
Acinetobacter baumannii and Klebsiella pneumoniae Isolates Obtained from Intensive Care Unit Patients in 2024: General Characterization, Prophages, Depolymerases and Esterases of Phage Origin

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Isolates and Culturing
2.2. Whole-Genome Sequencing and Assembly
2.3. Identification of Capsule Synthesis Loci and Multilocus Sequence Types
2.4. Antimicrobial Susceptibility
2.5. Search and Annotation of Prophages and Search for Phage Tail Depolymerases and Esterases
2.6. Bioinformatic Analysis
3. Results
3.1. General Characterization of A. baumannii and K. pneumoniae Isolates
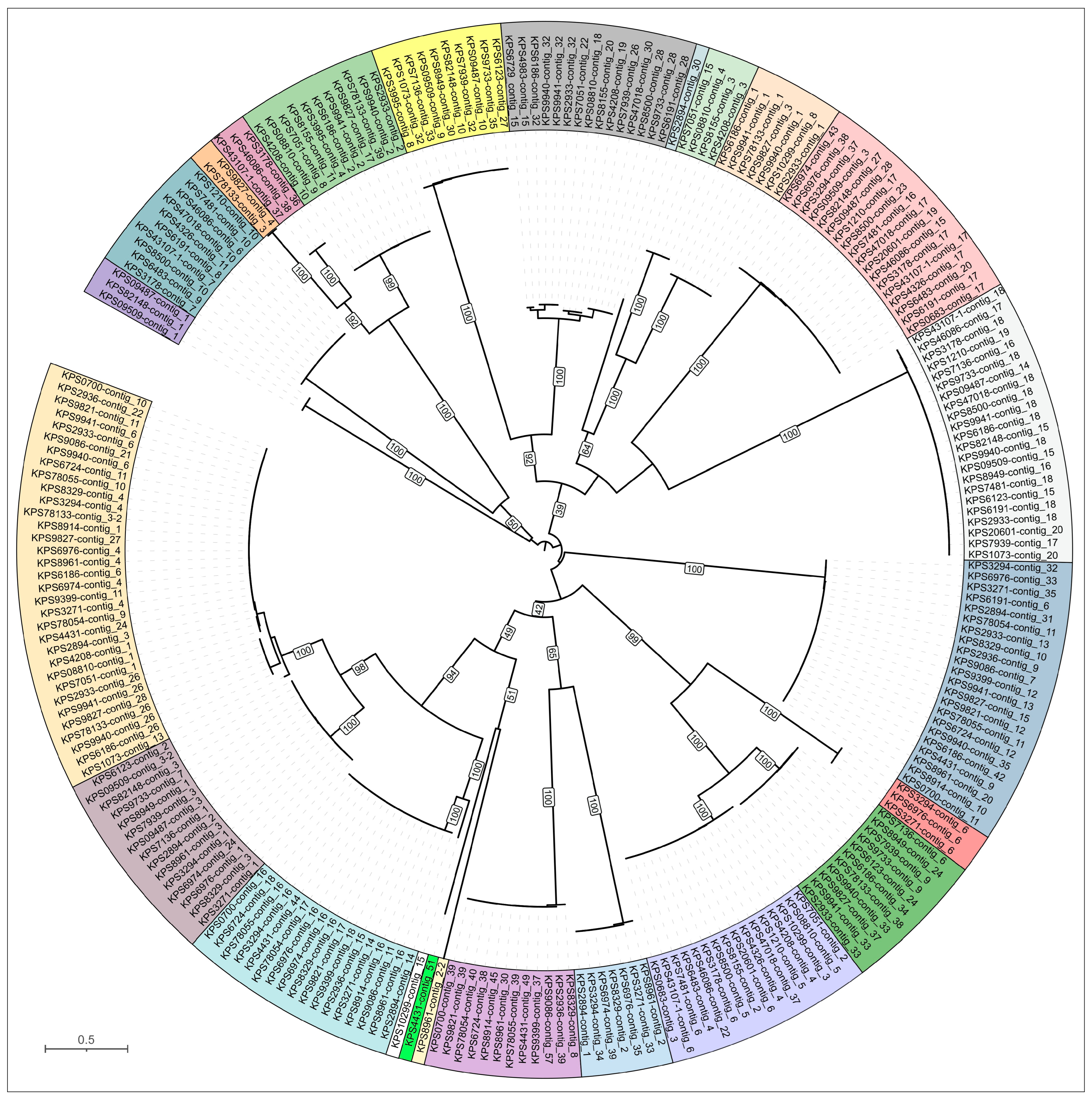
3.2. Phylogenomic Characterization of Isolates
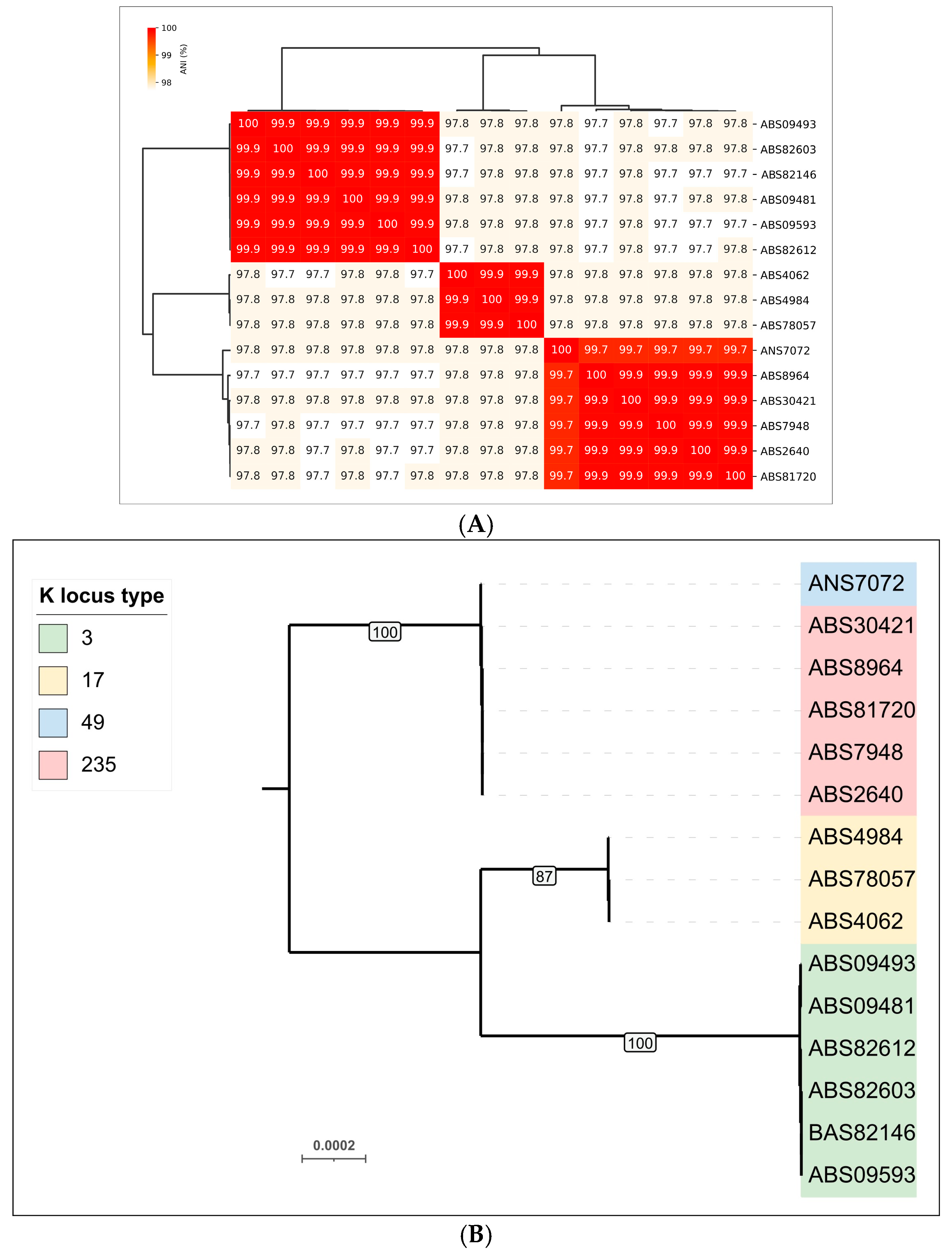
3.2.1. Phylogenomic Characterization of A. baumannii Isolates
3.2.2. Phylogenomic Characterization of K. pneumoniae Isolates
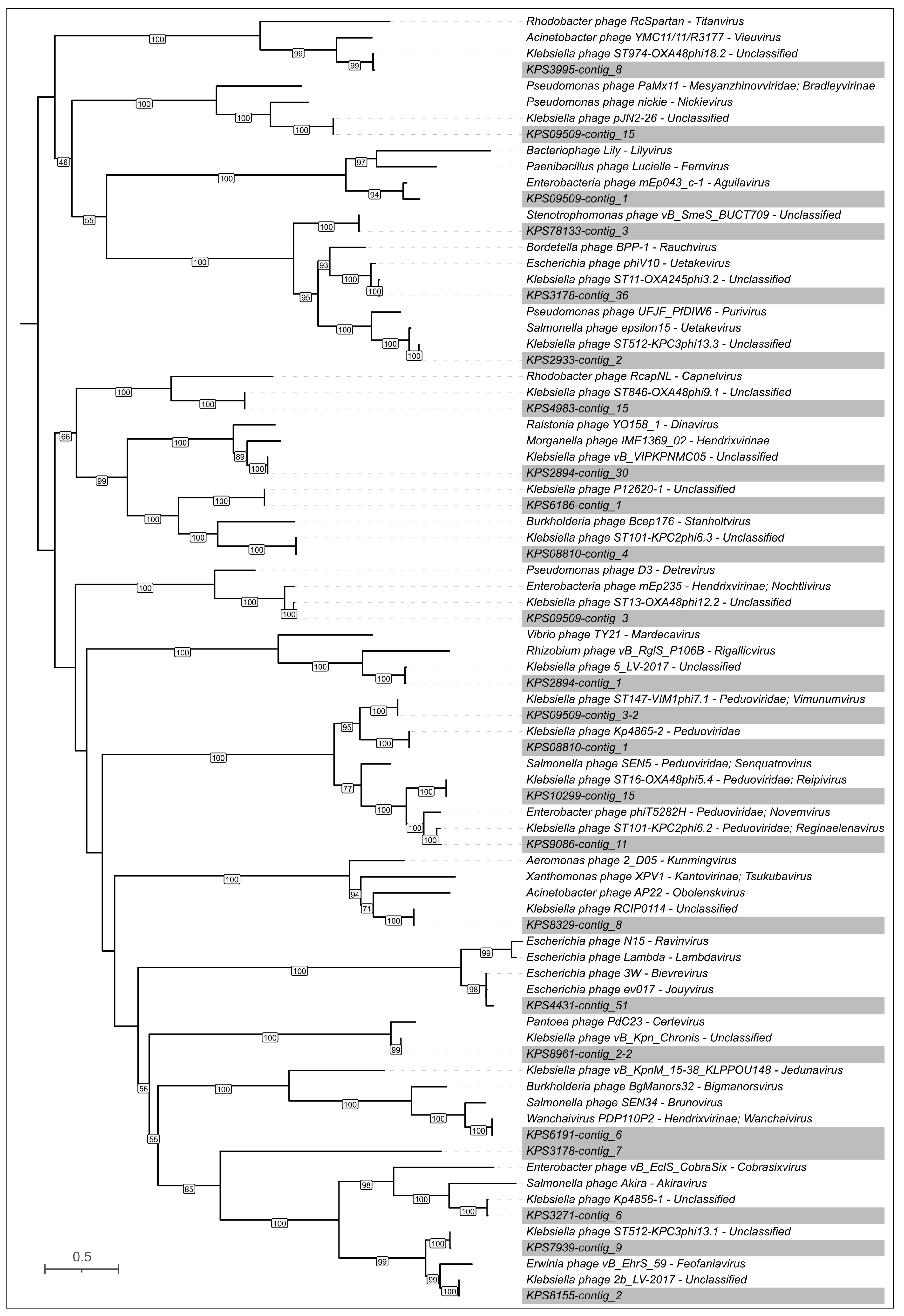
3.3. Identification of Prophage Regions and Phage Tail Depolymerases and Esterases in Bacterial Genomes
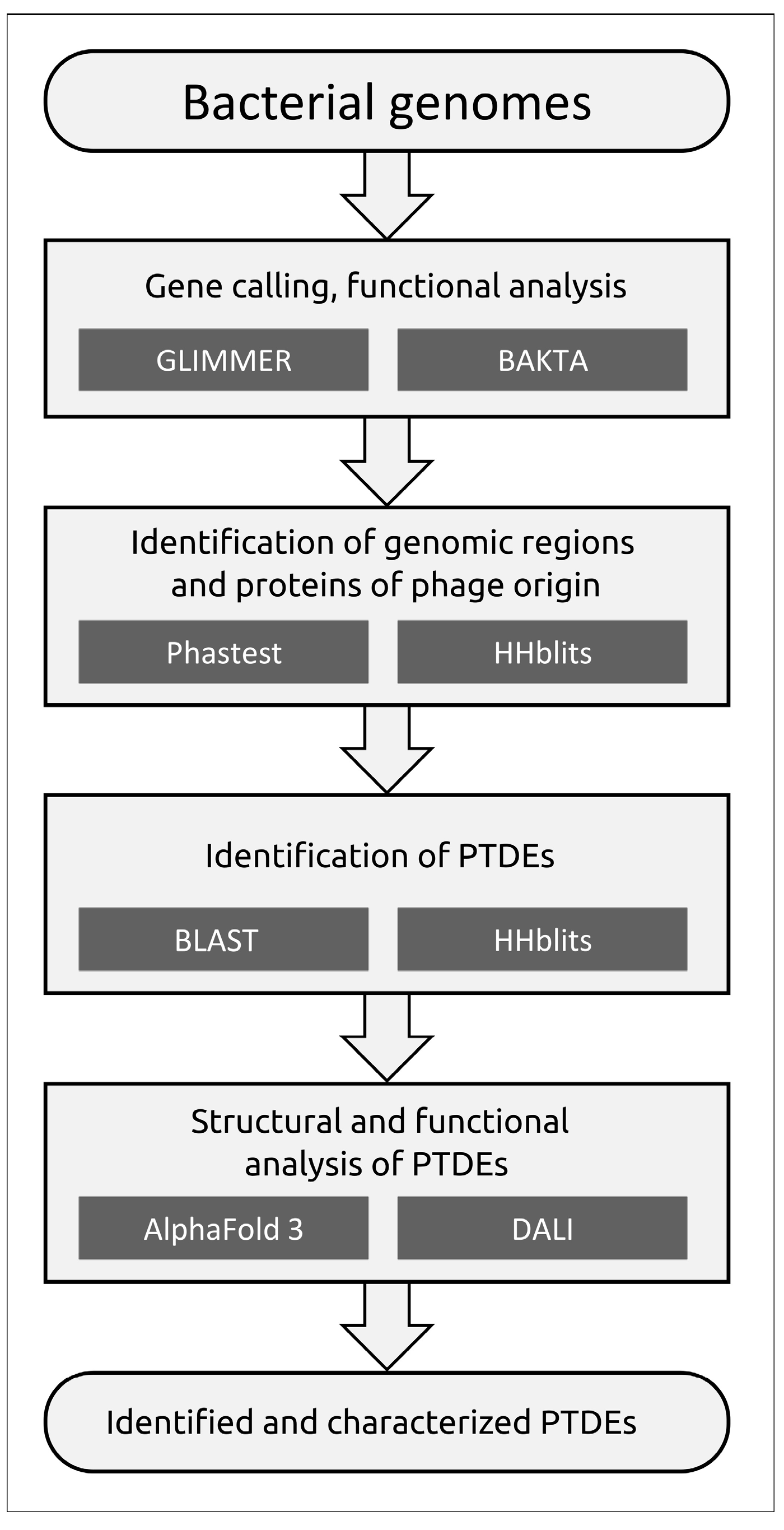
3.3.1. Protocol
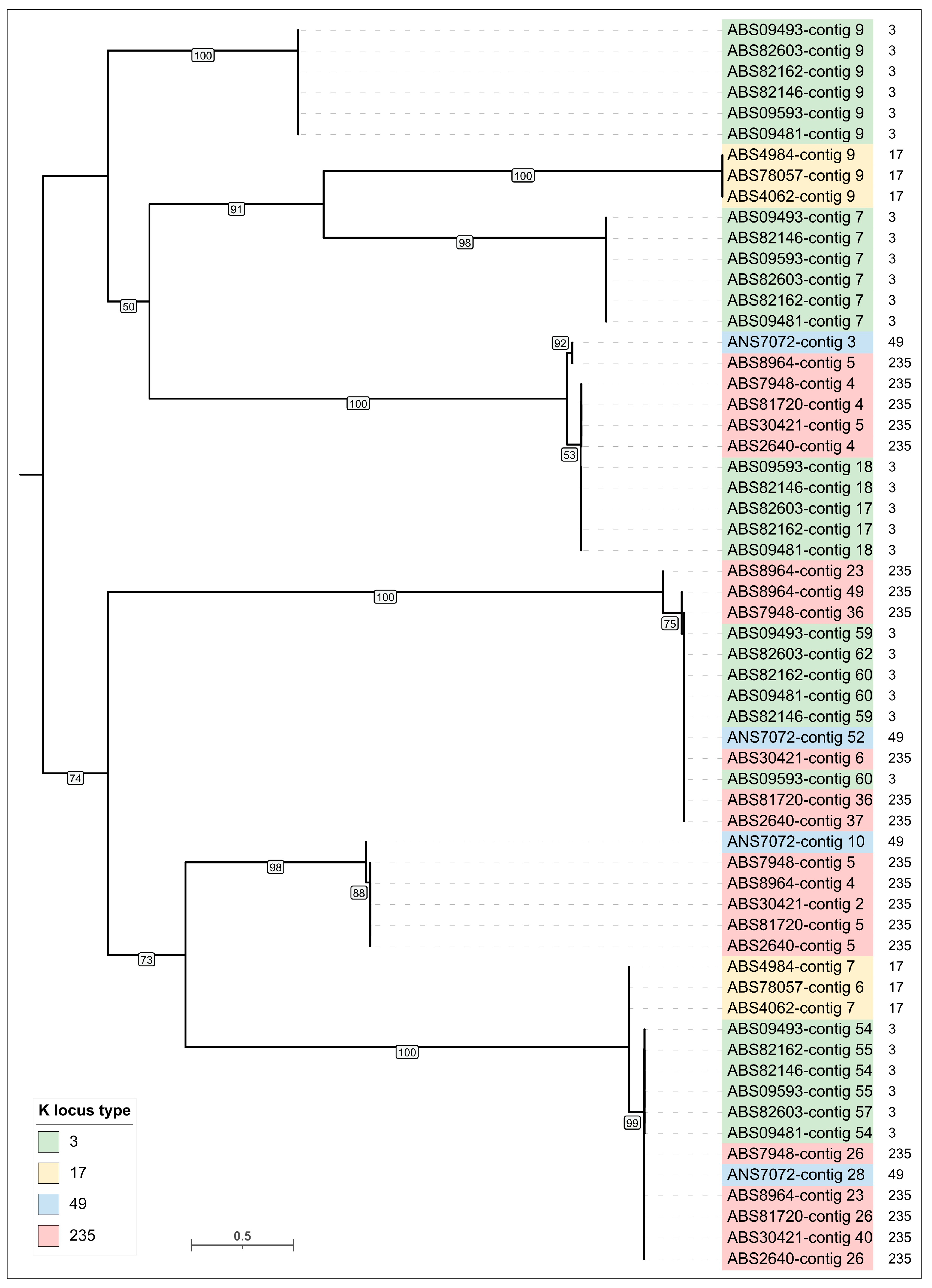
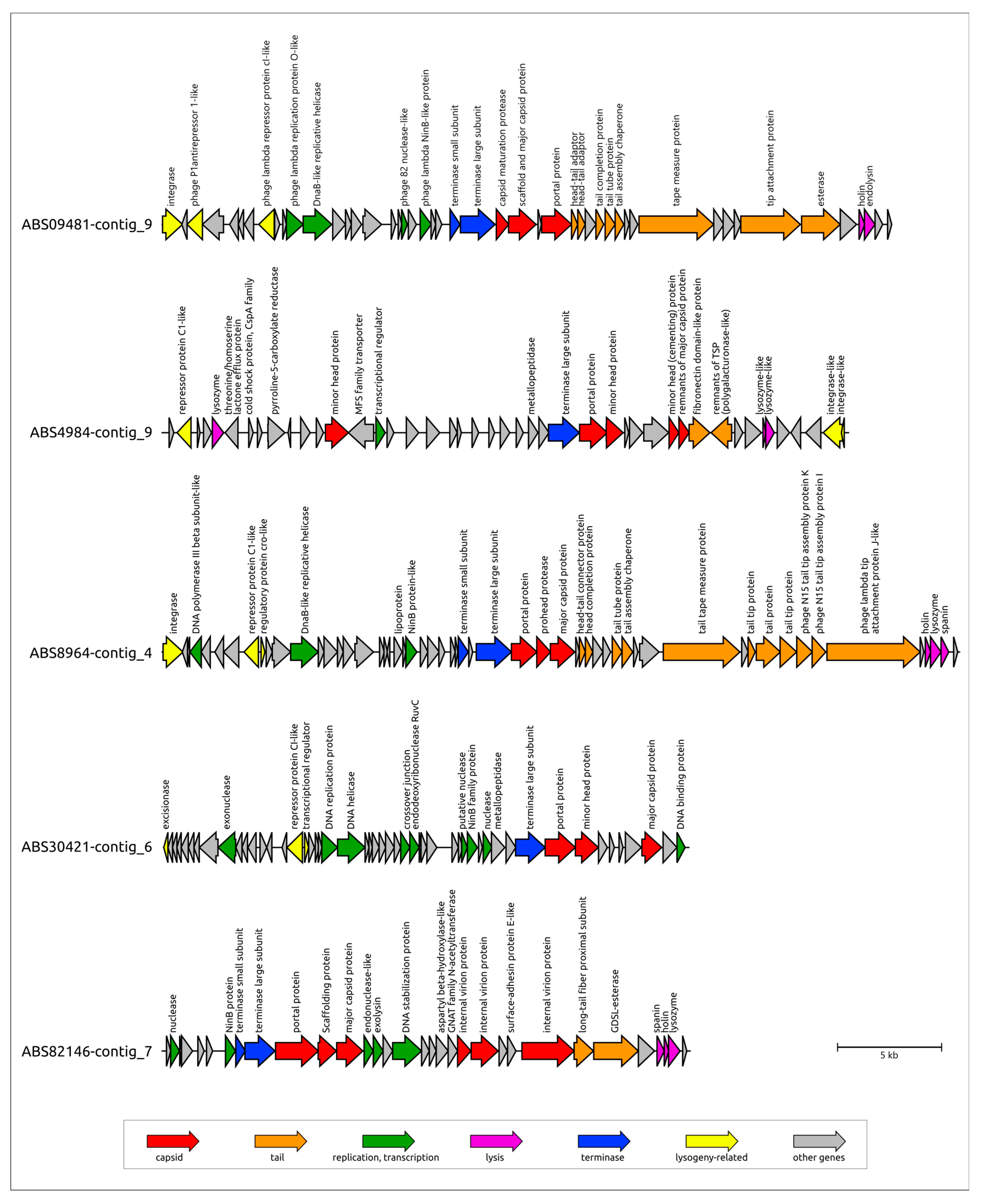
3.3.2. General Characterization of Prophage Regions in A. baumannii Genomes
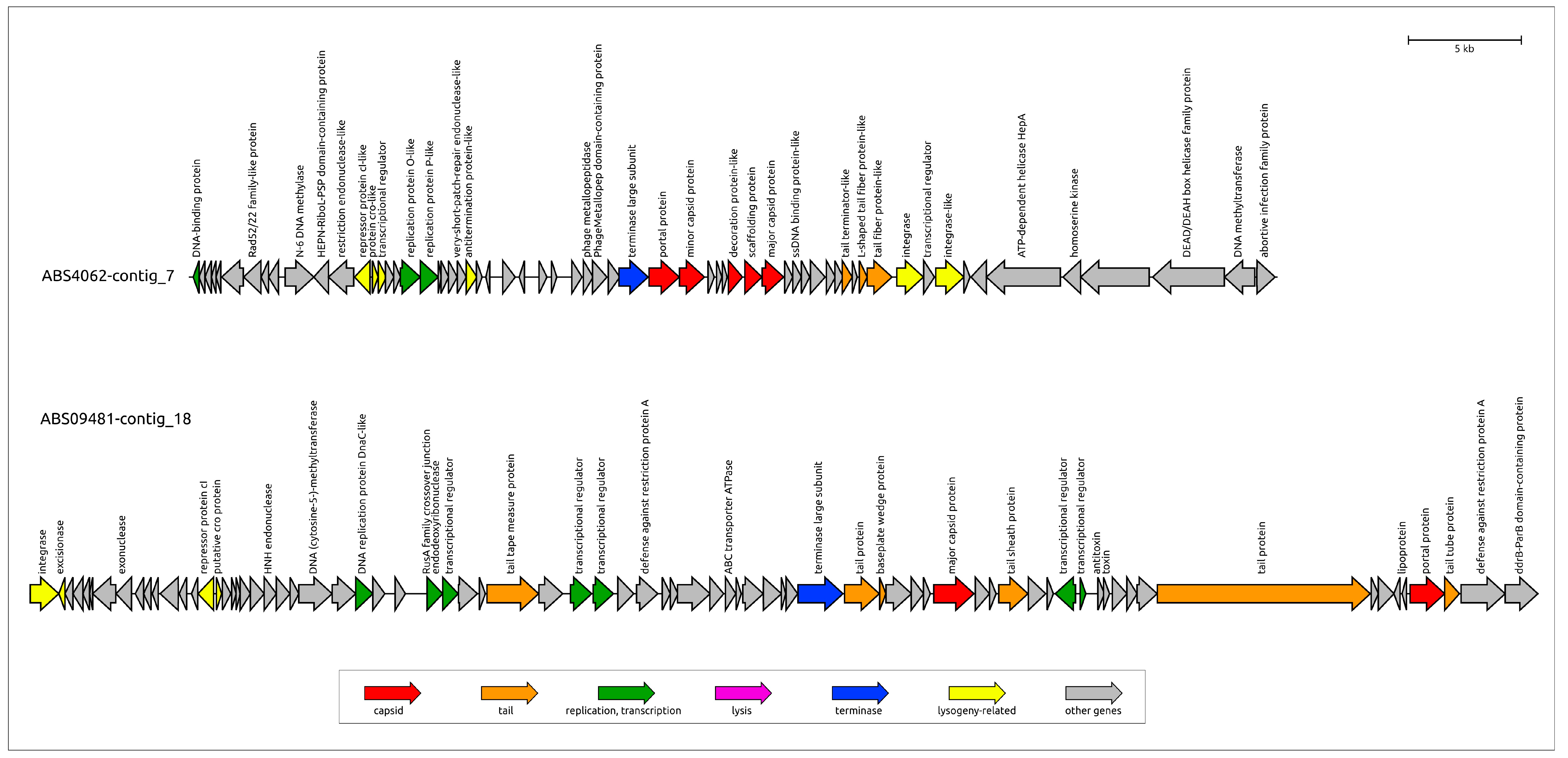
3.3.3. General Characterization of Prophage Regions in K. pneumoniae Genomes
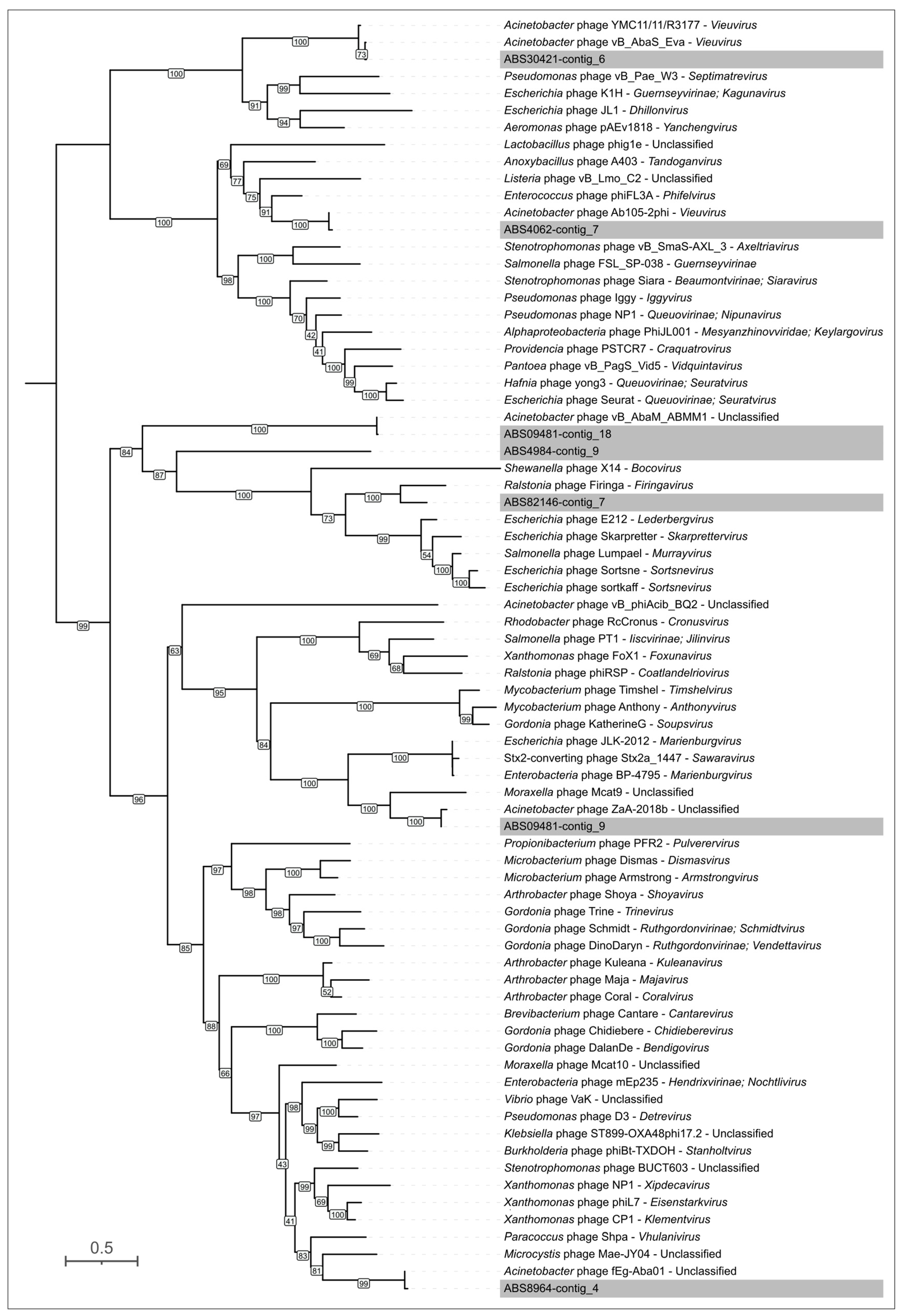
3.4. Analysis of Identification of PTDEs in A. baumannii and K. pneumoniae Genomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| bp | base pair |
| MCP | Major capsid protein |
| MDR | Multidrug-resistant |
| PDR | Pandrug resistant |
| PI | Pairwise identity |
| PTDE | Phage tail depolymerase and esterase |
| RBP | Receptor-binding protein |
| TLS | Terminase large subunit |
| TFP | Tail fiber protein |
| TSP | Tailspike protein |
| XDR | Extensively drug-resistant |
| ZOT | Zonular occludens toxin |
References
- Ayoub Moubareck, C.; Hammoudi Halat, D. Insights into Acinetobacter baumannii: A review of microbiological, virulence, and resistance traits in a threatening nosocomial pathogen. Antibiotics 2020, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Runtuvuori-Salmela, A.; Kunttu, H.M.T.; Laanto, E.; Almeida, G.M.F.; Mäkelä, K.; Middelboe, M.; Sundberg, L.-R. Prevalence of genetically similar Flavobacterium columnare phages across aquaculture environments reveals a strong potential for pathogen control. Environ. Microbiol. 2022, 24, 2404–2420. [Google Scholar] [CrossRef] [PubMed]
- Santajit, S.; Indrawattana, N. Mechanisms of antimicrobial resistance in ESKAPE pathogens. BioMed Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef]
- Howard, A.; O’Donoghue, M.; Feeney, A.; Sleator, R.D. Acinetobacter baumannii: An emerging opportunistic pathogen. Virulence 2012, 3, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Sarshar, M.; Behzadi, P.; Scribano, D.; Palamara, A.T.; Ambrosi, C. Acinetobacter baumannii: An ancient commensal with weapons of a pathogen. Pathogens 2021, 10, 387. [Google Scholar] [CrossRef]
- Abbas, R.; Chakkour, M.; Zein El Dine, H.; Obaseki, E.F.; Obeid, S.T.; Jezzini, A.; Ghssein, G.; Ezzeddine, Z. General overview of Klebsiella pneumonia: Epidemiology and the role of siderophores in its pathogenicity. Biology 2024, 13, 78. [Google Scholar] [CrossRef]
- Corona, A.; De Santis, V.; Agarossi, A.; Prete, A.; Cattaneo, D.; Tomasini, G.; Bonetti, G.; Patroni, A.; Latronico, N. Antibiotic therapy strategies for treating gram-negative severe infections in the critically Ill: A narrative review. Antibiotics 2023, 12, 1262. [Google Scholar] [CrossRef]
- Harding, C.M.; Hennon, S.W.; Feldman, M.F. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 2018, 16, 91–102. [Google Scholar] [CrossRef]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial antibiotic resistance: The most critical pathogens. Pathogens 2021, 10, 1310. [Google Scholar] [CrossRef]
- WHO Bacterial Priority Pathogens List 2024: Bacterial Pathogens of Public Health Importance, to Guide Research, Development, and Strategies to Prevent and Control Antimicrobial Resistance, 1st ed.; World Health Organization: Geneva, Switzerland, 2024; ISBN 978-92-4-009346-1.
- Russo, T.A.; Luke, N.R.; Beanan, J.M.; Olson, R.; Sauberan, S.L.; MacDonald, U.; Schultz, L.W.; Umland, T.C.; Campagnari, A.A. The K1 capsular polysaccharide of Acinetobacter baumannii strain 307-0294 is a major virulence factor. Infect. Immun. 2010, 78, 3993–4000. [Google Scholar] [CrossRef]
- Roshini, J.; Patro, L.P.P.; Sundaresan, S.; Rathinavelan, T. Structural diversity among Acinetobacter baumannii K-antigens and its implication in the in silico serotyping. Front. Microbiol. 2023, 14, 1191542. [Google Scholar] [CrossRef] [PubMed]
- Cahill, S.M.; Hall, R.M.; Kenyon, J.J. An update to the database for Acinetobacter baumannii capsular polysaccharide locus typing extends the extensive and diverse repertoire of genes found at and outside the K locus. Microb. Genom. 2022, 8, mgen000878. [Google Scholar] [CrossRef]
- Volozhantsev, N.V.; Shpirt, A.M.; Borzilov, A.I.; Komisarova, E.V.; Krasilnikova, V.M.; Shashkov, A.S.; Verevkin, A.V.; Knirel, Y.A. Characterization and therapeutic potential of bacteriophage-encoded polysaccharide depolymerases with β galactosidase activity against Klebsiella pneumoniae K57 capsular type. Antibiotics 2020, 9, 732. [Google Scholar] [CrossRef]
- Evseev, P.V.; Sukhova, A.S.; Tkachenko, N.A.; Skryabin, Y.P.; Popova, A.V. Lytic capsule-specific Acinetobacter bacteriophages encoding polysaccharide-degrading enzymes. Viruses 2024, 16, 771. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, M.J.; Huo, Y.; Stroyakovski, M.; Cheng, L.; Wan, D.; Dell, A.; Santini, J.M. Specificity and diversity of Klebsiella pneumoniae phage-encoded capsule depolymerases. Essays Biochem. 2024, 68, 661–677. [Google Scholar] [CrossRef]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef]
- Drobiazko, A.Y.; Kasimova, A.A.; Evseev, P.V.; Shneider, M.M.; Klimuk, E.I.; Shashkov, A.S.; Dmitrenok, A.S.; Chizhov, A.O.; Slukin, P.V.; Skryabin, Y.P.; et al. Capsule-targeting depolymerases derived from Acinetobacter baumannii prophage regions. Int. J. Mol. Sci. 2022, 23, 4971. [Google Scholar] [CrossRef]
- Kolupaeva, L.V.; Kolupaeva, N.V.; Lebedeva, A.Y.; Solomentsev, V.I.; Sizova, A.A.; Fursov, M.V.; Lazareva, E.B.; Chernenkaya, T.V.; Popova, A.V.; Volozhantsev, N.V. Genomic sequences of Acinetobacter baumannii and Klebsiella pneumoniae isolates obtained from the blood of patients of intensive care unit in 2024. Bacteriology 2025, 10, 116–118. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Chklovski, A.; Parks, D.H.; Woodcroft, B.J.; Tyson, G.W. CheckM2: A rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods. 2023, 20, 1203–1212. [Google Scholar] [CrossRef]
- Lam, M.M.C.; Wick, R.R.; Judd, L.M.; Holt, K.E.; Wyres, K.L. Kaptive 2.0: Updated capsule and lipopolysaccharide locus typing for the Klebsiella pneumoniae species complex. Microb. Genom. 2022, 8, 000800. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef]
- Wishart, D.S.; Han, S.; Saha, S.; Oler, E.; Peters, H.; Grant, J.R.; Stothard, P.; Gautam, V. PHASTEST: Faster than PHASTER, better than PHAST. Nucleic Acids Res. 2023, 51, W443–W450. [Google Scholar] [CrossRef]
- Schwengers, O.; Jelonek, L.; Dieckmann, M.A.; Beyvers, S.; Blom, J.; Goesmann, A. Bakta: Rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb. Genom. 2021, 7, 000685. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & Clustermap.Js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: Memory friendly classification with the genome taxonomy database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Holm, L. Dali server: Structural unification of protein families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- Troshin, K.; Sykilinda, N.; Shuraleva, S.; Tokmakova, A.; Tkachenko, N.; Kurochkina, L.; Miroshnikov, K.; Suzina, N.; Brzhozovskaya, E.; Petrova, K.; et al. Pseudomonas phage lydia and the evolution of the mesyanzhinovviridae family. Viruses 2025, 17, 369. [Google Scholar] [CrossRef]
- Peng, W.; Zeng, F.; Wu, Z.; Jin, Z.; Li, W.; Zhu, M.; Wang, Q.; Tong, Y.; Chen, L.; Bai, Q. Isolation and genomic analysis of temperate phage 5W targeting multidrug-resistant Acinetobacter baumannii. Arch. Microbiol. 2021, 204, 58. [Google Scholar] [CrossRef] [PubMed]
- Rastegar, S.; Skurnik, M.; Niaz, H.; Tadjrobehkar, O.; Samareh, A.; Hosseini-Nave, H.; Sabouri, S. Isolation, characterization, and potential application of Acinetobacter baumannii phages against extensively drug-resistant strains. Virus Genes 2024, 60, 725–736. [Google Scholar] [CrossRef]
- Mardiana, M.; Teh, S.-H.; Tsai, Y.-C.; Yang, H.-H.; Lin, L.-C.; Lin, N.-T. Characterization of a novel and active temperate phage vB_AbaM_ABMM1 with antibacterial activity against Acinetobacter baumannii infection. Sci. Rep. 2023, 13, 11347. [Google Scholar] [CrossRef]
- Badawy, S.; Pajunen, M.I.; Haiko, J.; Baka, Z.A.M.; Abou-Dobara, M.I.; El-Sayed, A.K.A.; Skurnik, M. Identification and functional analysis of temperate siphoviridae bacteriophages of Acinetobacter baumannii. Viruses 2020, 12, 604. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Wand, M.E.; Sutton, J.M.; Centron, D.; Kropinski, A.M.; Reynolds, D.M. Genome sequence of vB_AbaS_TRS1, a viable prophage isolated from Acinetobacter baumannii strain A118. Genome Announc. 2016, 4, e01051-16. [Google Scholar] [CrossRef]
- Trotereau, A.; Boyer, C.; Bornard, I.; Pécheur, M.J.B.; Schouler, C.; Torres-Barceló, C. High genomic diversity of novel phages infecting the plant pathogen ralstonia solanacearum, isolated in mauritius and reunion islands. Sci. Rep. 2021, 11, 5382. [Google Scholar] [CrossRef] [PubMed]
- Ariff, A.; Wise, M.J.; Kahler, C.M.; Tay, C.Y.; Peters, F.; Perkins, T.T.; Chang, B.J. Novel Moraxella catarrhalis prophages display hyperconserved non-structural genes despite their genomic diversity. BMC Genom. 2015, 16, 860. [Google Scholar] [CrossRef]
- Arellano-Maciel, D.; Hurtado-Ramírez, J.M.; Camelo-Valera, L.C.; Castillo-Ramírez, S.; Reyes, A.; López-Leal, G. Geographic variation in abundance and diversity of Acinetobacter baumannii vieuvirus bacteriophages. Front. Microbiol. 2025, 16, 1522711. [Google Scholar] [CrossRef]
- Moon, K.; Kang, I.; Kim, S.; Kim, S.-J.; Cho, J.-C. Genomic and ecological study of two distinctive freshwater bacteriophages infecting a comamonadaceae bacterium. Sci. Rep. 2018, 8, 7989. [Google Scholar] [CrossRef]
- Jeon, J.; D’Souza, R.; Pinto, N.; Ryu, C.-M.; Park, J.; Yong, D.; Lee, K. Complete Genome Sequence of the Siphoviral Bacteriophage Βϕ-R3177, Which lyses an OXA-66-producing carbapenem-resistant Acinetobacter baumannii isolate. Arch. Virol. 2015, 160, 3157–3160. [Google Scholar] [CrossRef]
- Popova, A.V.; Zhilenkov, E.L.; Myakinina, V.P.; Krasilnikova, V.M.; Volozhantsev, N.V. Isolation and characterization of wide host range lytic bacteriophage AP22 infecting Acinetobacter baumannii. FEMS Microbiol. Lett. 2012, 332, 40–46. [Google Scholar] [CrossRef]
- Evseev, P.V.; Shneider, M.M.; Kolupaeva, L.V.; Kasimova, A.A.; Timoshina, O.Y.; Perepelov, A.V.; Shpirt, A.M.; Shelenkov, A.A.; Mikhailova, Y.V.; Suzina, N.E.; et al. New Obolenskvirus phages Brutus and Scipio: Biology, evolution, and phage-host interaction. Int. J. Mol. Sci. 2024, 25, 2074. [Google Scholar] [CrossRef]
- Hayes, S.; Erker, C.; Horbay, M.A.; Marciniuk, K.; Wang, W.; Hayes, C. Phage Lambda P protein: Trans-activation, inhibition phenotypes and their suppression. Viruses 2013, 5, 619–653. [Google Scholar] [CrossRef]
- Tarkowski, T.A.; Mooney, D.; Thomason, L.C.; Stahl, F.W. Gene products encoded in the ninr region of phage Lambda participate in red-mediated recombination. Genes Cells 2002, 7, 351–363. [Google Scholar] [CrossRef]
- Dydecka, A.; Bloch, S.; Necel, A.; Topka, G.; Węgrzyn, A.; Tong, J.; Donaldson, L.W.; Węgrzyn, G.; Nejman-Faleńczyk, B. The Ea22 gene of lambdoid phages: Preserved prolysogenic function despite of high sequence diversity. Virus Genes 2020, 56, 266–277. [Google Scholar] [CrossRef]
- Schwarzkopf, J.M.F.; Mehner-Breitfeld, D.; Brüser, T.A. Dimeric holin/antiholin complex controls lysis by phage T4. Front. Microbiol. 2024, 15, 1419106. [Google Scholar] [CrossRef]
- Ide, D.; Gorelik, A.; Illes, K.; Nagar, B. Structural analysis of mammalian sialic acid esterase. J. Mol. Biol. 2024, 436, 168801. [Google Scholar] [CrossRef]
- Vieira, P.S.; Bonfim, I.M.; Araujo, E.A.; Melo, R.R.; Lima, A.R.; Fessel, M.R.; Paixão, D.A.A.; Persinoti, G.F.; Rocco, S.A.; Lima, T.B.; et al. Xyloglucan processing machinery in Xanthomonas pathogens and its role in the transcriptional activation of virulence factors. Nat. Commun. 2021, 12, 4049. [Google Scholar] [CrossRef]
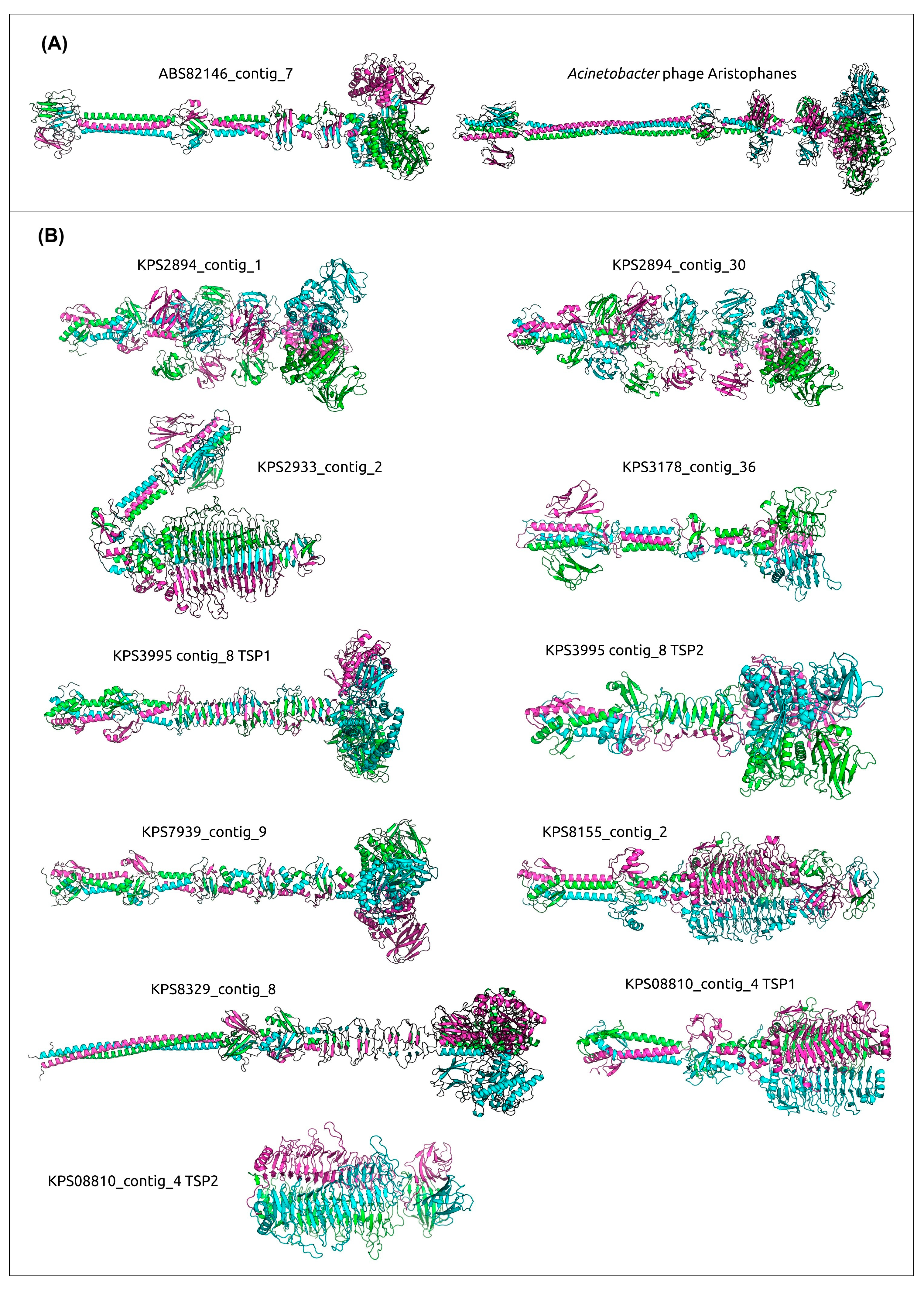
- Timoshina, O.Y.; Shneider, M.M.; Evseev, P.V.; Shchurova, A.S.; Shelenkov, A.A.; Mikhaylova, Y.V.; Sokolova, O.S.; Kasimova, A.A.; Arbatsky, N.P.; Dmitrenok, A.S.; et al. Novel Acinetobacter baumannii bacteriophage aristophanes encoding structural polysaccharide deacetylase. Viruses 2021, 13, 1688. [Google Scholar] [CrossRef]
- Latka, A.; Leiman, P.G.; Drulis-Kawa, Z.; Briers, Y. Modeling the architecture of depolymerase-containing receptor binding proteins in Klebsiella phages. Front. Microbiol. 2019, 10, 2649. [Google Scholar] [CrossRef]
- Wyres, K.L.; Wick, R.R.; Judd, L.M.; Froumine, R.; Tokolyi, A.; Gorrie, C.L.; Lam, M.M.C.; Duchêne, S.; Jenney, A.; Holt, K.E. Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. 2019, 15, e1008114. [Google Scholar] [CrossRef]
- Fraser, J.S.; Maxwell, K.L.; Davidson, A.R. Immunoglobulin-like domains on bacteriophage: Weapons of modest damage? Curr. Opin. Microbiol. 2007, 10, 382–387. [Google Scholar] [CrossRef]
- Xu, J.; Wang, D.; Gui, M.; Xiang, Y. Structural assembly of the tailed bacteriophage Φ29. Nat. Commun. 2019, 10, 2366. [Google Scholar] [CrossRef]
- Pell, L.G.; Gasmi-Seabrook, G.M.C.; Morais, M.; Neudecker, P.; Kanelis, V.; Bona, D.; Donaldson, L.W.; Edwards, A.M.; Howell, P.L.; Davidson, A.R.; et al. The solution structure of the c-terminal ig-like domain of the bacteriophage λ Tail tube protein. J. Mol. Biol. 2010, 403, 468–479. [Google Scholar] [CrossRef]
- Evseev, P.; Shneider, M.; Miroshnikov, K. Evolution of phage tail sheath protein. Viruses 2022, 14, 1148. [Google Scholar] [CrossRef]
- Williams, A.H.; Veyrier, F.J.; Bonis, M.; Michaud, Y.; Raynal, B.; Taha, M.-K.; White, S.W.; Haouz, A.; Boneca, I.G. Visualization of a substrate-induced productive conformation of the catalytic triad of the Neisseria meningitidis peptidoglycan O-acetylesterase reveals mechanistic conservation in SGNH esterase family members. Acta Cryst. 2014, 70, 2631–2639. [Google Scholar] [CrossRef]
- Huang, T.; Zhang, Z.; Tao, X.; Shi, X.; Lin, P.; Liao, D.; Ma, C.; Cai, X.; Lin, W.; Jiang, X.; et al. Structural and functional basis of bacteriophage K64-ORF41 depolymerase for capsular polysaccharide degradation of Klebsiella pneumoniae K64. Int. J. Biol. Macromol. 2024, 265, 130917. [Google Scholar] [CrossRef]
- Maciejewska, B.; Squeglia, F.; Latka, A.; Privitera, M.; Olejniczak, S.; Switala, P.; Ruggiero, A.; Marasco, D.; Kramarska, E.; Drulis-Kawa, Z.; et al. Klebsiella phage KP34gp57 capsular depolymerase structure and function: From a serendipitous finding to the design of active mini-enzymes against K. pneumoniae. mBio 2023, 14, e0132923. [Google Scholar] [CrossRef]
- Squeglia, F.; Maciejewska, B.; Łątka, A.; Ruggiero, A.; Briers, Y.; Drulis-Kawa, Z.; Berisio, R. Structural and functional studies of a Klebsiella phage capsule depolymerase tailspike: Mechanistic insights into capsular degradation. Structure 2020, 28, 613–624.e4. [Google Scholar] [CrossRef]
- Greenfield, J.; Shang, X.; Luo, H.; Zhou, Y.; Heselpoth, R.D.; Nelson, D.C.; Herzberg, O. Structure and tailspike glycosidase machinery of ORF212 from E. coli O157:H7 phage CBA120 (TSP3). Sci. Rep. 2019, 9, 7349. [Google Scholar] [CrossRef]
- Zhang, L.; Yan, Y.; Gan, Q.; She, Z.; Zhu, K.; Wang, J.; Gao, Z.; Dong, Y.; Gong, Y. Structural and functional characterization of the deep-sea thermophilic bacteriophage GVE2 tailspike protein. Int. J. Biol. Macromol. 2020, 164, 4415–4422. [Google Scholar] [CrossRef]
- Álvarez, V.E.; Quiroga, M.P.; Centrón, D. Identification of a specific biomarker of Acinetobacter baumannii global clone 1 by machine learning and PCR related to metabolic fitness of ESKAPE pathogens. mSystems 2023, 8, e00734. [Google Scholar] [CrossRef]
- Hamidian, M.; Nigro, S.J. Emergence, molecular mechanisms and global spread of carbapenem-resistant Acinetobacter baumannii. Microb. Genom. 2019, 5, e000306. [Google Scholar] [CrossRef]
- Gu, D.; Wu, Y.; Chen, K.; Zhang, Y.; Ju, X.; Yan, Z.; Xie, M.; Chan, E.W.C.; Chen, S.; Ruan, Z.; et al. Recovery and genetic characterization of clinically-relevant ST2 carbapenem-resistant Acinetobacter baumannii isolates from untreated hospital sewage in Zhejiang Province, China. Sci. Total Environ. 2024, 916, 170058. [Google Scholar] [CrossRef]
- Chen, P.K.; Liu, C.-Y.; Kuo, H.-Y.; Lee, Y.-T.; Liu, Y.-H.; Zhang, Y.-Z.; Kao, C.-Y. Emergence of extensively-drug-resistant hypervirulent Acinetobacter baumannii isolated from patients with bacteraemia: Bacterial phenotype and virulence analysis. Int. J. Antimicrob. Agents 2024, 64, 107358. [Google Scholar] [CrossRef]
- Giannouli, M.; Antunes, L.C.S.; Marchetti, V.; Triassi, M.; Visca, P.; Zarrilli, R. Virulence-related traits of epidemic Acinetobacter baumannii strains belonging to the international clonal lineages I-III and to the emerging genotypes ST25 and ST78. BMC Infect. Dis. 2013, 13, 282. [Google Scholar] [CrossRef]
- Pfeifer, Y.; Hunfeld, K.-P.; Borgmann, S.; Maneg, D.; Blobner, W.; Werner, G.; Higgins, P.G. Carbapenem-resistant Acinetobacter baumannii ST78 with OXA-72 carbapenemase and ESBL gene blaCTX-M-115. J. Antimicrob. Chemother. 2016, 71, 1426–1428. [Google Scholar] [CrossRef]
- Alekseeva, A.E.; Brusnigina, N.F.; Makhova, M.A. Genetic profile of carbapenem-resistant Acinetobacter baumannii strains. Russ. J. Infect. Immun. 2024, 14, 681–689. [Google Scholar] [CrossRef]
- Peirano, G.; Chen, L.; Kreiswirth, B.N.; Pitout, J.D.D. Emerging Antimicrobial-resistant high-risk Klebsiella pneumoniae clones ST307 and ST147. Antimicrob. Agents Chemother. 2020, 64, e01148-20. [Google Scholar] [CrossRef]
- de Sales, R.O.; Leaden, L.; Migliorini, L.B.; Severino, P. A Comprehensive genomic analysis of the emergent Klebsiella pneumoniae ST16 lineage: Virulence, antimicrobial resistance and a comparison with the clinically relevant ST11 strain. Pathogens 2022, 11, 1394. [Google Scholar] [CrossRef]
- Roe, C.C.; Vazquez, A.J.; Esposito, E.P.; Zarrilli, R.; Sahl, J.W. Diversity, virulence, and antimicrobial resistance in isolates from the newly emerging Klebsiella pneumoniae ST101 lineage. Front. Microbiol. 2019, 10, 542. [Google Scholar] [CrossRef]
- Arcari, G.; Carattoli, A. Global spread and evolutionary convergence of multidrug-resistant and hypervirulent Klebsiella pneumoniae high-risk clones. Pathog. Glob. Health 2023, 117, 328–341. [Google Scholar] [CrossRef]
- Fursova, N.K.; Astashkin, E.I.; Gabrielyan, N.I.; Novikova, T.S.; Fedyukina, G.N.; Kubanova, M.K.; Esenova, N.M.; Sharapchenko, S.O.; Volozhantsev, N.V. Emergence of five genetic lines ST395NDM-1, ST13OXA-48, ST3346OXA-48, ST39CTX-M-14, and novel ST3551OXA-48 of multidrug-resistant clinical Klebsiella pneumoniae in Russia. Microb. Drug Resist. 2020, 26, 924–933. [Google Scholar] [CrossRef]
- Arato, V.; Raso, M.M.; Gasperini, G.; Berlanda Scorza, F.; Micoli, F. Prophylaxis and treatment against Klebsiella pneumoniae: Current insights on this emerging anti-microbial resistant global threat. Int. J. Mol. Sci. 2021, 22, 4042. [Google Scholar] [CrossRef]
- Choi, M.; Hegerle, N.; Nkeze, J.; Sen, S.; Jamindar, S.; Nasrin, S.; Sen, S.; Permala-Booth, J.; Sinclair, J.; Tapia, M.D.; et al. The diversity of lipopolysaccharide (O) and capsular polysaccharide (K) antigens of invasive Klebsiella pneumoniae in a multi-country collection. Front. Microbiol. 2020, 11, 1249. [Google Scholar] [CrossRef]
- Narancic, J.; Gavric, D.; Kostanjsek, R.; Knezevic, P. First characterization of Acinetobacter baumannii specific filamentous phages. Viruses 2024, 16, 857. [Google Scholar] [CrossRef]
- Klonowska, A.; Ardley, J.; Moulin, L.; Zandberg, J.; Patrel, D.; Gollagher, M.; Marinova, D.; Reddy, T.B.K.; Varghese, N.; Huntemann, M.; et al. Discovery of a novel filamentous prophage in the genome of the Mimosa Pudica microsymbiont Cupriavidus taiwanensis STM 6018. Front. Microbiol. 2023, 14, 1082107. [Google Scholar] [CrossRef]
- Shapiro, J.W.; Putonti, C. UPΦ phages, a new group of filamentous phages found in several members of Enterobacteriales. Virus Evol. 2020, 6, veaa030. [Google Scholar] [CrossRef]
- Evseev, P.; Bocharova, J.; Shagin, D.; Chebotar, I. Analysis of Pseudomonas aeruginosa isolates from patients with cystic fibrosis revealed novel groups of filamentous bacteriophages. Viruses 2023, 15, 2215. [Google Scholar] [CrossRef]
- Hay, I.D.; Lithgow, T. Filamentous Phages: Masters of a microbial sharing economy. EMBO Rep. 2019, 20, e47427. [Google Scholar] [CrossRef]
- Loh, B.; Chen, J.; Manohar, P.; Yu, Y.; Hua, X.; Leptihn, S. A biological inventory of prophages in A. baumannii genomes reveal distinct distributions in classes, length, and genomic positions. Front. Microbiol. 2020, 11, 579802. [Google Scholar] [CrossRef]
- Raposo, M.L.; Pimentel, A.C.; Manageiro, V.; Duarte, A.; Caniça, M.; Vale, F.F. Identifying phage lysins through genomic analysis of prophages from Acinetobacter baumannii. Front. Microbiol. 2025, 16, 1532950. [Google Scholar] [CrossRef]
- Marques, A.T.; Tanoeiro, L.; Duarte, A.; Gonçalves, L.; Vítor, J.M.B.; Vale, F.F. Genomic analysis of prophages from Klebsiella pneumoniae clinical isolates. Microorganisms 2021, 9, 2252. [Google Scholar] [CrossRef]
- Shen, J.; Zhou, J.; Xu, Y.; Xiu, Z. Prophages contribute to genome plasticity of Klebsiella pneumoniae and may involve the chromosomal integration of ARGs in CG258. Genomics 2020, 112, 998–1010. [Google Scholar] [CrossRef]
- de Sousa, J.A.M.; Buffet, A.; Haudiquet, M.; Rocha, E.P.C.; Rendueles, O. Modular prophage interactions driven by capsule serotype select for capsule loss under phage predation. ISME J. 2020, 14, 2980–2996. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Strain Designation | Data of Isolation | Source of Isolation | GenBank Accession Number | KL | MLST | SCPM-Obolensk Accession Number | Antibiotic Resistance Phenotype |
|---|---|---|---|---|---|---|---|---|
| A. baumannii | ||||||||
| 1 | ABS4984 | 07.02.2024 | blood | JBMMJM000000000 | 17 | 19 | B-15821 | XDR |
| 2 | ABS4062 | 12.02.2024 | blood | JBMMJL000000000 | 17 | 19 | B-15822 | XDR |
| 3 | ANS7072 * | 23.04.2024 | blood | JBMMJD000000000 | 49 | 2 | B-16457 | XDR |
| 4 | ABS09493 | 02.05.2024 | blood | JBMMJJ000000000 | 3 | 78 | B-16451 | XDR |
| 5 | ABS09481 | 02.05.2024 | blood | JBMMJI000000000 | 3 | 78 | B-16452 | XDR |
| 6 | ABS09593 | 03.05.2024 | blood | JBMMJF000000000 | 3 | 78 | B-16455 | XDR |
| 7 | ABS78057 | 04.05.2024 | blood | JBMMJG000000000 | 17 | 19 | B-16454 | XDR |
| 8 | ABS82612 | 13.05.2024 | blood | JBMMJK000000000 | 3 | 78 | B-16450 | XDR |
| 9 | ABS82603 | 13.05.2024 | blood | JBMMJH000000000 | 3 | 78 | B-16453 | XDR |
| 10 | ABS82146 | 13.05.2024 | blood | JBMMJE000000000 | 3 | 78 | B-16456 | XDR |
| 11 | ABS30421 | 20.05.2024 | blood | JBMMHS000000000 | 235 | 2 | B-17190 | XDR |
| 12 | ABS2640 | 06.06.2024 | blood | JBMMHQ000000000 | 235 | 2 | B-17192 | XDR |
| 13 | ABS7948 | 07.06.2024 | blood | JBMMHR000000000 | 235 | 2 | B-17191 | XDR |
| 14 | ABS1720 | 06.07.2024 | blood | JBMMHO000000000 | 235 | 2 | B-17194 | XDR |
| 15 | ABS8964 | 29.07.2024 | blood | JBMMHP000000000 | 235 | 2 | B-17193 | XDR |
| K. pneumoniae | ||||||||
| 1 | KPS0683 | 09.01.2024 | blood | JBMMJP000000000 | 23 | 39 | B-15818 | PDR |
| 2 | KPS0700 | 10.01.2024 | blood | JBMMJO000000000 | 48 | 395 | B-15819 | PDR |
| 3 | KPS4326 | 15.01.2024 | blood | JBMMJN000000000 | 23 | 39 | B-15820 | PDR |
| 4 | KPS9399 | 26.01.2024 | blood | JBMMJC000000000 | 48 | 395 | B-16458 | PDR |
| 5 | KPS9821 | 29.01.2024 | blood | JBMMKC000000000 | 48 | 395 | B-15805 | PDR |
| 6 | KPS4208 | 30.01.2024 | blood | JBMMJZ000000000 | 17 | 101 | B-15808 | XDR |
| 7 | KPS9827 | 31.01.2024 | blood | JBMMKA000000000 | 107 | 512 | B-15807 | XDR |
| 8 | KPS6724 | 01.02.2024 | blood | JBMMKB000000000 | 48 | 395 | B-15806 | PDR |
| 9 | KPS7051 | 01.02.2024 | blood | JBMMIL000000000 | 17 | 101 | B-16476 | XDR |
| 10 | KPS6729 | 03.02.2024 | blood | JBMMJY000000000 | 1 | 23 | B-15809 | PDR |
| 11 | KPS4983 | 05.02.2024 | blood | JBMMJX000000000 | 1 | 23 | B-15810 | PDR |
| 12 | KPS3995 | 12.02.2024 | blood | JBMMJW000000000 | 57 | 218 | B-15811 | XDR |
| 13 | KPS2933 | 17.02.2024 | blood | JBMMJV000000000 | 107 | 512 | B-15812 | XDR |
| 14 | KPS7481 | 17.02.2024 | blood | JBMMJU000000000 | 23 | 39 | B-15813 | XDR |
| 15 | KPS1210 | 19.02.2024 | blood | JBMMJR000000000 | 23 | 39 | B-15816 | XDR |
| 16 | KPS7939 | 29.02.2024 | blood | JBMMJT000000000 | 64 | 39 | B-15814 | XDR |
| 17 | KPS9733 | 04.03.2024 | blood | JBMMJS000000000 | 64 | 147 | B-15815 | XDR |
| 18 | KPS8329 | 06.03.2024 | blood | JBMMJQ000000000 | 39 | 395 | B-15817 | XDR |
| 19 | KPS6974 | 16.03.2024 | blood | JBMMIP000000000 | 2 | 395 | B-16471 | XDR |
| 20 | KPS6976 | 25.03.2024 | blood | JBMMIQ000000000 | 2 | 395 | B-16470 | XDR |
| 21 | KPS6483 | 30.03.2024 | blood | JBMMIJ000000000 | 23 | 39 | B-16478 | XDR |
| 22 | KPS09487 | 02.04.2024 | blood | JBMMIT000000000 | 20 | 147 | B-16467 | XDR |
| 23 | KPS9086 | 04.04.2024 | blood | JBMMII000000000 | 48 | 395 | B-16479 | XDR |
| 24 | KPS1073 | 11.04.2024 | blood | JBMMIO000000000 | 112 | 15 | B-16472 | XDR |
| 25 | KPS2936 | 11.04.2024 | blood | JBMMIM000000000 | 48 | 395 | B-16475 | XDR |
| 26 | KPS3294 | 15.04.2024 | blood | JBMMIR000000000 | 2 | 395 | B-16469 | XDR |
| 27 | KPS3178 | 15.04.2024 | blood | JBMMIN000000000 | 23 | 39 | B-16473 | XDR |
| 28 | KPS3271 | 15.04.2024 | blood | JBMMIK000000000 | 2 | 395 | B-16477 | XDR |
| 29 | KPS82148 | 13.05.2024 | blood | JBMMJA000000000 | 20 | 147 | B-16460 | XDR |
| 30 | KPS47018 | 22.04.2024 | blood | JBMMIZ000000000 | 23 | 39 | B-16461 | XDR |
| 31 | KPS43107-1 | 22.04.2024 | blood | JBMMIV000000000 | 23 | 39 | B-16465 | XDR |
| 32 | KPS46086 | 24.04.2024 | blood | JBMMJB000000000 | 23 | 39 | B-16459 | XDR |
| 33 | KPS78055 | 02.05.2024 | blood | JBMMIY000000000 | 48 | 395 | B-16462 | XDR |
| 34 | KPS09509 | 02.05.2024 | blood | JBMMIX000000000 | 20 | 147 | B-16463 | XDR |
| 35 | KPS78054 | 02.05.2024 | blood | JBMMIS000000000 | 48 | 395 | B-16468 | XDR |
| 36 | KPS78133 | 03.05.2024 | blood | JBMMIU000000000 | 107 | 512 | B-16466 | PDR |
| 37 | KPS08810 | 06.05.2024 | blood | JBMMIW000000000 | 17 | 101 | B-16464 | XDR |
| 38 | KPS20601 | 08.07.2024 | blood | JBMMIH000000000 | 23 | 39 | B-17173 | XDR |
| 39 | KPS2894 | 08.07.2024 | blood | JBMMIG000000000 | 39 | 39 | B-17174 | XDR |
| 40 | KPS8961 | 29.07.2024 | blood | JBMMIF000000000 | 2 | 395 | B-17175 | XDR |
| 41 | KPS9940 | 05.08.2024 | blood | JBMMIE000000000 | 107 | 512 | B-17177 | PDR |
| 42 | KPS9941 | 05.08.2024 | blood | JBMMID000000000 | 107 | 512 | B-17179 | PDR |
| 43 | KPS6123 | 12.08.2024 | blood | JBMMIB000000000 | 64 | 147 | B-17181 | XDR |
| 44 | KPS10299 | 12.08.2024 | blood | JBMMIC000000000 | 102 | 307 | B-17180 | XDR |
| 45 | KPS8949 | 15.08.2024 | blood | JBMMIA000000000 | 64 | 147 | B-17182 | XDR |
| 46 | KPS7136 | 19.08.2024 | blood | JBMMHZ000000000 | 64 | 147 | B-17183 | XDR |
| 47 | KPS8914 | 19.08.2024 | blood | JBMMHY000000000 | 48 | 395 | B-17184 | XDR |
| 48 | KPS8155 | 10.09.2024 | blood | JBMMHX000000000 | 17 | 101 | B-17185 | XDR |
| 49 | KPS8500 | 16.09.2024 | blood | JBMMHW000000000 | 23 | 39 | B-17186 | XDR |
| 50 | KPS6191 | 16.09.2024 | blood | JBMMHV000000000 | 64 | 39 | B-17187 | XDR |
| 51 | KPS6186 | 16.09.2024 | blood | JBMMHU000000000 | 107 | 512 | B-17188 | XDR |
| 52 | KPS4431 | 17.09.2024 | blood | JBMMHT000000000 | 48 | 395 | B-17189 | XDR |
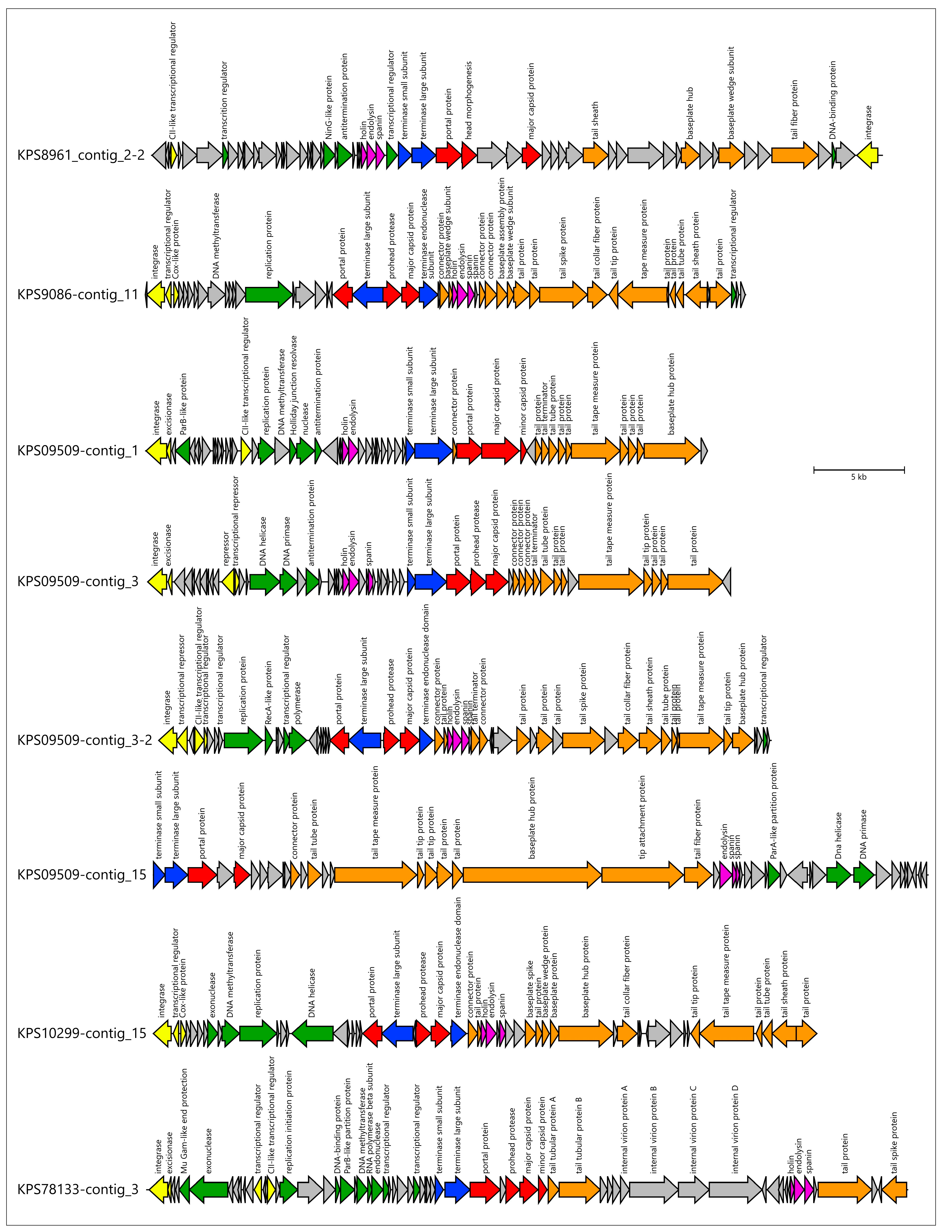
| Designation of Prophage Region | Size, bp | % GC | Related GenBank Phage Sequence (Taxonomy) |
|---|---|---|---|
| ABS09481-contig_18 | 66,919 | 37.0% | Acinetobacter phage vB_AbaM_ABMM1 (Unclassified) |
| ABS09481-contig_9 | 35,087 | 39.4% | Moraxella phage Mcat9 (Unclassified) |
| ABS30421-contig_6 | 25,324 | 39.8% | Acinetobacter phage vB_AbaS_Eva (Vieuvirus) |
| ABS4062-contig_7 | 48,276 | 36.7% | Acinetobacter phage Ab105-2phi (Vieuvirus) |
| ABS4984-contig_9 | 33,074 | 35.4% | Acinetobacter phage ZaA-2018b (Unclassified) |
| ABS82146-contig_7 | 25,454 | 41.5% | Ralstonia phage Firinga (Firingavirus) |
| ABS8964-contig_4 | 38,416 | 39.9% | Acinetobacter phage fEg-Aba01 (Unclassified) |
| Designation of Prophage Region | Size, bp | % GC | Related GenBank Phage Sequence (Taxonomy) |
|---|---|---|---|
| KPS08810-contig_1 | 36,056 | 50.8% | Klebsiella phage Kp4865-2 (Peduoviridae) |
| KPS08810-contig_4 | 37,471 | 50.6% | Klebsiella phage Kp4865-2 (Peduoviridae) |
| KPS09509-contig_1 | 31,076 | 50.2% | Enterobacteria phage mEp043_c-1 (Aguilavirus) |
| KPS09509-contig_15 | 42,861 | 50.7% | Klebsiella phage pJN2-26 (Unclassified) |
| KPS09509-contig_3 | 32,211 | 50.8% | Klebsiella phage ST13-OXA48phi12.2 (Unclassified) |
| KPS09509-contig_3-2 | 33,882 | 51.8% | Klebsiella phage ST147-VIM1phi7.1 (Peduoviridae; Vimunumvirus) |
| KPS10299-contig_15 | 36,702 | 49.8% | Klebsiella phage ST16-OXA48phi5.4 (Peduoviridae; Reipivirus) |
| KPS2894-contig_1 | 46,173 | 52.7% | Klebsiella phage 5_LV-2017 (Unclassified) |
| KPS2894-contig_30 | 26,753 | 51.1% | Klebsiella phage vB_VIPKPNMC05 (Unclassified) |
| KPS2933-contig_2 | 26,795 | 51.6% | Klebsiella phage ST512-KPC3phi13.3 (Unclassified) |
| KPS3178-contig_36 | 22,258 | 55.2% | Klebsiella phage ST11-OXA245phi3.2 (Unclassified) |
| KPS3178-contig_7 | 10,620 | 39.7% | Curvibacter phage P26059A (Unclassified) |
| KPS3271-contig_6 | 37,003 | 51.8% | Klebsiella phage Kp4856-1 (Unclassified) |
| KPS3995-contig_8 | 42,804 | 51.6% | Klebsiella phage ST974-OXA48phi18.2 (Unclassified) |
| KPS4431-contig_51 | 25,748 | 53.4% | Escherichia phage ev017 (Jouyvirus) |
| KPS4983-contig_15 | 38,044 | 53.5% | Klebsiella phage ST846-OXA48phi9.1 (Unclassified) |
| KPS6186-contig_1 | 22,351 | 51.8% | Klebsiella phage P12620-1 (Unclassified) |
| KPS6191-contig_6 | 38,650 | 52.7% | Escherichia phage PDP110_P2 (Hendrixvirinae; Wanchaivirus) |
| KPS78133-contig_3 | 42,102 | 50.7% | Stenotrophomonas phage vB_SmeS_BUCT709 (Unclassified) |
| KPS7939-contig_9 | 45,850 | 52.9% | Klebsiella phage ST512-KPC3phi13.1 (Unclassified) |
| KPS8155-contig_2 | 49,994 | 52.4% | Klebsiella phage 2b_LV-2017 (Unclassified) |
| KPS8329-contig_8 | 40,215 | 52.0% | Klebsiella phage RCIP0114 (Unclassified) |
| KPS8961_contig_2-2 | 40,432 | 52.3% | Klebsiella phage vB_Kpn_Chronis (Unclassified) |
| KPS9086-contig_11 | 33,216 | 55.5% | Klebsiella phage ST101-KPC2phi6.2 (Peduoviridae; Reginaelenavirus) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolupaeva, N.V.; Kolupaeva, L.V.; Evseev, P.V.; Skryabin, Y.P.; Lazareva, E.B.; Chernenkaya, T.V.; Volozhantsev, N.V.; Popova, A.V. Acinetobacter baumannii and Klebsiella pneumoniae Isolates Obtained from Intensive Care Unit Patients in 2024: General Characterization, Prophages, Depolymerases and Esterases of Phage Origin. Viruses 2025, 17, 623. https://doi.org/10.3390/v17050623
Kolupaeva NV, Kolupaeva LV, Evseev PV, Skryabin YP, Lazareva EB, Chernenkaya TV, Volozhantsev NV, Popova AV. Acinetobacter baumannii and Klebsiella pneumoniae Isolates Obtained from Intensive Care Unit Patients in 2024: General Characterization, Prophages, Depolymerases and Esterases of Phage Origin. Viruses. 2025; 17(5):623. https://doi.org/10.3390/v17050623
Chicago/Turabian StyleKolupaeva, Nadezhda V., Lyubov V. Kolupaeva, Peter V. Evseev, Yuriy P. Skryabin, Elena B. Lazareva, Tatyana V. Chernenkaya, Nikolay V. Volozhantsev, and Anastasia V. Popova. 2025. "Acinetobacter baumannii and Klebsiella pneumoniae Isolates Obtained from Intensive Care Unit Patients in 2024: General Characterization, Prophages, Depolymerases and Esterases of Phage Origin" Viruses 17, no. 5: 623. https://doi.org/10.3390/v17050623
APA StyleKolupaeva, N. V., Kolupaeva, L. V., Evseev, P. V., Skryabin, Y. P., Lazareva, E. B., Chernenkaya, T. V., Volozhantsev, N. V., & Popova, A. V. (2025). Acinetobacter baumannii and Klebsiella pneumoniae Isolates Obtained from Intensive Care Unit Patients in 2024: General Characterization, Prophages, Depolymerases and Esterases of Phage Origin. Viruses, 17(5), 623. https://doi.org/10.3390/v17050623