon genes |
| IFI16 | IFN-γ-inducible protein 16 |
| MDA5 | Melona differentiation-associated protein 5 |
| pDC | Plasmacytoid DC |
| cDC | Conventional DC |
| TCR | T cell receptor |
| TH1 | T helper 1 cell |
| TH2 | T helper 2 cell |
| TFH | T follicular helper cell |
| TRM | Tissue-resident memory T cell |
| TEM | Effector memory T cell |
| Treg | T regulatory cell |
| ACV | Acyclovir |
| TK | Thymidine kinase |
| RONS | Reactive oxygen and nitrogen species |
References
- Herpes Simplex Virus. 2024. Available online: https://www.who.int/news-room/fact-sheets/detail/herpes-simplex-virus (accessed on 6 December 2024).
- Zhu, S.; Viejo-Borbolla, A. Pathogenesis and virulence of herpes simplex virus. Virulence 2021, 12, 2670–2702. [Google Scholar] [CrossRef]
- Gopinath, D.; Koe, K.H.; Maharajan, M.K.; Panda, S. A Comprehensive Overview of Epidemiology, Pathogenesis and the Management of Herpes Labialis. Viruses 2023, 15, 225. [Google Scholar] [CrossRef]
- Bradshaw, M.J.; Venkatesan, A. Herpes Simplex Virus-1 Encephalitis in Adults: Pathophysiology, Diagnosis, and Management. Neurotherapeutics 2016, 13, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease: Increasing evidence for a major role of the virus. Front. Aging Neurosci. 2014, 6, 202. [Google Scholar] [CrossRef] [PubMed]
- Cairns Dana, M.; Rouleau, N.; Parker Rachael, N.; Walsh Katherine, G.; Gehrke, L.; Kaplan David, L. A 3D human brain–like tissue model of herpes-induced Alzheimer’s disease. Sci. Adv. 2020, 6, eaay8828. [Google Scholar] [CrossRef] [PubMed]
- Cernik, C.; Gallina, K.; Brodell, R.T. The Treatment of Herpes Simplex Infections: An Evidence-Based Review. Arch. Intern. Med. 2008, 168, 1137–1144. [Google Scholar] [CrossRef]
- Webre, J.M.; Hill, J.M.; Nolan, N.M.; Clement, C.; McFerrin, H.E.; Bhattacharjee, P.S.; Hsia, V.; Neumann, D.M.; Foster, T.P.; Lukiw, W.J.; et al. Rabbit and mouse models of HSV-1 latency, reactivation, and recurrent eye diseases. J. Biomed. Biotechnol. 2012, 2012, 612316. [Google Scholar] [CrossRef]
- Karasneh, G.A.; Shukla, D. Herpes simplex virus infects most cell types in vitro: Clues to its success. Virol. J. 2011, 8, 481. [Google Scholar] [CrossRef]
- Madavaraju, K.; Koganti, R.; Volety, I.; Yadavalli, T.; Shukla, D. Herpes Simplex Virus Cell Entry Mechanisms: An Update. Front. Cell. Infect. Microbiol. 2021, 10, 617578. [Google Scholar] [CrossRef]
- Hilterbrand Adam, T.; Daly Raecliffe, E.; Heldwein Ekaterina, E. Contributions of the Four Essential Entry Glycoproteins to HSV-1 Tropism and the Selection of Entry Routes. mBio 2021, 12, e00143-21. [Google Scholar] [CrossRef]
- St. Leger, A.J.; Peters, B.; Sidney, J.; Sette, A.; Hendricks, R.L. Defining the Herpes Simplex Virus-Specific CD8+ T Cell Repertoire in C57BL/6 Mice. J. Immunol. 2011, 186, 3927–3933. [Google Scholar] [CrossRef] [PubMed]
- BenMohamed, L.; Bertrand, G.; McNamara, C.D.; Gras-Masse, H.; Hammer, J.; Wechsler, S.L.; Nesburn, A.B. Identification of Novel Immunodominant CD4+ Th1-Type T-Cell Peptide Epitopes from Herpes Simplex Virus Glycoprotein D That Confer Protective Immunity. J. Virol. 2003, 77, 9463–9473. [Google Scholar] [CrossRef]
- Tebaldi, G.; Pritchard, S.M.; Nicola, A.V. Herpes Simplex Virus Entry by a Nonconventional Endocytic Pathway. J. Virol. 2020, 94, e01910-20. [Google Scholar] [CrossRef]
- Wolfstein, A.; Nagel, C.-H.; Radtke, K.; Döhner, K.; Allan, V.J.; Sodeik, B. The Inner Tegument Promotes Herpes Simplex Virus Capsid Motility Along Microtubules in vitro. Traffic 2006, 7, 227–237. [Google Scholar] [CrossRef]
- Fan, W.H.; Roberts, A.P.E.; McElwee, M.; Bhella, D.; Rixon, F.J.; Lauder, R. The Large Tegument Protein pUL36 Is Essential for Formation of the Capsid Vertex-Specific Component at the Capsid-Tegument Interface of Herpes Simplex Virus 1. J. Virol. 2015, 89, 1502–1511. [Google Scholar] [CrossRef]
- Yang, L.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; Zhang, S.; et al. Innate Immune Evasion of Alphaherpesvirus Tegument Proteins. Front. Immunol. 2019, 10, 2196. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Wang, M.; Cheng, A.; Jia, R.; Yang, Q.; Wu, Y.; Zhu, D.; Zhao, X.; Chen, S.; Liu, M.; et al. The Role of VP16 in the Life Cycle of Alphaherpesviruses. Front. Microbiol. 2020, 11, 1910. [Google Scholar] [CrossRef]
- Xing, J.; Ni, L.; Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes Simplex Virus 1-Encoded Tegument Protein VP16 Abrogates the Production of Beta Interferon (IFN) by Inhibiting NF-κB Activation and Blocking IFN Regulatory Factor 3 To Recruit Its Coactivator CBP. J. Virol. 2013, 87, 9788–9801. [Google Scholar] [CrossRef] [PubMed]
- Grosche, L.; Kummer, M.; Steinkasserer, A. What Goes Around, Comes Around—HSV-1 Replication in Monocyte-Derived Dendritic Cells. Front. Microbiol. 2017, 8, 2149. [Google Scholar] [CrossRef]
- Gao, Y.; Cheng, J.; Xu, X.; Li, X.; Zhang, J.; Ma, D.; Jiang, G.; Liao, Y.; Fan, S.; Niu, Z.; et al. HSV-1 Infection of Epithelial Dendritic Cells Is a Critical Strategy for Interfering with Antiviral Immunity. Viruses 2022, 14, 1046. [Google Scholar] [CrossRef]
- Miranda-Saksena, M.; Denes, C.E.; Diefenbach, R.J.; Cunningham, A.L. Infection and Transport of Herpes Simplex Virus Type 1 in Neurons: Role of the Cytoskeleton. Viruses 2018, 10, 92. [Google Scholar] [CrossRef] [PubMed]
- Nicola Anthony, V.; Hou, J.; Major Eugene, O.; Straus Stephen, E. Herpes Simplex Virus Type 1 Enters Human Epidermal Keratinocytes, but Not Neurons, via a pH-Dependent Endocytic Pathway. J. Virol. 2005, 79, 7609–7616. [Google Scholar] [CrossRef] [PubMed]
- Bearer, E.L.; Breakefield, X.O.; Schuback, D.; Reese, T.S.; LaVail, J.H. Retrograde axonal transport of herpes simplex virus: Evidence for a single mechanism and a role for tegument. Proc. Natl. Acad. Sci. USA 2000, 97, 8146–8150. [Google Scholar] [CrossRef]
- Grinde, B. Herpesviruses: Latency and reactivation—Viral strategies and host response. J. Oral Microbiol. 2013, 5, 22766. [Google Scholar] [CrossRef]
- Millhouse, S.; Wigdahl, B. Molecular circuitry regulating herpes simplex virus type 1 latency in neurons. J. Neurovirol. 2000, 6, 6–24. [Google Scholar] [CrossRef]
- Harkness Justine, M.; Kader, M.; DeLuca Neal, A. Transcription of the Herpes Simplex Virus 1 Genome during Productive and Quiescent Infection of Neuronal and Nonneuronal Cells. J. Virol. 2014, 88, 6847–6861. [Google Scholar] [CrossRef] [PubMed]
- Kenny, J.J.; Millhouse, S.; Wotring, M.; Wigdahl, B. Upstream Stimulatory Factor Family Binds to the Herpes Simplex Virus Type 1 Latency-Associated Transcript Promoter. Virology 1997, 230, 381–391. [Google Scholar] [CrossRef]
- Millhouse, S.; Kenny, J.J.; Quinn, P.G.; Lee, V.; Wigdahl, B. ATF/CREB elements in the herpes simplex virus type 1 latency-associated transcript promoter interact with members of the ATF/CREB and AP-1 transcription factor families. J. Biomed. Sci. 1998, 5, 451–464. [Google Scholar] [CrossRef]
- Wang, S.; Song, X.; Rajewski, A.; Santiskulvong, C.; Ghiasi, H. Stacking the odds: Multiple sites for HSV-1 latency. Sci. Adv. 2023, 9, eadf4904. [Google Scholar] [CrossRef]
- Nicoll, M.P.; Hann, W.; Shivkumar, M.; Harman, L.E.R.; Connor, V.; Coleman, H.M.; Proença, J.T.; Efstathiou, S. The HSV-1 Latency-Associated Transcript Functions to Repress Latent Phase Lytic Gene Expression and Suppress Virus Reactivation from Latently Infected Neurons. PLoS Pathog. 2016, 12, e1005539. [Google Scholar] [CrossRef]
- Wigdahl, B.; Scheck, A.C.; Ziegler, R.J.; De Clercq, E.; Rapp, F. Analysis of the herpes simplex virus genome during in vitro latency in human diploid fibroblasts and rat sensory neurons. J. Virol. 1984, 49, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Cliffe Anna, R.; Garber David, A.; Knipe David, M. Transcription of the Herpes Simplex Virus Latency-Associated Transcript Promotes the Formation of Facultative Heterochromatin on Lytic Promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef]
- Wang, Q.-Y.; Zhou, C.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar] [CrossRef]
- Tormanen, K.; Allen, S.; Mott, K.R.; Ghiasi, H. The Latency-Associated Transcript Inhibits Apoptosis via Downregulation of Components of the Type I Interferon Pathway during Latent Herpes Simplex Virus 1 Ocular Infection. J. Virol. 2019, 93, e00103-19. [Google Scholar] [CrossRef] [PubMed]
- Jones, C. Intimate Relationship Between Stress and Human Alpha-Herpes Virus 1 (HSV-1) Reactivation from Latency. Curr. Clin. Microbiol. Rep. 2023, 10, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Suzich, J.B.; Cliffe, A.R. Strength in diversity: Understanding the pathways to herpes simplex virus reactivation. Virology 2018, 522, 81–91. [Google Scholar] [CrossRef]
- Wigdahl, B.L.; Isom, H.C.; Rapp, F. Repression and activation of the genome of herpes simplex viruses in human cells. Proc. Natl. Acad. Sci. USA 1981, 78, 6522–6526. [Google Scholar] [CrossRef]
- Wigdahl, B.L.; Scheck, A.C.; De Clercq, E.; Rapp, F. High Efficiency Latency and Activation of Herpes Simplex Virus in Human Cells. Science 1982, 217, 1145–1146. [Google Scholar] [CrossRef]
- Bucher, L.J.; Wigdahl, B.; Rapp, F. Maintenance of human cytomegalovirus genome in human diploid fibroblast cells. Virology 1983, 130, 269–271. [Google Scholar] [CrossRef]
- Wigdahl, B.L.; Isom, H.C.; Clercq, E.D.; Rapp, F. Activation of herpes simplex virus (HSV) type 1 genome by temperature-sensitive mutants of HSV type 2. Virology 1982, 116, 468–479. [Google Scholar] [CrossRef]
- Holland David, J.; Miranda-Saksena, M.; Boadle Ross, A.; Armati, P.; Cunningham Anthony, L. Anterograde Transport of Herpes Simplex Virus Proteins in Axons of Peripheral Human Fetal Neurons: An Immunoelectron Microscopy Study. J. Virol. 1999, 73, 8503–8511. [Google Scholar] [CrossRef] [PubMed]
- DuRaine, G.; Johnson, D.C. Anterograde transport of α-herpesviruses in neuronal axons. Virology 2021, 559, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Wigdahl, B.L.; Ziegler, R.J.; Sneve, M.; Rapp, F. Herpes simplex virus latency and reactivation in isolated rat sensory neurons. Virology 1983, 127, 159–167. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef]
- Ostler Jeffery, B.; Harrison Kelly, S.; Schroeder, K.; Thunuguntla, P.; Jones, C. The Glucocorticoid Receptor (GR) Stimulates Herpes Simplex Virus 1 Productive Infection, in Part Because the Infected Cell Protein 0 (ICP0) Promoter Is Cooperatively Transactivated by the GR and Krüppel-Like Transcription Factor 15. J. Virol. 2019, 93, e02063-18. [Google Scholar] [CrossRef]
- Harrison Kelly, S.; Wijesekera, N.; Robinson Anastasia, G.J.; Santos Vanessa, C.; Oakley Robert, H.; Cidlowski John, A.; Jones, C. Impaired glucocorticoid receptor function attenuates herpes simplex virus 1 production during explant-induced reactivation from latency in female mice. J. Virol. 2023, 97, e01305–e01323. [Google Scholar] [CrossRef]
- Du, T.; Zhou, G.; Roizman, B. Induction of apoptosis accelerates reactivation of latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc. Natl. Acad. Sci. USA 2012, 109, 14616–14621. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.P.; Wu, S.; Wigdahl, B.; Jennings, S.R. Immunological control of herpes simplex virus infections. J. Neurovirol. 2013, 19, 328–345. [Google Scholar] [CrossRef]
- Wu, R.-Q.; Zhang, D.-F.; Tu, E.; Chen, Q.-M.; Chen, W. The mucosal immune system in the oral cavity—An orchestra of T cell diversity. Int. J. Oral Sci. 2014, 6, 125–132. [Google Scholar] [CrossRef]
- Pelaez-Prestel, H.F.; Sanchez-Trincado, J.L.; Lafuente, E.M.; Reche, P.A. Immune Tolerance in the Oral Mucosa. Int. J. Mol. Sci. 2021, 22, 12149. [Google Scholar] [CrossRef]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef] [PubMed]
- Piipponen, M.; Li, D.; Landén, N.X. The Immune Functions of Keratinocytes in Skin Wound Healing. Int. J. Mol. Sci. 2020, 21, 8790. [Google Scholar] [CrossRef] [PubMed]
- Heath, W.R.; Carbone, F.R. The skin-resident and migratory immune system in steady state and memory: Innate lymphocytes, dendritic cells and T cells. Nat. Immunol. 2013, 14, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Chessa, C.; Bodet, C.; Jousselin, C.; Wehbe, M.; Lévêque, N.; Garcia, M. Antiviral and Immunomodulatory Properties of Antimicrobial Peptides Produced by Human Keratinocytes. Front. Microbiol. 2020, 11, 1155. [Google Scholar] [CrossRef]
- Kubo, A.; Nagao, K.; Yokouchi, M.; Sasaki, H.; Amagai, M. External antigen uptake by Langerhans cells with reorganization of epidermal tight junction barriers. J. Exp. Med. 2009, 206, 2937–2946. [Google Scholar] [CrossRef]
- Hovav, A.H. Dendritic cells of the oral mucosa. Mucosal Immunol. 2014, 7, 27–37. [Google Scholar] [CrossRef]
- Capucha, T.; Mizraji, G.; Segev, H.; Blecher-Gonen, R.; Winter, D.; Khalaileh, A.; Tabib, Y.; Attal, T.; Nassar, M.; Zelentsova, K.; et al. Distinct Murine Mucosal Langerhans Cell Subsets Develop from Pre-dendritic Cells and Monocytes. Immunity 2015, 43, 369–381. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Paludan, S.R.; Bowie, A.G.; Horan, K.A.; Fitzgerald, K.A. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 2011, 11, 143–154. [Google Scholar] [CrossRef]
- Danastas, K.; Miranda-Saksena, M.; Cunningham, A.L. Herpes Simplex Virus Type 1 Interactions with the Interferon System. Int. J. Mol. Sci. 2020, 21, 5150. [Google Scholar] [CrossRef]
- Sutter, J.; Bruggeman, P.J.; Wigdahl, B.; Krebs, F.C.; Miller, V. Manipulation of Oxidative Stress Responses by Non-Thermal Plasma to Treat Herpes Simplex Virus Type 1 Infection and Disease. Int. J. Mol. Sci. 2023, 24, 4673. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, Y.; Zhu, L.; Wang, X.; Richers, B.; Leung, D.Y.M.; Bin, L. Interferon Kappa Is Important for Keratinocyte Host Defense against Herpes Simplex Virus-1. J. Immunol. Res. 2020, 2020, 5084682. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, L.H.; Wilson, S.C.; Morrison, H.M.; Karalis, V.; Chung, J.-Y.J.; Chen, K.J.; Bateup, H.S.; Szpara, M.L.; Lee, A.Y.; Cox, J.S.; et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat. Commun. 2020, 11, 3382. [Google Scholar] [CrossRef]
- Danastas, K.; Guo, G.; Merjane, J.; Hong, N.; Larsen, A.; Miranda-Saksena, M.; Cunningham, A.L. Interferon inhibits the release of herpes simplex virus-1 from the axons of sensory neurons. mBio 2023, 14, e01818–e01823. [Google Scholar] [CrossRef]
- Dumas, A.; Knaus, U.G. Raising the ‘Good’ Oxidants for Immune Protection. Front. Immunol. 2021, 12, 698042. [Google Scholar] [CrossRef]
- Martinez-Martin, N.; Viejo-Borbolla, A. Toll-like receptor-mediated recognition of herpes simplex virus. Front. Biosci. 2010, 2, 718–729. [Google Scholar] [CrossRef]
- Leoni, V.; Gianni, T.; Salvioli, S.; Campadelli-Fiume, G. Herpes Simplex Virus Glycoproteins gH/gL and gB Bind Toll-Like Receptor 2, and Soluble gH/gL Is Sufficient to Activate NF-κB. J. Virol. 2012, 86, 6555–6562. [Google Scholar] [CrossRef]
- Cai, M.; Li, M.; Wang, K.; Wang, S.; Lu, Q.; Yan, J.; Mossman, K.L.; Lin, R.; Zheng, C. The Herpes Simplex Virus 1-Encoded Envelope Glycoprotein B Activates NF-κB through the Toll-Like Receptor 2 and MyD88/TRAF6-Dependent Signaling Pathway. PLoS ONE 2013, 8, e54586. [Google Scholar] [CrossRef]
- Sato, A.; Linehan, M.M.; Iwasaki, A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17343–17348. [Google Scholar] [CrossRef]
- Zolini, G.P.; Lima, G.K.; Lucinda, N.; Silva, M.A.; Dias, M.F.; Pessoa, N.L.; Coura, B.P.; Cartelle, C.T.; Arantes, R.M.E.; Kroon, E.G.; et al. Defense against HSV-1 in a murine model is mediated by iNOS and orchestrated by the activation of TLR2 and TLR9 in trigeminal ganglia. J. Neuroinflamm. 2014, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Luker, G.D.; Barchet, W.; Leib, D.A.; Akira, S.; Colonna, M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 2004, 103, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Bowen, G.N.; Zhou, S.; Cerny, A.; Zacharia, A.; Knipe, D.M.; Finberg, R.W.; Kurt-Jones, E.A. Role of Specific Innate Immune Responses in Herpes Simplex Virus Infection of the Central Nervous System. J. Virol. 2012, 86, 2273–2281. [Google Scholar] [CrossRef]
- Sørensen, L.N.; Reinert, L.S.; Malmgaard, L.; Bartholdy, C.; Thomsen, A.R.; Paludan, S.R. TLR2 and TLR9 Synergistically Control Herpes Simplex Virus Infection in the Brain1. J. Immunol. 2008, 181, 8604–8612. [Google Scholar] [CrossRef]
- Lind, N.A.; Rael, V.E.; Pestal, K.; Liu, B.; Barton, G.M. Regulation of the nucleic acid-sensing Toll-like receptors. Nat. Rev. Immunol. 2022, 22, 224–235. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of Adaptor TRIF in the MyD88-Independent Toll-Like Receptor Signaling Pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Mak, T.W.; Sen, G.; Li, X. Toll-like receptor 3-mediated activation of NF-κB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-β. Proc. Natl. Acad. Sci. USA 2004, 101, 3533–3538. [Google Scholar] [CrossRef]
- Latif, M.B.; Raja, R.; Kessler, P.M.; Sen, G.C. Relative Contributions of the cGAS-STING and TLR3 Signaling Pathways to Attenuation of Herpes Simplex Virus 1 Replication. J. Virol. 2020, 94, e01717-19. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 Deficiency in Patients with Herpes Simplex Encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Ma, F.; Li, B.; Liu, S.-y.; Iyer, S.S.; Yu, Y.; Wu, A.; Cheng, G. Positive Feedback Regulation of Type I IFN Production by the IFN-Inducible DNA Sensor cGAS. J. Immunol. 2015, 194, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Reinert, L.S.; Lopušná, K.; Winther, H.; Sun, C.; Thomsen, M.K.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vægter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the cGAS–STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 2016, 7, 13348. [Google Scholar] [CrossRef]
- Rasmussen, S.B.; Jensen, S.B.; Nielsen, C.; Quartin, E.; Kato, H.; Chen, Z.J.; Silverman, R.H.; Akira, S.; Paludan, S.R. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene-like receptors, which synergize to induce type I interferon production. J. Gen. Virol. 2009, 90 Pt 1, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-H.; MacMillan, J.B.; Chen, Z.J. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Wojtasiak, M.; Pickett, D.L.; Tate, M.D.; Bedoui, S.; Job, E.R.; Whitney, P.G.; Brooks, A.G.; Reading, P.C. Gr-1+ cells, but not neutrophils, limit virus replication and lesion development following flank infection of mice with herpes simplex virus type-1. Virology 2010, 407, 143–151. [Google Scholar] [CrossRef]
- Tumpey, T.M.; Chen, S.H.; Oakes, J.E.; Lausch, R.N. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J. Virol. 1996, 70, 898–904. [Google Scholar] [CrossRef]
- Hor, J.L.; Heath, W.R.; Mueller, S.N. Neutrophils are dispensable in the modulation of T cell immunity against cutaneous HSV-1 infection. Sci. Rep. 2017, 7, 41091. [Google Scholar] [CrossRef]
- Lee, D.H.; Ghiasi, H. Roles of M1 and M2 Macrophages in Herpes Simplex Virus 1 Infectivity. J. Virol. 2017, 91, e00578-17. [Google Scholar] [CrossRef]
- Lang, J.; Bohn, P.; Bhat, H.; Jastrow, H.; Walkenfort, B.; Cansiz, F.; Fink, J.; Bauer, M.; Olszewski, D.; Ramos-Nascimento, A.; et al. Acid ceramidase of macrophages traps herpes simplex virus in multivesicular bodies and protects from severe disease. Nat. Commun. 2020, 11, 1338. [Google Scholar] [CrossRef]
- Lang, P.A.; Recher, M.; Honke, N.; Scheu, S.; Borkens, S.; Gailus, N.; Krings, C.; Meryk, A.; Kulawik, A.; Cervantes-Barragan, L.; et al. Tissue Macrophages Suppress Viral Replication and Prevent Severe Immunopathology in an Interferon-I-Dependent Manner in Mice. Hepatology 2010, 52, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Baraz, L.; Khazanov, E.; Condiotti, R.; Kotler, M.; Nagler, A. Natural killer (NK) cells prevent virus production in cell culture. Bone Marrow Transplant. 1999, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Björkström, N.K.; Strunz, B.; Ljunggren, H.-G. Natural killer cells in antiviral immunity. Nat. Rev. Immunol. 2022, 22, 112–123. [Google Scholar] [CrossRef]
- York, I.A.; Roop, C.; Andrews, D.W.; Riddell, S.R.; Graham, F.L.; Johnson, D.C. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 1994, 77, 525–535. [Google Scholar] [CrossRef]
- Pietra, G.; Semino, C.; Cagnoni, F.; Boni, L.; Cangemi, G.; Frumento, G.; Melioli, G. Natural killer cells lyse autologous herpes simplex virus infected targets using cytolytic mechanisms distributed clonotypically. J. Med. Virol. 2000, 62, 354–363. [Google Scholar] [CrossRef]
- Samudio, I.; Rezvani, K.; Shaim, H.; Hofs, E.; Ngom, M.; Bu, L.; Liu, G.; Lee, J.T.C.; Imren, S.; Lam, V.; et al. UV-inactivated HSV-1 potently activates NK cell killing of leukemic cells. Blood 2016, 127, 2575–2586. [Google Scholar] [CrossRef] [PubMed]
- Habu, S.; Akamatsu, K.; Tamaoki, N.; Okumura, K. In vivo significance of NK cell on resistance against virus (HSV-1) infections in mice. J. Immunol. 1986, 133, 2743–2747. [Google Scholar] [CrossRef]
- Bukowski, J.F.; Welsh, R.M. The role of natural killer cells and interferon in resistance to acute infection of mice with herpes simplex virus type 1. J. Immunol. 1986, 9, 3481–3485. [Google Scholar] [CrossRef]
- Reading, P.C.; Whitney, P.G.; Barr, D.P.; Wojtasiak, M.; Mintern, J.D.; Waithman, J.; Brooks, A.G. IL-18, but not IL-12, Regulates NK Cell Activity following Intranasal Herpes Simplex Virus Type 1 Infection1. J. Immunol. 2007, 179, 3214–3221. [Google Scholar] [CrossRef]
- Nandakumar, S.; Woolard Stacie, N.; Yuan, D.; Rouse Barry, T.; Kumaraguru, U. Natural Killer Cells as Novel Helpers in Anti-Herpes Simplex Virus Immune Response. J. Virol. 2008, 82, 10820–10831. [Google Scholar] [CrossRef]
- Brandt, C.R.; Salkowski, C.A. Activation of NK cells in mice following corneal infection with herpes simplex virus type-1. Investig. Ophthalmol. Vis. Sci. 1992, 33, 113–120. [Google Scholar]
- Jamali, A.; Hu, K.; Sendra, V.G.; Blanco, T.; Lopez, M.J.; Ortiz, G.; Qazi, Y.; Zheng, L.; Turhan, A.; Harris, D.L.; et al. Characterization of Resident Corneal Plasmacytoid Dendritic Cells and Their Pivotal Role in Herpes Simplex Keratitis. Cell Rep. 2020, 32, 108099. [Google Scholar] [CrossRef]
- Welner, R.S.; Pelayo, R.; Nagai, Y.; Garrett, K.P.; Wuest, T.R.; Carr, D.J.; Borghesi, L.A.; Farrar, M.A.; Kincade, P.W. Lymphoid precursors are directed to produce dendritic cells as a result of TLR9 ligation during herpes infection. Blood 2008, 112, 3753–3761. [Google Scholar] [CrossRef]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like Receptors on Hematopoietic Progenitor Cells Stimulate Innate Immune System Replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef]
- Del Prete, A.; Salvi, V.; Soriani, A.; Laffranchi, M.; Sozio, F.; Bosisio, D.; Sozzani, S. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell. Mol. Immunol. 2023, 20, 432–447. [Google Scholar] [CrossRef]
- Vogel, K.; Thomann, S.; Vogel, B.; Schuster, P.; Schmidt, B. Both plasmacytoid dendritic cells and monocytes stimulate natural killer cells early during human herpes simplex virus type 1 infections. Immunology 2014, 143, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, H.; Bosnjak, L.; Harman, A.N.; Marsden, V.; Tyring, S.K.; Meng, T.-C.; Cunningham, A.L. Role for Plasmacytoid Dendritic Cells in the Immune Control of Recurrent Human Herpes Simplex Virus Infection. J. Virol. 2009, 83, 1952–1961. [Google Scholar] [CrossRef]
- Kassim, S.H.; Rajasagi, N.K.; Ritz, B.W.; Pruett, S.B.; Gardner, E.M.; Chervenak, R.; Jennings, S.R. Dendritic Cells Are Required for Optimal Activation of Natural Killer Functions following Primary Infection with Herpes Simplex Virus Type 1. J. Virol. 2009, 83, 3175–3186. [Google Scholar] [CrossRef] [PubMed]
- Frank, G.M.; Buela, K.-A.G.; Maker, D.M.; Harvey, S.A.K.; Hendricks, R.L. Early Responding Dendritic Cells Direct the Local NK Response to Control Herpes Simplex Virus 1 Infection within the Cornea. J. Immunol. 2012, 188, 1350–1359. [Google Scholar] [CrossRef]
- Kittan, N.A.; Bergua, A.; Haupt, S.; Donhauser, N.; Schuster, P.; Korn, K.; Harrer, T.; Schmidt, B. Impaired Plasmacytoid Dendritic Cell Innate Immune Responses in Patients with Herpes Virus-Associated Acute Retinal Necrosis1. J. Immunol. 2007, 179, 4219–4230. [Google Scholar] [CrossRef]
- Shah, K.; Al-Haidari, A.; Sun, J.; Kazi, J.U. T cell receptor (TCR) signaling in health and disease. Signal Transduct. Target. Ther. 2021, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-R.; Byeon, Y.; Kim, D.; Park, S.-G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 2020, 52, 750–761. [Google Scholar] [CrossRef] [PubMed]
- van Lint, A.; Ayers, M.; Brooks, A.G.; Coles, R.M.; Heath, W.R.; Carbone, F.R. Herpes Simplex Virus-Specific CD8+ T Cells Can Clear Established Lytic Infections from Skin and Nerves and Can Partially Limit the Early Spread of Virus after Cutaneous Inoculation1. J. Immunol. 2004, 172, 392–397. [Google Scholar] [CrossRef]
- Johnson, A.J.; Chu, C.-F.; Milligan, G.N. Effector CD4+ T-Cell Involvement in Clearance of Infectious Herpes Simplex Virus Type 1 from Sensory Ganglia and Spinal Cords. J. Virol. 2008, 82, 9678–9688. [Google Scholar] [CrossRef]
- Wallace, M.E.; Keating, R.; Heath, W.R.; Carbone, F.R. The Cytotoxic T-Cell Response to Herpes Simplex Virus Type 1 Infection of C57BL/6 Mice Is Almost Entirely Directed against a Single Immunodominant Determinant. J. Virol. 1999, 73, 7619–7626. [Google Scholar] [CrossRef]
- Belz, G.T.; Smith, C.M.; Eichner, D.; Shortman, K.; Karupiah, G.; Carbone, F.R.; Heath, W.R. Cutting Edge: Conventional CD8α+ Dendritic Cells Are Generally Involved in Priming CTL Immunity to Viruses 1. J. Immunol. 2004, 172, 1996–2000. [Google Scholar] [CrossRef]
- Allan, R.S.; Smith, C.M.; Belz, G.T.; van Lint, A.L.; Wakim, L.M.; Heath, W.R.; Carbone, F.R. Epidermal Viral Immunity Induced by CD8α+ Dendritic Cells But Not by Langerhans Cells. Science 2003, 301, 1925–1928. [Google Scholar] [CrossRef]
- Sigal, L.J. Activation of CD8 T Lymphocytes during Viral Infections. In Encyclopedia of Immunobiology; Ratcliffe, M.J.H., Ed.; Academic Press: Oxford, UK, 2016; pp. 286–290. [Google Scholar]
- Koh, C.-H.; Lee, S.; Kwak, M.; Kim, B.-S.; Chung, Y. CD8 T-cell subsets: Heterogeneity, functions, and therapeutic potential. Exp. Mol. Med. 2023, 55, 2287–2299. [Google Scholar] [CrossRef] [PubMed]
- Zöphel, D.; Angenendt, A.; Kaschek, L.; Ravichandran, K.; Hof, C.; Janku, S.; Hoth, M.; Lis, A. Faster cytotoxicity with age: Increased perforin and granzyme levels in cytotoxic CD8+ T cells boost cancer cell elimination. Aging Cell 2022, 21, e13668. [Google Scholar] [CrossRef]
- Waterhouse, N.J.; Sutton, V.R.; Sedelies, K.A.; Ciccone, A.; Jenkins, M.; Turner, S.J.; Bird, P.I.; Trapani, J.A. Cytotoxic T lymphocyte–induced killing in the absence of granzymes A and B is unique and distinct from both apoptosis and perforin-dependent lysis. J. Cell Biol. 2006, 173, 133–144. [Google Scholar] [CrossRef]
- Cullen, S.P.; Brunet, M.; Martin, S.J. Granzymes in cancer and immunity. Cell Death Differ. 2010, 17, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Sutton, V.R.; Davis, J.E.; Cancilla, M.; Johnstone, R.W.; Ruefli, A.A.; Sedelies, K.; Browne, K.A.; Trapani, J.A. Initiation of Apoptosis by Granzyme B Requires Direct Cleavage of Bid, but Not Direct Granzyme B–Mediated Caspase Activation. J. Exp. Med. 2000, 192, 1403–1414. [Google Scholar] [CrossRef]
- Barry, M.; Heibein, J.A.; Pinkoski, M.J.; Lee, S.-F.; Moyer, R.W.; Green, D.R.; Bleackley, R.C. Granzyme B Short-Circuits the Need for Caspase 8 Activity during Granule-Mediated Cytotoxic T-Lymphocyte Killing by Directly Cleaving Bid. Mol. Cell. Biol. 2000, 20, 3781–3794. [Google Scholar] [CrossRef] [PubMed]
- Hosking, M.P.; Flynn, C.T.; Whitton, J.L. Antigen-Specific Naive CD8+ T Cells Produce a Single Pulse of IFN-γ In Vivo within Hours of Infection, but without Antiviral Effect. J. Immunol. 2014, 193, 1873–1885. [Google Scholar] [CrossRef]
- Krummel, M.F.; Mahale, J.N.; Uhl, L.F.K.; Hardison, E.A.; Mujal, A.M.; Mazet, J.M.; Weber, R.J.; Gartner, Z.J.; Gérard, A. Paracrine costimulation of IFN-γ signaling by integrins modulates CD8 T cell differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, 11585–11590. [Google Scholar] [CrossRef] [PubMed]
- Bhat, P.; Leggatt, G.; Waterhouse, N.; Frazer, I.H. Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 2017, 8, e2836. [Google Scholar] [CrossRef]
- Lee, H.-G.; Cho, M.-J.; Choi, J.-M. Bystander CD4+ T cells: Crossroads between innate and adaptive immunity. Exp. Mol. Med. 2020, 52, 1255–1263. [Google Scholar] [CrossRef]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T Cells: Differentiation and Functions. J. Immunol. Res. 2012, 2012, 925135. [Google Scholar] [CrossRef]
- Chentoufi, A.A.; Binder, N.R.; Berka, N.; Durand, G.; Nguyen, A.; Bettahi, I.; Maillère, B.; BenMohamed, L. Asymptomatic Human CD4+ Cytotoxic T-Cell Epitopes Identified from Herpes Simplex Virus Glycoprotein B. J. Virol. 2008, 82, 11792–11802. [Google Scholar] [CrossRef]
- Heller, K.N.; Gurer, C.; Münz, C. Virus-specific CD4+ T cells: Ready for direct attack. J. Exp. Med. 2006, 203, 805–808. [Google Scholar] [CrossRef]
- Sparks-Thissen, R.L.; Braaten, D.C.; Kreher, S.; Speck, S.H.; Virgin, H.W. An Optimized CD4 T-Cell Response Can Control Productive and Latent Gammaherpesvirus Infection. J. Virol. 2004, 78, 6827–6835. [Google Scholar] [CrossRef] [PubMed]
- Sester, M.; Sester, U.; Gartner, B.; Heine, G.; Girndt, M.; Mueller-Lantzsch, N.; Meyerhans, A.; Kohler, H. Levels of virus-specific CD4 T cells correlate with cytomegalovirus control and predict virus-induced disease after renal transplantation1. Transplantation 2001, 71, 1287–1294. [Google Scholar] [CrossRef]
- Yun, H.; Rowe, A.M.; Lathrop, K.L.; Harvey, S.A.K.; Hendricks, R.L. Reversible Nerve Damage and Corneal Pathology in Murine Herpes Simplex Stromal Keratitis. J. Virol. 2014, 88, 7870–7880. [Google Scholar] [CrossRef]
- Egan, K.P.; Allen, A.G.; Wigdahl, B.; Jennings, S.R. Modeling the pathology, immune responses, and kinetics of HSV-1 replication in the lip scarification model. Virology 2018, 514, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Chatzileontiadou, D.S.M.; Sloane, H.; Nguyen, A.T.; Gras, S.; Grant, E.J. The Many Faces of CD4+ T Cells: Immunological and Structural Characteristics. Int. J. Mol. Sci. 2021, 22, 73. [Google Scholar] [CrossRef] [PubMed]
- Boeren, M.; Meysman, P.; Laukens, K.; Ponsaerts, P.; Ogunjimi, B.; Delputte, P. T cell immunity in HSV-1- and VZV-infected neural ganglia. Trends Microbiol. 2023, 31, 51–61. [Google Scholar] [CrossRef]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding roles for CD4+ T cells in immunity to viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar] [CrossRef]
- Harpur, C.M.; Kato, Y.; Dewi, S.T.; Stankovic, S.; Johnson, D.N.; Bedoui, S.; Whitney, P.G.; Lahoud, M.H.; Caminschi, I.; Heath, W.R.; et al. Classical Type 1 Dendritic Cells Dominate Priming of Th1 Responses to Herpes Simplex Virus Type 1 Skin Infection. J. Immunol. 2019, 202, 653–663. [Google Scholar] [CrossRef]
- Heiligenhaus, A.; Bauer, D.; Zheng, M.; Mrzyk, S.; Steuhl, K.-P. CD4+ T-cell type 1 and type 2 cytokines in the HSV-1 infected cornea. Graefe’s Arch. Clin. Exp. Ophthalmol. 1999, 237, 399–406. [Google Scholar] [CrossRef]
- Ghiasi, H.; Cai, S.; Perng, G.-C.; Nesburn, A.B.; Wechsler, S.L. Both CD4+ and CD8+ T cells are involved in protection against HSV-1 induced corneal scarring. Br. J. Ophthalmol. 2000, 84, 408–412. [Google Scholar] [CrossRef]
- Stankovic, S.; Harpur, C.M.; Macleod, B.L.; Whitney, P.G.; Gebhardt, T.; Brooks, A.G. Limited Internodal Migration of T Follicular Helper Cells after Peripheral Infection with Herpes Simplex Virus-1. J. Immunol. 2015, 195, 4892–4899. [Google Scholar] [CrossRef] [PubMed]
- van Velzen, M.; Jing, L.; Osterhaus, A.D.M.E.; Sette, A.; Koelle, D.M.; Verjans, G.M.G.M. Local CD4 and CD8 T-Cell Reactivity to HSV-1 Antigens Documents Broad Viral Protein Expression and Immune Competence in Latently Infected Human Trigeminal Ganglia. PLoS Pathog. 2013, 9, e1003547. [Google Scholar] [CrossRef]
- Khanna, K.M.; Bonneau, R.H.; Kinchington, P.R.; Hendricks, R.L. Herpes Simplex Virus-Specific Memory CD8+ T Cells Are Selectively Activated and Retained in Latently Infected Sensory Ganglia. Immunity 2003, 18, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Treat, B.R.; Bidula, S.M.; Leger, A.J.S.; Hendricks, R.L.; Kinchington, P.R. Herpes Simplex Virus 1-Specific CD8+ T Cell Priming and Latent Ganglionic Retention Are Shaped by Viral Epitope Promoter Kinetics. J. Virol. 2020, 94, e01193-19. [Google Scholar] [CrossRef]
- Liu, T.; Khanna, K.M.; Chen, X.; Fink, D.J.; Hendricks, R.L. Cd8+ T Cells Can Block Herpes Simplex Virus Type 1 (HSV-1) Reactivation from Latency in Sensory Neurons. J. Exp. Med. 2000, 191, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Cheng, A.-C.; Wang, M.-S.; Jia, R.-Y.; Sun, K.-F.; Yang, Q.; Wu, Y.; Zhu, D.; Chen, S.; Liu, M.-F.; et al. The suppression of apoptosis by α-herpesvirus. Cell Death Dis. 2017, 8, e2749. [Google Scholar] [CrossRef]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef]
- Derfuss, T.; Segerer, S.; Herberger, S.; Sinicina, I.; Hüfner, K.; Ebelt, K.; Knaus, H.-G.; Steiner, I.; Meinl, E.; Dornmair, K.; et al. Presence of HSV-1 Immediate Early Genes and Clonally Expanded T-cells with a Memory Effector Phenotype in Human Trigeminal Ganglia. Brain Pathol. 2007, 17, 389–398. [Google Scholar] [CrossRef]
- Ma, J.Z.; Russell, T.A.; Spelman, T.; Carbone, F.R.; Tscharke, D.C. Lytic Gene Expression Is Frequent in HSV-1 Latent Infection and Correlates with the Engagement of a Cell-Intrinsic Transcriptional Response. PLoS Pathog. 2014, 10, e1004237. [Google Scholar] [CrossRef]
- Pereira, R.A.; Simmons, A. Cell Surface Expression of H2 Antigens on Primary Sensory Neurons in Response to Acute but Not Latent Herpes Simplex Virus Infection In Vivo. J. Virol. 1999, 73, 6484–6489. [Google Scholar] [CrossRef]
- Medana, I.M.; Gallimore, A.; Oxenius, A.; Martinic, M.M.A.; Wekerle, H.; Neumann, H. MHC class I-restricted killing of neurons by virus-specific CD8+ T lymphocytes is effected through the Fas/FasL, but not the perforin pathway. Eur. J. Immunol. 2000, 30, 3623–3633. [Google Scholar] [CrossRef]
- Unger, P.-P.A.; Oja, A.E.; Khemai-Mehraban, T.; Ouwendijk, W.J.D.; Hombrink, P.; Verjans, G.M.G.M. T-cells in human trigeminal ganglia express canonical tissue-resident memory T-cell markers. J. Neuroinflamm. 2022, 19, 249. [Google Scholar] [CrossRef]
- Verjans, G.M.G.M.; Hintzen, R.Q.; van Dun, J.M.; Poot, A.; Milikan, J.C.; Laman, J.D.; Langerak, A.W.; Kinchington, P.R.; Osterhaus, A.D.M.E. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc. Natl. Acad. Sci. USA 2007, 104, 3496–3501. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef]
- Himmelein, S.; St Leger, A.J.; Knickelbein, J.E.; Rowe, A.; Freeman, M.L.; Hendricks, R.L. Circulating herpes simplex type 1 (HSV-1)-specific CD8+T cells do not access HSV-1 latently infected trigeminal ganglia. Herpesviridae 2011, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Masopust, D. Tissue-Resident Memory T Cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef]
- Wakim, L.M.; Waithman, J.; van Rooijen, N.; Heath, W.R.; Carbone, F.R. Dendritic Cell-Induced Memory T Cell Activation in Nonlymphoid Tissues. Science 2008, 319, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Smolders, J.; Heutinck, K.M.; Fransen, N.L.; Remmerswaal, E.B.M.; Hombrink, P.; ten Berge, I.J.M.; van Lier, R.A.W.; Huitinga, I.; Hamann, J. Tissue-resident memory T cells populate the human brain. Nat. Commun. 2018, 9, 4593. [Google Scholar] [CrossRef]
- Mackay, L.K.; Stock, A.T.; Ma, J.Z.; Jones, C.M.; Kent, S.J.; Mueller, S.N.; Heath, W.R.; Carbone, F.R.; Gebhardt, T. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl. Acad. Sci. USA 2012, 109, 7037–7042. [Google Scholar] [CrossRef]
- Lucinda, N.; Figueiredo, M.M.; Pessoa, N.L.; Santos, B.S.Á.d.S.; Lima, G.K.; Freitas, A.M.; Machado, A.M.V.; Kroon, E.G.; Antonelli, L.R.d.V.; Campos, M.A. Dendritic cells, macrophages, NK and CD8+ T lymphocytes play pivotal roles in controlling HSV-1 in the trigeminal ganglia by producing IL1-beta, iNOS and granzyme B. Virol. J. 2017, 14, 37. [Google Scholar] [CrossRef]
- Theil, D.; Derfuss, T.; Paripovic, I.; Herberger, S.; Meinl, E.; Schueler, O.; Strupp, M.; Arbusow, V.; Brandt, T. Latent Herpesvirus Infection in Human Trigeminal Ganglia Causes Chronic Immune Response. Am. J. Pathol. 2003, 163, 2179–2184. [Google Scholar] [CrossRef]
- Kiefer, R.; Haas, C.A.; Kreutzberg, G.W. Gamma interferon-like immunoreactive material in rat neurons: Evidence against a close relationship to gamma interferon. Neuroscience 1991, 45, 551–560. [Google Scholar] [CrossRef]
- Decman, V.; Kinchington, P.R.; Harvey, S.A.K.; Hendricks, R.L. Gamma Interferon Can Block Herpes Simplex Virus Type 1 Reactivation from Latency, Even in the Presence of Late Gene Expression. J. Virol. 2005, 79, 10339–10347. [Google Scholar] [CrossRef] [PubMed]
- Knickelbein, J.E.; Khanna, K.M.; Yee, M.B.; Baty, C.J.; Kinchington, P.R.; Hendricks, R.L. Noncytotoxic Lytic Granule–Mediated CD8+ T Cell Inhibition of HSV-1 Reactivation from Neuronal Latency. Science 2008, 322, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Gerada, C.; Steain, M.; Campbell, T.M.; McSharry, B.; Slobedman, B.; Abendroth, A. Granzyme B Cleaves Multiple Herpes Simplex Virus 1 and Varicella-Zoster Virus (VZV) Gene Products, and VZV ORF4 Inhibits Natural Killer Cell Cytotoxicity. J. Virol. 2019, 93, e01140-19. [Google Scholar] [CrossRef] [PubMed]
- Perng, G.-C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; et al. Virus-Induced Neuronal Apoptosis Blocked by the Herpes Simplex Virus Latency-Associated Transcript. Science 2000, 287, 1500–1503. [Google Scholar] [CrossRef]
- Vagaska, B.; New, S.E.P.; Alvarez-Gonzalez, C.; D’Acquisto, F.; Gomez, S.G.; Bulstrode, N.W.; Madrigal, A.; Ferretti, P. MHC-class-II are expressed in a subpopulation of human neural stem cells in vitro in an IFNγ–independent fashion and during development. Sci. Rep. 2016, 6, 24251. [Google Scholar] [CrossRef]
- Dhanushkodi Nisha, R.; Prakash, S.; Srivastava, R.; Coulon Pierre-Gregoire, A.; Arellano, D.; Kapadia Rayomand, V.; Fahim, R.; Suzer, B.; Jamal, L.; Vahed, H.; et al. Antiviral CD19+CD27+ Memory B Cells Are Associated with Protection from Recurrent Asymptomatic Ocular Herpesvirus Infection. J. Virol. 2022, 96, e02057-21. [Google Scholar] [CrossRef]
- Su, C.; Zhan, G.; Zheng, C. Evasion of host antiviral innate immunity by HSV-1, an update. Virol. J. 2016, 13, 38. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, C. The Race between Host Antiviral Innate Immunity and the Immune Evasion Strategies of Herpes Simplex Virus 1. Microbiol. Mol. Biol. Rev. 2020, 84, e00099-20. [Google Scholar] [CrossRef]
- Daubeuf, S.; Singh, D.; Tan, Y.; Liu, H.; Federoff, H.J.; Bowers, W.J.; Tolba, K. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 2009, 113, 3264–3275. [Google Scholar] [CrossRef]
- Lint, A.L.v.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes Simplex Virus Immediate-Early ICP0 Protein Inhibits Toll-Like Receptor 2-Dependent Inflammatory Responses and NF-κB Signaling. J. Virol. 2010, 84, 10802–10811. [Google Scholar] [CrossRef]
- Peri, P.; Mattila, R.K.; Kantola, H.; Broberg, E.; Karttunen, H.S.; Waris, M.; Vuorinen, T.; Hukkanen, V. Herpes Simplex Virus Type 1 Us3 Gene Deletion Influences Toll-like Receptor Responses in Cultured Monocytic Cells. Virol. J. 2008, 5, 140. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; Most, R.v.d.; Ahmed, R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N.; Ahmed, R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2009, 106, 8623–8628. [Google Scholar] [CrossRef]
- Chentoufi, A.A.; Kritzer, E.; Tran, M.V.; Dasgupta, G.; Lim, C.H.; Yu, D.C.; Afifi, R.E.; Jiang, X.; Carpenter, D.; Osorio, N.; et al. The Herpes Simplex Virus 1 Latency-Associated Transcript Promotes Functional Exhaustion of Virus-Specific CD8+ T Cells in Latently Infected Trigeminal Ganglia: A Novel Immune Evasion Mechanism. J. Virol. 2011, 85, 9127–9138. [Google Scholar] [CrossRef]
- Allen, S.J.; Hamrah, P.; Gate, D.; Mott, K.R.; Mantopoulos, D.; Zheng, L.; Town, T.; Jones, C.; Andrian, U.H.v.; Freeman, G.J.; et al. The Role of LAT in Increased CD8+ T Cell Exhaustion in Trigeminal Ganglia of Mice Latently Infected with Herpes Simplex Virus 1. J. Virol. 2011, 85, 4184–4197. [Google Scholar] [CrossRef]
- Wang, S.; Jaggi, U.; Katsumata, M.; Ghiasi, H. The importance of IFNα2A (Roferon-A) in HSV-1 latency and T cell exhaustion in ocularly infected mice. PLoS Pathog. 2024, 20, e1012612. [Google Scholar] [CrossRef]
- Canova, P.N.; Charron, A.J.; Leib, D.A. Models of Herpes Simplex Virus Latency. Viruses 2024, 16, 747. [Google Scholar] [CrossRef] [PubMed]
- Mark, K.E.; Wald, A.; Magaret, A.S.; Selke, S.; Olin, L.; Huang, M.-L.; Corey, L. Rapidly Cleared Episodes of Herpes Simplex Virus Reactivation in Immunocompetent Adults. J. Infect. Dis. 2008, 198, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Walton, A.H.; Muenzer, J.T.; Rasche, D.; Boomer, J.S.; Sato, B.; Brownstein, B.H.; Pachot, A.; Brooks, T.L.; Deych, E.; Shannon, W.D.; et al. Reactivation of Multiple Viruses in Patients with Sepsis. PLoS ONE 2014, 9, e98819. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Geng, S.; Suo, Y.; Wei, X.; Cai, Q.; Wu, B.; Zhou, X.; Shi, Y.; Wang, B. Critical Role of Regulatory T Cells in the Latency and Stress-Induced Reactivation of HSV-1. Cell Rep. 2018, 25, 2379–2389.e3. [Google Scholar] [CrossRef]
- Bonneau, R.H.; Sheridan, J.F.; Feng, N.; Glaser, R. Stress-induced modulation of the primary cellular immune response to herpes simplex virus infection is mediated by both adrenal-dependent and independent mechanisms. J. Neuroimmunol. 1993, 42, 167–176. [Google Scholar] [CrossRef]
- Freeman, M.L.; Sheridan, B.S.; Bonneau, R.H.; Hendricks, R.L. Psychological Stress Compromises CD8+ T Cell Control of Latent Herpes Simplex Virus Type 1 Infections1. J. Immunol. 2007, 179, 322–328. [Google Scholar] [CrossRef]
- Hendricks, R.L.; Busch, J.L.; Cherpes, T.L. Female Sex Hormones Inhibit the Function of HSV-Specific CD8+ T Cells in Latently Infected Trigeminal Ganglia and Induce HSV-1 Reactivation from Latency. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2828. [Google Scholar]
- Prakash, S.; Roy, S.; Srivastava, R.; Coulon, P.-G.; Dhanushkodi, N.R.; Vahed, H.; Jankeel, A.; Geertsema, R.; Amezquita, C.; Nguyen, L.; et al. Unique molecular signatures of antiviral memory CD8+ T cells associated with asymptomatic recurrent ocular herpes. Sci. Rep. 2020, 10, 13843. [Google Scholar] [CrossRef]
- Goldsmith, K.; Chen, W.; Johnson, D.C.; Hendricks, R.L. Infected Cell Protein (ICP)47 Enhances Herpes Simplex Virus Neurovirulence by Blocking the CD8+ T Cell Response. J. Exp. Med. 1998, 187, 341–348. [Google Scholar] [CrossRef]
- Çuburu, N.; Graham, B.S.; Buck, C.B.; Kines, R.C.; Pang, Y.-Y.S.; Day, P.M.; Lowy, D.R.; Schiller, J.T. Intravaginal immunization with HPV vectors induces tissue-resident CD8+ T cell responses. J. Clin. Investig. 2012, 122, 4606–4620. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Clark, R.A.; Liu, L.; Wagers, A.J.; Fuhlbrigge, R.C.; Kupper, T.S. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 2012, 483, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Vezys, V.; Marzo, A.L.; Lefrançois, L. Preferential Localization of Effector Memory Cells in Nonlymphoid Tissue. Science 2001, 291, 2413–2417. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Chong, B.; Mirchandani, N.; Brinster, N.K.; Yamanaka, K.-i.; Dowgiert, R.K.; Kupper, T.S. The Vast Majority of CLA+ T Cells Are Resident in Normal Skin1. J. Immunol. 2006, 176, 4431–4439. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Koelle, D.M.; Cao, J.; Vazquez, J.; Huang, M.L.; Hladik, F.; Wald, A.; Corey, L. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J. Exp. Med. 2007, 204, 595–603. [Google Scholar] [CrossRef]
- Hondowicz, B.D.; Kim, K.S.; Ruterbusch, M.J.; Keitany, G.J.; Pepper, M. IL-2 is required for the generation of viral-specific CD4+ Th1 tissue-resident memory cells and B cells are essential for maintenance in the lung. Eur. J. Immunol. 2018, 48, 80–86. [Google Scholar] [CrossRef]
- Gehlhausen, J.R.; Iwasaki, A. B cells join T cell clusters in the host response to recurrent herpes simplex virus 2 infection. J. Clin. Investig. 2021, 131, e148300. [Google Scholar] [CrossRef]
- Iijima, N.; Linehan, M.M.; Zamora, M.; Butkus, D.; Dunn, R.; Kehry, M.R.; Laufer, T.M.; Iwasaki, A. Dendritic cells and B cells maximize mucosal Th1 memory response to herpes simplex virus. J. Exp. Med. 2008, 205, 3041–3052. [Google Scholar] [CrossRef]
- Elion, G.B. Mechanism of action and selectivity of acyclovir. Am. J. Med. 1982, 73, 7–13. [Google Scholar] [CrossRef]
- Jiang, Y.-C.; Feng, H.; Lin, Y.-C.; Guo, X.-R. New strategies against drug resistance to herpes simplex virus. Int. J. Oral Sci. 2016, 8, 1–6. [Google Scholar] [CrossRef]
- Mertz, G.J.; Eron, L.; Kaufman, R.; Goldberg, L.; Raab, B.; Conant, M.; Mills, J.; Kurtz, T.; Davis, L.G. Prolonged continuous versus intermittent oral acyclovir treatment in normal adults with frequently recurring genital herpes simplex virus infection. Am. J. Med. 1988, 85, 14–19. [Google Scholar] [PubMed]
- Mindel, A.; Carney, O.; Freris, M.; Faherty, A.; Patou, G.; Williams, P. Dosage and safety of long-term suppressive acyclovir therapy for recurrent genital herpes. Lancet 1988, 331, 926–928. [Google Scholar] [CrossRef]
- Spruance, S.L.; Stewart, J.C.B.; Rowe, N.H.; McKeough, M.B.; Wenerstrom, G.; Freeman, D.J. Treatment of Recurrent Herpes Simplex Labialis with Oral Acyclovir. J. Infect. Dis. 1990, 161, 185–190. [Google Scholar] [CrossRef]
- Raborn, G.W.; McGaw, W.T.; Grace, M.; Tyrrell, L.D.; Samuels, S.M. Oral acyclovir and herpes labialis: A randomized, double-blind, placebo-controlled study. J. Am. Dent. Assoc. 1987, 115, 38–42. [Google Scholar] [CrossRef]
- Van Vloten, W.A.; Swart, R.N.J.; Pot, F. Topical acyclovir therapy in patients with recurrent orofacial herpes simplex infections. J. Antimicrob. Chemother. 1983, 12, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Spruance, S.L.; Schnipper, L.E.; Overall, J.C., Jr.; Kern, E.R.; Wester, B.; Modlin, J.; Wenerstrom, G.; Burton, C.; Arndt, K.A.; Chiu, G.L.; et al. Treatment of Herpes Simplex Labialis with Topical Acyclovir in Polyethylene Glycol. J. Infect. Dis. 1982, 146, 85–90. [Google Scholar] [CrossRef]
- Spruance, S.L.; Jones, T.M.; Blatter, M.M.; Vargas-Cortes, M.; Barber, J.; Hill, J.; Goldstein, D.; Schultz, M. High-Dose, Short-Duration, Early Valacyclovir Therapy for Episodic Treatment of Cold Sores: Results of Two Randomized, Placebo-Controlled, Multicenter Studies. Antimicrob. Agents Chemother. 2003, 47, 1072–1080. [Google Scholar] [CrossRef]
- Smith, R.L.; Morroni, J.; Wilcox, C.L. Lack of effect of treatment with penciclovir or acyclovir on the establishment of latent HSV-1 in primary sensory neurons in culture. Antivir. Res. 2001, 52, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, L.G.; Baker, D.; Gelb, L.; Blythe, J.; Hale, R.; Frost, P.; Crumpacker, C.; Rabinovich, S.; Peacock, J.E., Jr.; Herndon, J.; et al. Prolonged Continuous Acyclovir Treatment of Normal Adults with Frequently Recurring Genital Herpes Simplex Virus Infection. JAMA 1991, 265, 747–751. [Google Scholar] [CrossRef]
- Raborn, G.W.; Martel, A.Y.; Grace, M.G.A.; McGaw, W.T. Oral acyclovir in prevention of herpes labialis: A randomized, double-blind, multi-centered clinical trial. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1998, 85, 55–59. [Google Scholar] [CrossRef]
- Pachnio, A.; Begum, J.; Fox, A.; Moss, P. Acyclovir Therapy Reduces the CD4+ T Cell Response against the Immunodominant pp65 Protein from Cytomegalovirus in Immune Competent Individuals. PLoS ONE 2015, 10, e0125287. [Google Scholar] [CrossRef] [PubMed]
- Erlich, K.S.; Hauer, L.; Mills, J. Effects of long-term acyclovir chemosuppression on serum IgG antibody to herpes simplex virus. J. Med. Virol. 1988, 26, 33–39. [Google Scholar] [CrossRef]
- Lafferty, W.E.; Brewer, L.A.; Corey, L. Alteration of lymphocyte transformation response to herpes simplex virus infection by acyclovir therapy. Antimicrob. Agents Chemother. 1984, 26, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sato, H.; Fukuda, Y.; Kurokawa, M.; Kageyama, S.; Kawana, T.; Shiraki, K. Acyclovir Treatment of Skin Lesions Results in Immune Deviation in Mice Infected Cutaneously with Herpes Simplex Virus. Antivir. Chem. Chemother. 1999, 10, 251–257. [Google Scholar] [CrossRef]
- Mohamed, H.; Esposito, R.A.; Kutzler, M.A.; Wigdahl, B.; Krebs, F.C.; Miller, V. Nonthermal plasma as part of a novel strategy for vaccination. Plasma Process. Polym. 2020, 17, 2000051. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Charlebois, E.D.; Sigouroudinia, M.; Goldbeck, C.; Hartog, K.; Sekulovich, R.E.; Langenberg, A.G.M.; Burke, R.L. Limited Antibody-Dependent Cellular Cytotoxicity Antibody Response Induced by a Herpes Simplex Virus Type 2 Subunit Vaccine. J. Infect. Dis. 2000, 181, 335–339. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Flechtner, J.B.; McNeil, L.K.; Heineman, T.; Oliphant, T.; Tasker, S.; Wald, A.; Hetherington, S. Therapeutic HSV-2 vaccine decreases recurrent virus shedding and recurrent genital herpes disease. Vaccine 2019, 37, 3443–3450. [Google Scholar] [CrossRef]
- Egan, K.P.; Awasthi, S.; Tebaldi, G.; Hook, L.M.; Naughton, A.M.; Fowler, B.T.; Beattie, M.; Alameh, M.-G.; Weissman, D.; Cohen, G.H.; et al. A Trivalent HSV-2 gC2, gD2, gE2 Nucleoside-Modified mRNA-LNP Vaccine Provides Outstanding Protection in Mice against Genital and Non-Genital HSV-1 Infection, Comparable to the Same Antigens Derived from HSV-1. Viruses 2023, 15, 1483. [Google Scholar] [CrossRef]
- Awasthi, S.; Knox, J.J.; Desmond, A.; Alameh, M.-G.; Gaudette, B.T.; Lubinski, J.M.; Naughton, A.; Hook, L.M.; Egan, K.P.; Tam, Y.K.; et al. Trivalent nucleoside-modified mRNA vaccine yields durable memory B cell protection against genital herpes in preclinical models. J. Clin. Investig. 2021, 131, e152310. [Google Scholar] [CrossRef]
- Egan, K.P.; Hook, L.M.; Naughton, A.; Pardi, N.; Awasthi, S.; Cohen, G.H.; Weissman, D.; Friedman, H.M. An HSV-2 nucleoside-modified mRNA genital herpes vaccine containing glycoproteins gC, gD, and gE protects mice against HSV-1 genital lesions and latent infection. PLoS Pathog. 2020, 16, e1008795. [Google Scholar] [CrossRef]
- Royer, D.J.; Hendrix, J.F.; Larabee, C.M.; Reagan, A.M.; Sjoelund, V.H.; Robertson, D.M.; Carr, D.J.J. Vaccine-induced antibodies target sequestered viral antigens to prevent ocular HSV-1 pathogenesis, preserve vision, and preempt productive neuronal infection. Mucosal Immunol. 2019, 12, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.J.J.; Gmyrek, G.B.; Filiberti, A.; Berube, A.N.; Browne, W.P.; Gudgel, B.M.; Sjoelund, V.H. Distinguishing Features of High- and Low-Dose Vaccine against Ocular HSV-1 Infection Correlates with Recognition of Specific HSV-1–Encoded Proteins. Immunohorizons 2020, 4, 608–626. [Google Scholar] [CrossRef]
- Carr, D.J.J.; Berube, A.; Gershburg, E. The Durability of Vaccine Efficacy against Ocular HSV-1 Infection Using ICP0 Mutants 0∆NLS and 0∆RING Is Lost over Time. Pathogens 2021, 10, 1470. [Google Scholar] [CrossRef]
- Naidu, S.K.; Nabi, R.; Cheemarla, N.R.; Stanfield, B.A.; Rider, P.J.; Jambunathan, N.; Chouljenko, V.N.; Carter, R.; Del Piero, F.; Langohr, I.; et al. Intramuscular vaccination of mice with the human herpes simplex virus type-1(HSV-1) VC2 vaccine, but not its parental strain HSV-1(F) confers full protection against lethal ocular HSV-1 (McKrae) pathogenesis. PLoS ONE 2020, 15, e0228252. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Cardin, R.D.; Smith, G.A.; Pickard, G.E.; Sollars, P.J.; Dixon, D.A.; Pasula, R.; Bravo, F.J. The R2 non-neuroinvasive HSV-1 vaccine affords protection from genital HSV-2 infections in a guinea pig model. NPJ Vaccines 2020, 5, 104. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Han, L.; Shi, C.; Li, Y.; Qian, S.; Feng, Z.; Yu, L. An updated review of HSV-1 infection-associated diseases and treatment, vaccine development, and vector therapy application. Virulence 2024, 15, 2425744. [Google Scholar] [CrossRef]
- von Woedtke, T.; Emmert, S.; Metelmann, H.-R.; Rupf, S.; Weltmann, K.-D. Perspectives on cold atmospheric plasma (CAP) applications in medicine. Phys. Plasmas 2020, 27, 070601. [Google Scholar] [CrossRef]
- Lin, A.; Chernets, N.; Han, J.; Alicea, Y.; Dobrynin, D.; Fridman, G.; Freeman, T.A.; Fridman, A.; Miller, V. Non-Equilibrium Dielectric Barrier Discharge Treatment of Mesenchymal Stem Cells: Charges and Reactive Oxygen Species Play the Major Role in Cell Death. Plasma Process. Polym. 2015, 12, 1117–1127. [Google Scholar] [CrossRef]
- von Woedtke, T.; Schmidt, A.; Bekeschus, S.; Wende, K.; Weltmann, K.D. Plasma Medicine: A Field of Applied Redox Biology. In Vivo 2019, 33, 1011–1026. [Google Scholar] [CrossRef]
- Mohamed, H.; Nayak, G.; Rendine, N.; Wigdahl, B.; Krebs, F.C.; Bruggeman, P.J.; Miller, V. Non-Thermal Plasma as a Novel Strategy for Treating or Preventing Viral Infection and Associated Disease. Front. Phys. 2021, 9, 286. [Google Scholar] [CrossRef]
- Filipić, A.; Primc, G.; Zaplotnik, R.; Mehle, N.; Gutierrez-Aguirre, I.; Ravnikar, M.; Mozetič, M.; Žel, J.; Dobnik, D. Cold Atmospheric Plasma as a Novel Method for Inactivation of Potato Virus Y in Water Samples. Food Environ. Virol. 2019, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Nayak, G.; Aboubakr, H.A.; Goyal, S.M.; Bruggeman, P.J. Reactive species responsible for the inactivation of feline calicivirus by a two-dimensional array of integrated coaxial microhollow dielectric barrier discharges in air. Plasma Process. Polym. 2018, 15, 1700119. [Google Scholar] [CrossRef]
- Aboubakr, H.A.; Williams, P.; Gangal, U.; Youssef, M.M.; El-Sohaimy, S.A.A.; Bruggeman, P.J.; Goyal, S.M. Virucidal effect of cold atmospheric gaseous plasma on feline calicivirus, a surrogate for human norovirus. Appl. Environ. Microbiol. 2015, 81, 3612–3622. [Google Scholar] [CrossRef]
- Shi, X.M.; Zhang, G.J.; Wu, X.L.; Peng, Z.Y.; Zhang, Z.H.; Shao, X.J.; Chang, Z.S. Effect of Low-Temperature Plasma on Deactivation of Hepatitis B Virus. IEEE Trans. Plasma Sci. 2012, 40, 2711–2716. [Google Scholar] [CrossRef]
- Mohamed, H.; Berman, R.; Connors, J.; Haddad, E.K.; Miller, V.; Nonnemacher, M.R.; Dampier, W.; Wigdahl, B.; Krebs, F.C. Immunomodulatory Effects of Non-Thermal Plasma in a Model for Latent HIV-1 Infection: Implications for an HIV-1-Specific Immunotherapy. Biomedicines 2023, 11, 122. [Google Scholar] [CrossRef]
- Mohamed, H.; Clemen, R.; Freund, E.; Lackmann, J.-W.; Wende, K.; Connors, J.; Haddad, E.K.; Dampier, W.; Wigdahl, B.; Miller, V.; et al. Non-thermal plasma modulates cellular markers associated with immunogenicity in a model of latent HIV-1 infection. PLoS ONE 2021, 16, e0247125. [Google Scholar] [CrossRef]
- Sutter, J.; Brettschneider, J.; Wigdahl, B.; Bruggeman, P.J.; Krebs, F.C.; Miller, V. Non-Thermal Plasma Reduces HSV-1 Infection of and Replication in HaCaT Keratinocytes In Vitro. Int. J. Mol. Sci. 2024, 25, 3839. [Google Scholar] [CrossRef]
- Alekseev, O.; Donovan, K.; Limonnik, V.; Azizkhan-Clifford, J. Nonthermal Dielectric Barrier Discharge (DBD) Plasma Suppresses Herpes Simplex Virus Type 1 (HSV-1) Replication in Corneal Epithelium. Transl. Vis. Sci. Technol. 2014, 3, 2. [Google Scholar] [CrossRef]
- Brun, P.; Brun, P.; Vono, M.; Venier, P.; Tarricone, E.; Deligianni, V.; Martines, E.; Zuin, M.; Spagnolo, S.; Cavazzana, R.; et al. Disinfection of Ocular Cells and Tissues by Atmospheric-Pressure Cold Plasma. PLoS ONE 2012, 7, e33245. [Google Scholar] [CrossRef]
- Kalghatgi, S.; Kelly, C.M.; Cerchar, E.; Torabi, B.; Alekseev, O.; Fridman, A.; Friedman, G.; Azizkhan-Clifford, J. Effects of Non-Thermal Plasma on Mammalian Cells. PLoS ONE 2011, 6, e16270. [Google Scholar] [CrossRef]
- Abduvokhidov, D.; Yusupov, M.; Shahzad, A.; Attri, P.; Shiratani, M.; Oliveira, M.C.; Razzokov, J. Unraveling the Transport Properties of RONS across Nitro-Oxidized Membranes. Biomolecules 2023, 13, 1043. [Google Scholar] [CrossRef] [PubMed]
- Volotskova, O.; Dubrovsky, L.; Keidar, M.; Bukrinsky, M. Cold Atmospheric Plasma Inhibits HIV-1 Replication in Macrophages by Targeting Both the Virus and the Cells. PLoS ONE 2016, 11, e0165322. [Google Scholar] [CrossRef]
- Moseley, R.; Waddington, R.J. Modification of gingival proteoglycans by reactive oxygen species: Potential mechanism of proteoglycan degradation during periodontal diseases. Free Radic. Res. 2021, 55, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhou, R.; Zhou, R.; Li, W.; Weerasinghe, J.; Chen, S.; Rehm, B.H.A.; Zhao, L.; Frentiu, F.D.; Zhang, Z.; et al. Cold atmospheric plasma for preventing infection of viruses that use ACE2 for entry. Theranostics 2022, 12, 2811–2832. [Google Scholar] [CrossRef]
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Investig. 2001, 108, 503–510. [Google Scholar] [CrossRef]
- Bunz, O.; Mese, K.; Funk, C.; Wulf, M.; Bailer, S.M.; Piwowarczyk, A.; Ehrhardt, A. Cold atmospheric plasma as antiviral therapy—effect on human herpes simplex virus type 1. J. Gen. Virol. 2020, 101, 208–215. [Google Scholar] [CrossRef]
- Lee, H.-G.; Choi, J.-H.; Jang, Y.-S.; Kim, U.-K.; Kim, G.-C.; Hwang, D.-S. Non-thermal plasma accelerates the healing process of peripheral nerve crush injury in rats. Int. J. Med. Sci. 2020, 17, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Do, C.B.; Jaiswal, M.S.; Jang, Y.-S.; Kim, U.-K.; Kim, G.-C.; Hwang, D.-S. Non-thermal plasma directly accelerates neuronal proliferation by stimulating axon formation. Sci. Rep. 2022, 12, 15868. [Google Scholar] [CrossRef]
- Jang, J.-Y.; Hong, Y.J.; Lim, J.; Choi, J.S.; Choi, E.H.; Kang, S.; Rhim, H. Cold atmospheric plasma (CAP), a novel physicochemical source, induces neural differentiation through cross-talk between the specific RONS cascade and Trk/Ras/ERK signaling pathway. Biomaterials 2018, 156, 258–273. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kang, S.U.; Kim, K.I.; Kang, S.; Shin, Y.S.; Chang, J.W.; Yang, S.S.; Lee, K.; Lee, J.-S.; Moon, E.; et al. Nonthermal Plasma Induces Apoptosis in ATC Cells: Involvement of JNK and p38 MAPK-Dependent ROS. Yonsei Med. J. 2014, 55, 1640–1647. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Coulon, P.-G.; Roy, S.; Prakash, S.; Srivastava, R.; Dhanushkodi, N.; Salazar, S.; Amezquita, C.; Nguyen, L.; Vahed, H.; Nguyen, A.M.; et al. Upregulation of Multiple CD8+ T Cell Exhaustion Pathways Is Associated with Recurrent Ocular Herpes Simplex Virus Type 1 Infection. J. Immunol. 2020, 205, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.G.; Xiang, B.; Merlino, D.J.; Baybutt, T.R.; Sahu, J.; Fridman, A.; Snook, A.E.; Miller, V. Non-thermal plasma induces immunogenic cell death in vivo in murine CT26 colorectal tumors. OncoImmunology 2018, 7, e1484978. [Google Scholar] [CrossRef]
- Van Loenhout, J.; Flieswasser, T.; Freire Boullosa, L.; De Waele, J.; Van Audenaerde, J.; Marcq, E.; Jacobs, J.; Lin, A.; Lion, E.; Dewitte, H.; et al. Cold Atmospheric Plasma-Treated PBS Eliminates Immunosuppressive Pancreatic Stellate Cells and Induces Immunogenic Cell Death of Pancreatic Cancer Cells. Cancers 2019, 11, 1597. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.B.; Choi, H.G.; Gurmessa, S.K.; Jang, I.-T.; Kumar, N.; Jiang, Z.; Kaushik, N.K.; Kim, H.-J. Enhancing antitumor immunity in Lewis lung cancer through plasma-treated medium-induced activation of dendritic cells. Cancer Cell Int. 2024, 24, 389. [Google Scholar] [CrossRef]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Friedman, P.C.; Miller, V.; Fridman, G.; Fridman, A. Use of cold atmospheric pressure plasma to treat warts: A potential therapeutic option. Clin. Exp. Dermatol. 2019, 44, 459–461. [Google Scholar] [CrossRef]
- Friedman, P.C.; Fridman, G.; Fridman, A. Using cold plasma to treat warts in children: A case series. Pediatr. Dermatol. 2020, 37, 706–709. [Google Scholar] [CrossRef]
- Roberts, C.M.; Pfister, J.R.; Spear, S.J. Increasing Proportion of Herpes Simplex Virus Type 1 as a Cause of Genital Herpes Infection in College Students. Sex. Transm. Dis. 2003, 30, 797–800. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutter, J.; Hope, J.L.; Wigdahl, B.; Miller, V.; Krebs, F.C. Immunological Control of Herpes Simplex Virus Type 1 Infection: A Non-Thermal Plasma-Based Approach. Viruses 2025, 17, 600. https://doi.org/10.3390/v17050600
Sutter J, Hope JL, Wigdahl B, Miller V, Krebs FC. Immunological Control of Herpes Simplex Virus Type 1 Infection: A Non-Thermal Plasma-Based Approach. Viruses. 2025; 17(5):600. https://doi.org/10.3390/v17050600
Chicago/Turabian StyleSutter, Julia, Jennifer L. Hope, Brian Wigdahl, Vandana Miller, and Fred C. Krebs. 2025. "Immunological Control of Herpes Simplex Virus Type 1 Infection: A Non-Thermal Plasma-Based Approach" Viruses 17, no. 5: 600. https://doi.org/10.3390/v17050600
APA StyleSutter, J., Hope, J. L., Wigdahl, B., Miller, V., & Krebs, F. C. (2025). Immunological Control of Herpes Simplex Virus Type 1 Infection: A Non-Thermal Plasma-Based Approach. Viruses, 17(5), 600. https://doi.org/10.3390/v17050600