Abstract
Respiratory syncytial virus (RSV) is a leading cause of lower respiratory tract infections in infants, young children, and immunocompromised individuals. Currently, FDA-approved monoclonal antibody therapies are limited to infants and young children with severe RSV disease. As a result, there is an urgent need for comprehensive studies of RSV pathogenesis to support the development of new therapeutic strategies. Exchange proteins directly activated by cAMP (EPAC) have recently emerged as key regulators in various viral infections. Our previous work identified EPAC isoform 2 (EPAC2) as a critical factor in RSV replication and host innate immune responses. However, the molecular mechanisms underlying EPAC2’s role in RSV infection remain unclear. In this study, we investigated EPAC2-mediated RSV infection by identifying EPAC2-interacting proteins. Proteomics and immunoprecipitation analyses revealed that junction plakoglobin (JUP) interacts with EPAC2 in both mock- and RSV-infected cells, with this interaction notably enhanced during RSV infection. To determine JUP’s role in RSV infection, we compared viral replication in JUP-deficient and control cells. JUP downregulation significantly reduced the production of infectious RSV particles, likely by impairing viral budding and viral gene transcription. Moreover, our findings indicate that JUP is essential for an effective cellular immune response to RSV infection. Together, these results suggest that EPAC2 and JUP may cooperatively regulate RSV replication and dissemination.
1. Introduction
Respiratory syncytial virus (RSV) is a negative-strand RNA virus and the most common cause of lower respiratory tract infection (LRTI) in young children and infants [1]. Nearly 90% of infants are infected with RSV within the first two years of life, with the majority of hospitalizations occurring in otherwise healthy infants [1]. While vaccine development has shown promise, current efforts are primarily limited to protecting elderly individuals and pregnant women [2,3]. Nirsevimab remains the only available treatment, and its use is restricted to infants and select young children at increased risk for severe RSV disease [4]. Despite these advances, RSV continues to pose a significant global health challenge due to its considerable economic and clinical burden. This highlights the need for a comprehensive investigation into RSV disease mechanisms to identify therapeutic targets and develop effective strategies to reduce the severity of RSV infection.
Exchange proteins directly activated by cAMP (EPAC) are relatively novel guanine exchange factors (GEFs) that regulate a variety of biological processes [5]. EPAC proteins transduce signals primarily by activating small GTPases, predominantly Rap1 and Rap2, which influence diverse downstream targets [5]. Initially recognized for their roles in cancer, neurological disorders, diabetes, and inflammation [6,7,8,9,10,11,12], EPACs have more recently emerged as key players in viral infections, with essential roles identified in at least seven human viruses [13,14,15,16,17,18]. Two main isoforms of EPAC exist in eukaryotic cells: EPAC1 and EPAC2. EPAC1 has been shown to favor replication of viruses such as Ebola, Middle East respiratory syndrome coronavirus (MERS-CoV), SARS-CoV-2, and influenza [14,15,18]. Our previous studies identified EPAC2 as a promising therapeutic target for RSV infection; knockdown or knockout of EPAC2, as well as treatment with an EPAC2-specific inhibitor, significantly suppressed RSV replication and reduced the expression of pro-inflammatory cytokines and chemokines [13,16]. The critical role of EPAC2 in RSV infection was further validated in mouse models [17]. Beyond RSV, EPAC2 has also been implicated in pulmonary inflammation and tissue remodeling in response to cigarette smoke exposure, highlighting its importance in lung health [19]. However, the downstream targets of EPAC2 in the pulmonary setting remain largely unknown. Identifying these targets could provide valuable insights into RSV pathogenesis and inform the development of novel therapeutic strategies.
Recently, we conducted a proteomics study to identify proteins that interact with EPAC2 and discovered that junction plakoglobin (JUP) was among its associated partners. JUP, also known as γ-catenin, is a homolog to β-catenin and plays a key role in desmosome formation and cell-cell adhesion [20,21]. However, the functions of JUP in viral infection remain largely unexplored. Among the EPAC2-binding proteins identified, JUP exhibited the least enhancement in its interaction with EPAC2 by RSV. We chose to focus on the JUP-EPAC2 interaction based on the rationale that if this modest interaction could be experimentally validated, other EPAC2-associated proteins showing stronger binding might be even more biologically significant. JUP was also of particular interest due to its established functions. Silencing JUP has been shown to disrupt the structure of actin in the cell cytoskeleton21, and the cytoskeleton is known to play essential roles in multiple stages of viral infection, including the transport and assembly of viral proteins and particles, viral immune evasion, and cell-to-cell fusion [22,23,24,25]. Although JUP’s role in viral infection has not been defined, its influence on actin dynamics suggests it could impact RSV assembly and budding. Indeed, previous studies have shown that the disruption of actin inhibits RSV release [26], suggesting a potential link between JUP and viral egress. These findings suggest that EPAC2 may regulate RSV assembly and budding, at least in part, through its interaction with JUP.
In this study, we demonstrated that silencing JUP through small-interfering RNA (siRNA) transfection in A549 cells led to a significant decrease in infectious particle production, viral gene transcription, and immune cytokine/chemokine secretion as compared to control samples transfected with non-targeting control siRNA (siCN). In addition, JUP controls replication-independent cellular inflammatory responses. Together, these results indicate that JUP may play a role in EPAC-2-mediated pathways that influence RSV infection.
2. Materials and Methods
2.1. Cell Lines and Virus Preparation
Human alveolar type II-like epithelial A549 cell line and human epithelial type II cell line Hep 2 were purchased from ATCC (Manassas, VA, USA). A549 cells, a common respiratory virus infection model, were cultured and maintained in F12K media with 10% FBS (vol/vol), 100 IU/mL penicillin, and 100 µg/mL streptomycin, as previously described [13,27]. These cell lines were maintained in incubators at 37 degrees Celsius and 5% CO2. Viral titer was determined by immunostaining in Hep-2 cells using polyclonal biotin-conjugated goat anti-RSV antibody (7950-0104; Bio-Rad, Hercules, CA, USA) and streptavidin peroxidase polymer (Sigma, St Louis, MO, USA) sequentially, as described [28,29].
2.2. EPAC2-Associated Proteins Preparation
A549 cells in 50% confluence were transfected with Flag-tagged EPAC2 plasmid (a gift from Dr. Susumu Seino, Kobe University Graduate School of Medicine, Japan) using FuGENE 6® Reagent (Promega, Madison, Wi, USA), according to the manufacturer’s protocol. Empty vectors were used as controls. After 30 h post-transfection, the cells were mock-infected or infected with RSV at an MOI of 1 for 15 h. The cells were then lysed by buffer 1 from the Immunoprecipitation Kit (# 11719386001, Roche, Indianapolis, IN, USA), followed by nonspecific cleaning using Protein G-agarose. The proteins in the lysis buffer 1 were then mixed with the monoclonal anti-Flag M2 antibody (F1804, Sigma, Saint Louis, MO, USA) or control and incubated at 4 °C for 4 h on a rocking platform, followed by Protein G resin overnight at 4 °C. The pellet beads by gravity sedimentation were then sequentially washed by Immunoprecipitation Washing buffers 2 and 3, according to the manufacturer’s protocol. An aliquot of the complex was then loaded into an SDS-PAGE gel to validate Flag-tagged EPAC2 overexpression and immunoprecipitation.
The remaining protein complex was analyzed via mass spectrometry to Identify EPAC2-associated proteins. The beads were treated with sample loading buffer and subjected to SDS-PAGE using a 10% Bis-Tris gel. Protein bands were visualized using Coomassie Brilliant Blue staining, and the gel was segmented into four molecular weight fractions. Each gel fragment underwent in-gel digestion with 100 ng of trypsin in 20 µL of 50 mM NH4HCO3 at 37 °C overnight. Peptides were subsequently extracted with 100% acetonitrile, vacuum-dried, and reconstituted in 10 µL of 5% methanol containing 0.1% formic acid. The samples were then analyzed via nanoHPLC-MS/MS using an EASY-nLC1000 (Thermo Fisher Scientific, Waltham, MA, USA) coupled to an Orbitrap Fusion mass spectrometer (Thermo Scientific) with an electrospray ionization (ESI) source. A custom-built trap column packed with 3 µm Reprosil-Pur Basic C18 beads and a 5 cm × 150 µm capillary column packed with 1.9 µm Reprosil-Pur Basic C18 beads were employed for separation. The instrument, operated under Xcalibur software version 2.2 (Thermo Fisher Scientific), was set to data-dependent acquisition mode. MS/MS spectra were searched against a target-decoy Human RefSeq database (release 2015_06, containing 73,637 entries) using the Proteome Discoverer 1.4 interface (Thermo Fisher) with Mascot algorithm (Mascot 2.4, Matrix Science, Mount Prospect, IL, USA). The precursor ion mass tolerance was set to 20 ppm, with a fragment ion mass tolerance of 0.02 Da, allowing up to two missed cleavages. Peptide identifications were validated using Percolator with a false discovery rate (FDR) threshold of 1% based on q-values. Protein abundance was determined using iBAQ calculations derived from previously published methodologies [30]. Proteins that were not detected and lacked iBAQ values were imputed with half of the minimum detected value to facilitate p-value and fold-change calculations.
2.3. Western Blot
Western blot analysis was performed to validate EPAC-2-associated proteins, identified by the proteomics studies. In brief, Flag-tagged EPAC2 that had undergone immunoprecipitation using an anti-Flag antibody was loaded into an SDS-PAGE gel. The anti-JUP antibody (A303-718A-T, Bethyl, Montgomery, TX, USA) was used to detect JUP in the pull-down complex. The membranes were stripped and reprobed by an anti-Flag antibody to validate the IP. The immunoprecipitation input was also checked by loading the total cell lysates to SDS-PAGE, followed by checking the expression of EPAC2-Flag, JUP, and β-actin.
The endogenous interaction between EPAC2 and JUP was also examined. Briefly, A549 cells were either mock-infected or infected with RSV at a multiplicity of infection (MOI) of 1 for 15 h. Immunoprecipitation was performed as described above, using an anti-EPAC2 antibody (Cat# 19103-1-AP, Proteintech, Rosemont, IL, USA) for pulldown, followed by detection of JUP in the EPAC2 complex using an anti-JUP antibody (Cat# 13-8500, Thermo Fisher Scientific).
2.4. siRNA-Mediated Gene Silencing
A549 cells at 80-90% confluence were transfected with 100 nM siRNA specific for JUP (Table 1, Sigma) using Lipofectamine™ 2000 Transfection Reagent (Thermo Scientific) for 24 h or 48 h. Scrambled siRNAs were used as controls (Cat# SIC001, Sigma). The suppression efficiency of siRNAs was then confirmed by Western blot.

Table 1.
JUP siRNA Primers.
2.5. Cytokine/Chemokine Concentration Measurement
To quantify immune and inflammatory mediators in mock or RSV-infected samples, pellets were removed by centrifugation at 1000 rpm for 5 min. A 50 µL aliquot of the resulting supernatants was analyzed using the Bio-Plex multiplex system (Bio-Plex Pro Human Cytokine 27-plex Assay, Cat #M500KCAF0Y, Bio-Rad) to measure 27 kinds of cytokines and chemokines, including FGF-β, IL-2, IL-10, MIP-1α, Eotaxin, IL-4, IL-12, MIP-1β, G-CSF, IL-5, IL-13, PDGF-bb, GM-CSF, IL-6, IL-15, RANTES, IFN-γ, IL-7, IL-17A, TNF-α, IL-1β, IL-8, IP-10, VEGF, IL-1ra, IL-9, and MCP-1.
2.6. Virus Titration Assay
RSV harvested from A549 cell lysates or culture supernatants was diluted in a 5-fold serial dilution, followed by seeding 150 µL of it to Hep-2 cells, which were grown to confluence in 24-well plates (1.5 × 105 cells/well). Plates were then put in a shaker for 1 h at 37 °C, 5% CO2, and 50 rpm. After 1 h, 1 mL of MEM with 2% serum and 0.75% methylcellulose was added to each well, and the plates were kept in the incubator for 5 days (37 degrees Celsius, 5% CO2). After 5 days of incubation, the culturing gel solution was removed from the wells, and 0.5 mL of 10% formaldehyde was added to each well to fix the cells. After 30 min of incubation at room temperature, formaldehyde was removed from the wells, and 1 ml of 1% crystal violet (in EtOH) was added. After 30 min of incubation, the dye was removed, followed by plate washing and plaque counting to calculate the viral titers.
2.7. Quantitative Real-Time PCR (qRT-PCR)
To quantify RSV N gene expression, total cellular RNA was extracted by TRIzol reagents (Thermo Fisher Scientific). CDNA was synthesized with 1 μg of total RNA in a 20-μL reaction mixture using the TaqMan Reverse Transcription Reagents kit from ABI (catalog number N8080234; Applied Biosystems, Foster City, CA, USA). We used RT primer 5′-CTGCGATGAGTGGCAGGCTTTTTTTTTTTTAACTCAAAGCTC-3′. We incorporated a “tag” (underlined letters) as part of the assay due to self-priming exhibited by viral RNA. The tag sequence was derived from the bacterial chloramphenicol resistance (Cmr) gene. The sequence with bold letters is complementary to the poly(A) tails of the transcribed RSV N gene. The sequence in italics is N gene-specific. The reaction conditions were as follows: 25 °C for 10 min, 48 °C for 30 min, and 95 °C for 5 min. At a 25 °C annealing temperature, the 8 nucleotides (nt) matching N-specific sequences would not be sufficient for stable efficient priming of cDNA from the antigenome of hMPV. On the other hand, 20 nucleotides matching transcribed N (12 T’s and N gene-specific nucleotides) can attain stable annealing to the transcribed N gene. For Quantitative real-time PCR amplification, we used the RSV tag reverse primer CTGCGATGAGTGGCAGGC and the forward primer ACTACAGTGTATTAGACTTRACAGCAGAAG. The PCR was performed with 1 μL of cDNA in a total volume of 25 μL by using iTaq TM Universal SYBR Green Supermix (Cat# 1725124, Bio-rad, Hercules, CA, USA). The final concentration of the primers was 300 nM. 18S RNA was used as a housekeeping gene for normalization. PCR assays were run with the ABI Prism 7500 sequence detection system with the following conditions: 50 °C for 2 min, 95 °C for 10 min, and then 95 °C for 15 s and 60 °C for 1 min for 40 cycles. Duplicate cycle threshold (CT) values were analyzed in Microsoft Excel by the comparative CT (ΔΔCT) method according to the manufacturer’s instructions (Applied Biosystems). The amount of target (2−ΔΔCT) was obtained by normalization to the endogenous reference (18S) sample.
To quantify RSV antigenomic copies, synthetic transcripts of the genome were generated from the Topo plasmid containing N-P-M genes, using the T7 MegaScript kit, following the digestion with PmeI. The reaction mixture was then treated with Turbo Dnase and purified using the MegaScript kit. Primers were designed to span the N and P regions of the viral genome and incorporated a Cmr tag. First-strand cDNA was transcribed with a P-specific primer, 5′-CTGCGATGAGTGGCAGGCACTACAGTGTATTAGACTTRACAGCAGAAG-3′. For PCR assays, we used RSV tag primer CTGCGATGAGTGGCAGGC and primer RSV P GCATCTTCTCCATGRAATTCAGG.
2.8. Reporter Gene Assays
To investigate the role of JUP in inflammatory responses, A549 cells in a 24-well plate were transfected in triplicate with 0.05 µg/well luciferase reporter gene plasmids containing multiple copies of NF-κB binding sites (NF-κB-Luc) and 100 nM siRNAs, either scrambled or JUP-specific, using lipofectamine 2000 according to the manufacturer’s protocol. At 24 h post-transfection, the cells were treated with TNF-α at a 20 ng/mL concentration per well. Following 24 h of treatment, the cells were lysed to measure luciferase reporter activity, using a SpectraMax iD3 microplate reader (Molecular Devices, San Jose, CA, USA). Cells without treatment were used as controls.
We also investigated the function of JUP in mediating RSV-induced cellular responses. In brief, A549 cells in 24-well plates were transfected with 0.5 µg/well luciferase reporter plasmids containing multiple copies of IRF-3 binding sites of IFN-β (IRF3-Luc) and 100 nM siRNA, scrambled or JUP-specific, using lipofectamine 2000. A4 24 h post-transfection, the cells were mock-infected or infected with RSV at an MOI of 1 for 15 h, followed by cell lysis and luciferase activity measurement.
2.9. Statistical Analysis
The experimental results were analyzed using GraphPad Prism 5 software. An unpaired two-tailed t-test was employed to compare the difference. A p-value < 0.05 was considered to indicate a statistically significant difference. Single and two asterisks represent p-values of <0.05 and <0.01, respectively. Means ± standard errors (Ses) are shown.
3. Results
3.1. EPAC2-Associated Proteins
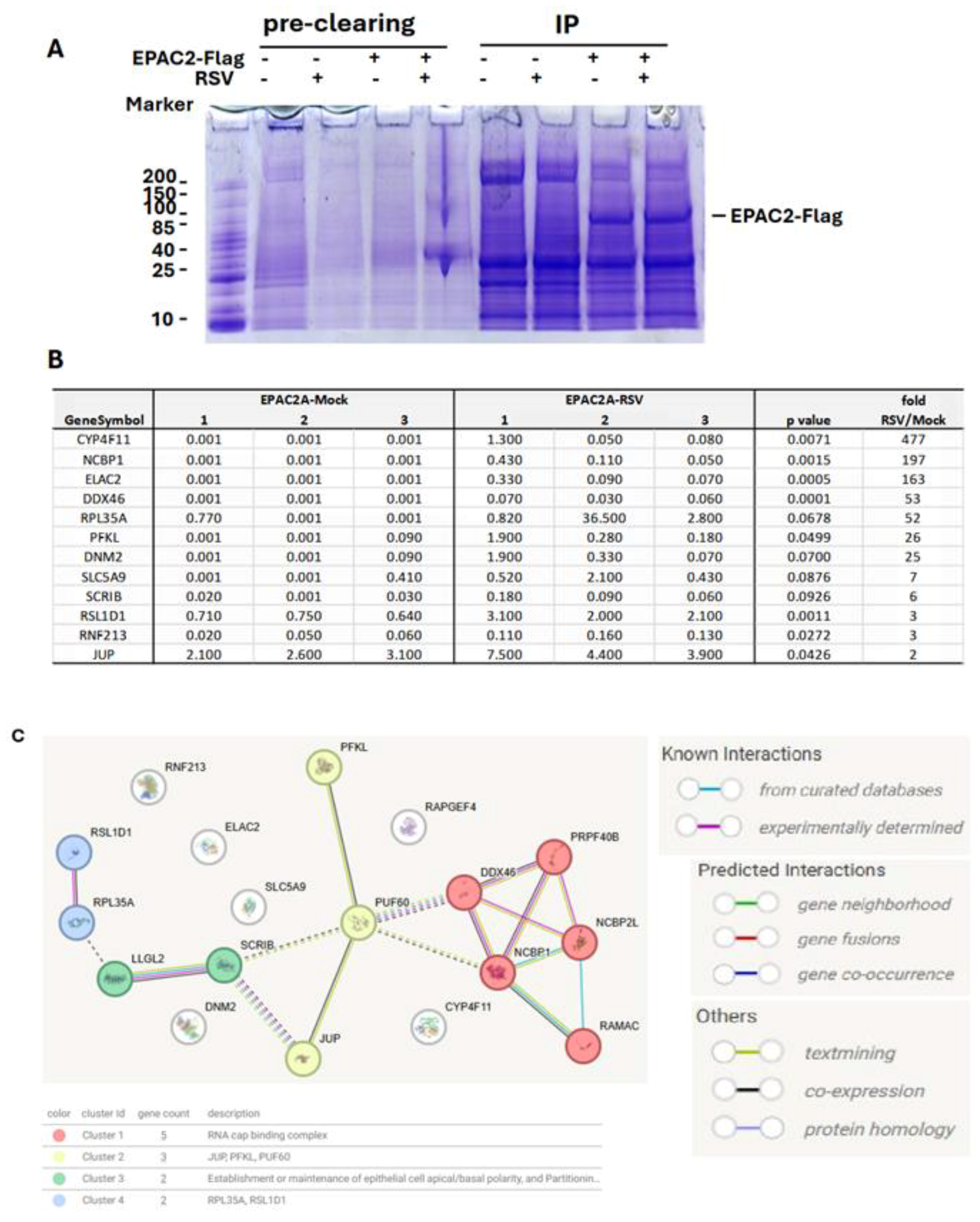
The role of EPAC in viral infections is an emerging area of study. In infections caused by MERS-CoV, SARS-CoV-2, or influenza, EPAC1 has been shown to promote viral replication [14,15,18]. Additionally, EPAC1 facilitates Ebola virus uptake into vascular endothelial cells via micropinocytosis. Whether EPAC2 contributes to viral infections remained unclear until our laboratory discovered its significant role in regulating both proinflammatory responses and RSV replication [13]. Most recently, in vivo experiments using RSV-infected mouse models further confirmed the critical involvement of EPAC2 in prompting RSV replication and RSV-induced pulmonary inflammation [17]. We also recently reported a similar role of EPAC2 in hMPV infection [16]. Despite these findings, the molecular mechanisms underlying EPAC2-mediated RSV infection remain largely unknown. To address this, we first employed a proteomics method to identify EPAC2-associated proteins. As shown in Figure 1A, EPAC2-Flag was successfully expressed and enriched using anti-Flag antibodies. Proteomic analysis identified eleven EPAC2-associated proteins; among them, eight exhibited significantly enhanced interaction with EPAC2 following RSV infection, while three showed reduced interaction (Figure 1B). Notably, all listed proteins displayed no detectable binding in cells lacking EPAC2-flag expression. Further STRING network analysis revealed four natural clusters, identified using the MCL clustering method (which detects clusters based on stochastic flow) (Figure 1C).
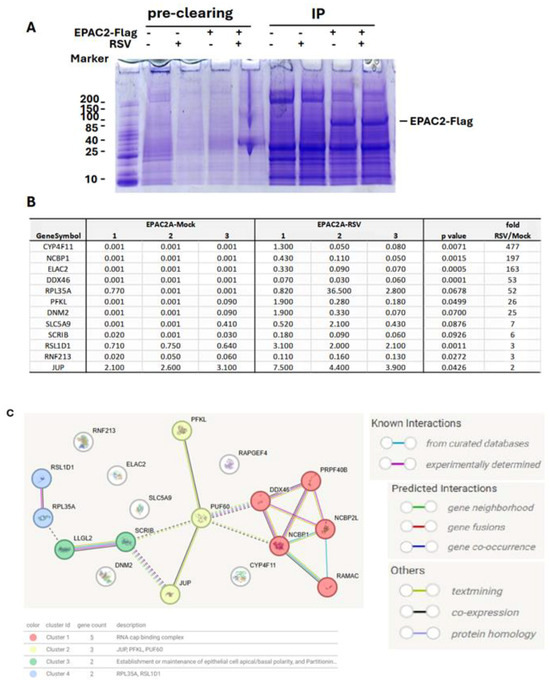
Figure 1.
EPAC2-associated proteins. Plasmid expressing Flag-tagged EPAC2 was transfected into A549 cells, followed by mock or RSV infection. Cells with empty vectors were used as controls. Cells were lysed and subsequently precleaned with Protein G-agarose. The proteins in lysis were then mixed with the monoclonal anti-Flag M2 antibody, followed by Protein G resin, and washed. (A). An aliquot of the protein G-agarose complex was then loaded into an SDS-PAGE gel to validate the overexpression and immunoprecipitation. (B) The list of EPAC2-associated proteins and the impact of RSV infection on their interactions. (C) MSL clustering analysis using STRING. A defined number of clusters is based on the stochastic flow. The edges indicate both functional and physical protein associations. The line color indicates the type of interaction evidence. The dashed edges represent inter-cluster interactions.
3.2. JUP-EPAC2 Interaction
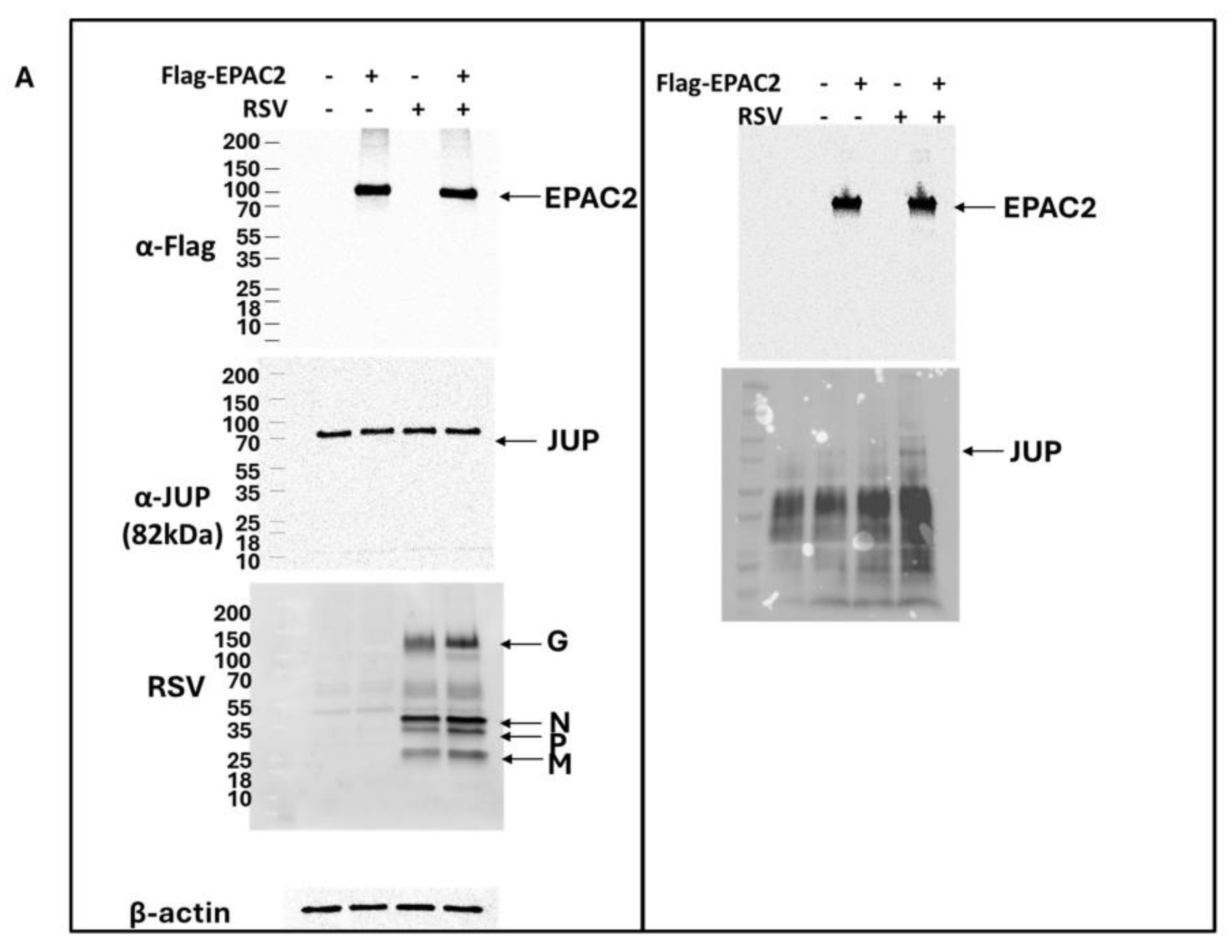
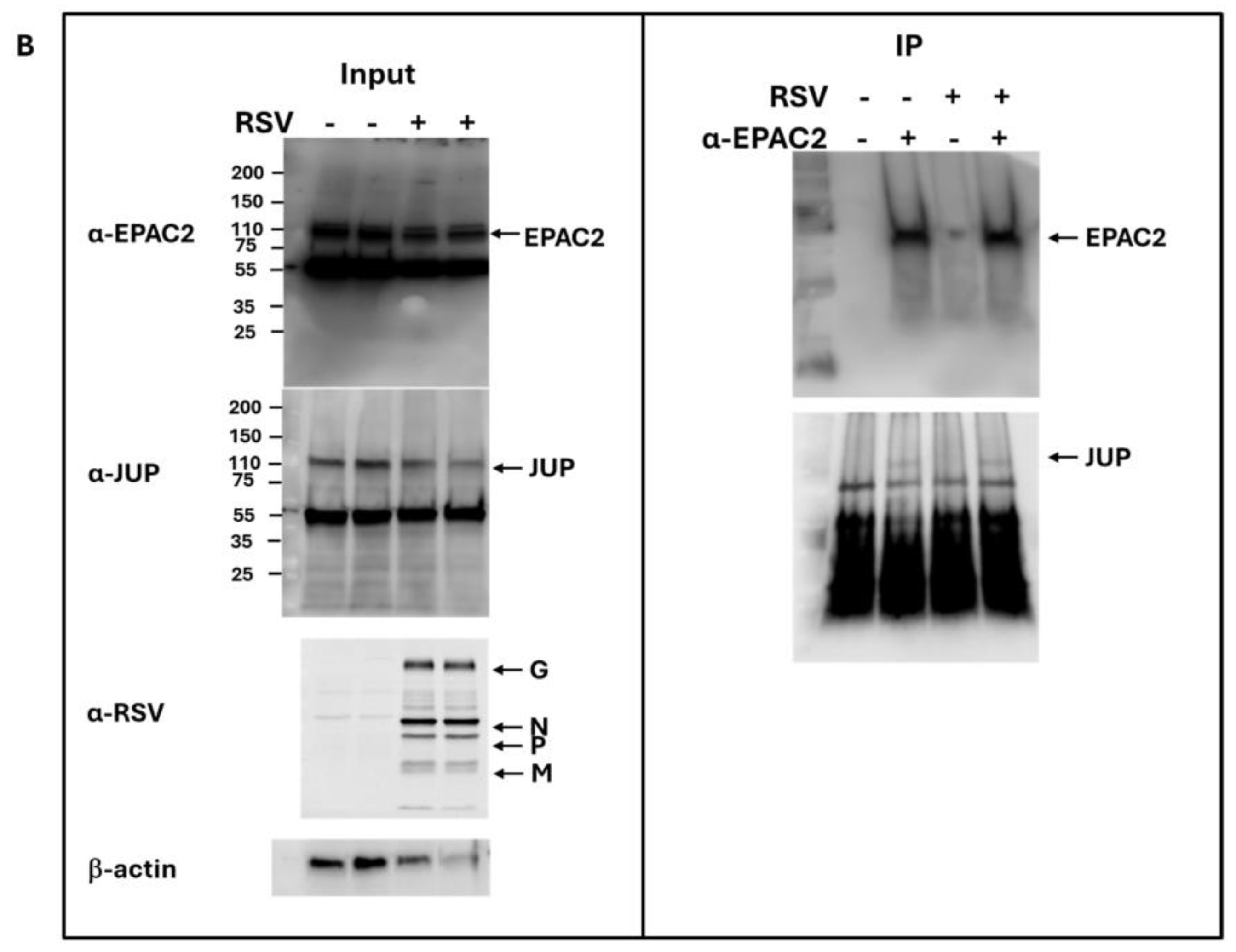
As shown in Figure 1B, JUP exhibited the least enhancement in its binding to EPAC2 following RSV infection compared to other EPAC2-associated proteins. Based on this observation, we chose to begin our experimental validation by investigating the JUP-EPAC2 interaction, reasoning that if this modest interaction could be confirmed, the stronger interactions observed for other proteins were likely to be biologically meaningful as well. An additional rationale for selecting JUP as our initial target was its known functional significance. Silencing JUP has been shown to disrupt actin organization within the cytoskeleton [21], and the cytoskeleton plays a critical role in multiple stages of viral infection, including the transport or assembly of viral proteins and particles, immune evasion, and cell-to-cell fusion [22,23,24,25]. As shown in Figure 2A. JUP was detected in the pull-down product using an anti-Flag antibody against Flag-tagged EPAC2, confirming the association of EPAC2 and JUP. This interaction was observed even in the absence of RSV infection (bottom-right panel, second column) and was increased by RSV infection (bottom-right panel, last column).
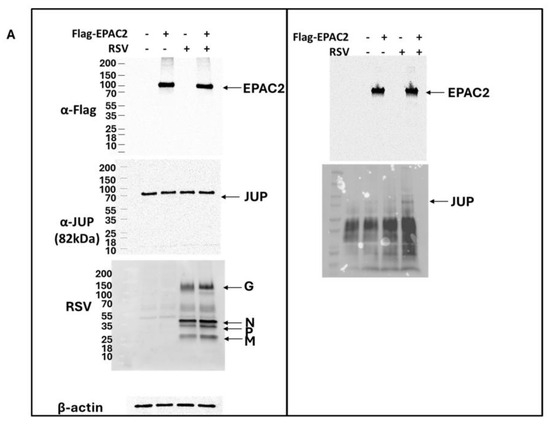
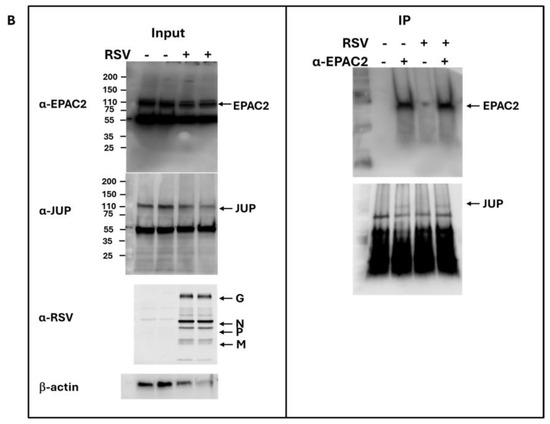
Figure 2.
Experimental confirmation of JUP-EPAC2 interaction. (A) The EPAC2-Flag overexpression and immunoprecipitation after mock or RSV infection were performed as described in Figure 1. (Left) panel: The input of overexpressed flag-tagged EPAC2, endogenous JUP, RSV proteins, and internal control β-actin were assessed by Western blot. (Right) panel: The presence of flag-tagged EPAC2 and JUP in the immune precipitation complex following anti-Flag antibody and IgG resin treatment was detected by Western blot. (B). EPAC2-JUP interaction was also investigated using an anti-EPAC2 antibody to pull down the endogenous EPAC2, followed by JUP detection in the IP complex. The IP experiments were repeated twice.
To further validate this interaction under endogenous conditions, we performed immunoprecipitation using an anti-EPAC2 antibody to pull down native EPAC2 and assess its association with JUP. As shown in Figure 2B, the EPAC2-JUP interaction was confirmed in both mock- and RSV-infected cells.
3.3. The Roles of JUP in RSV Infection
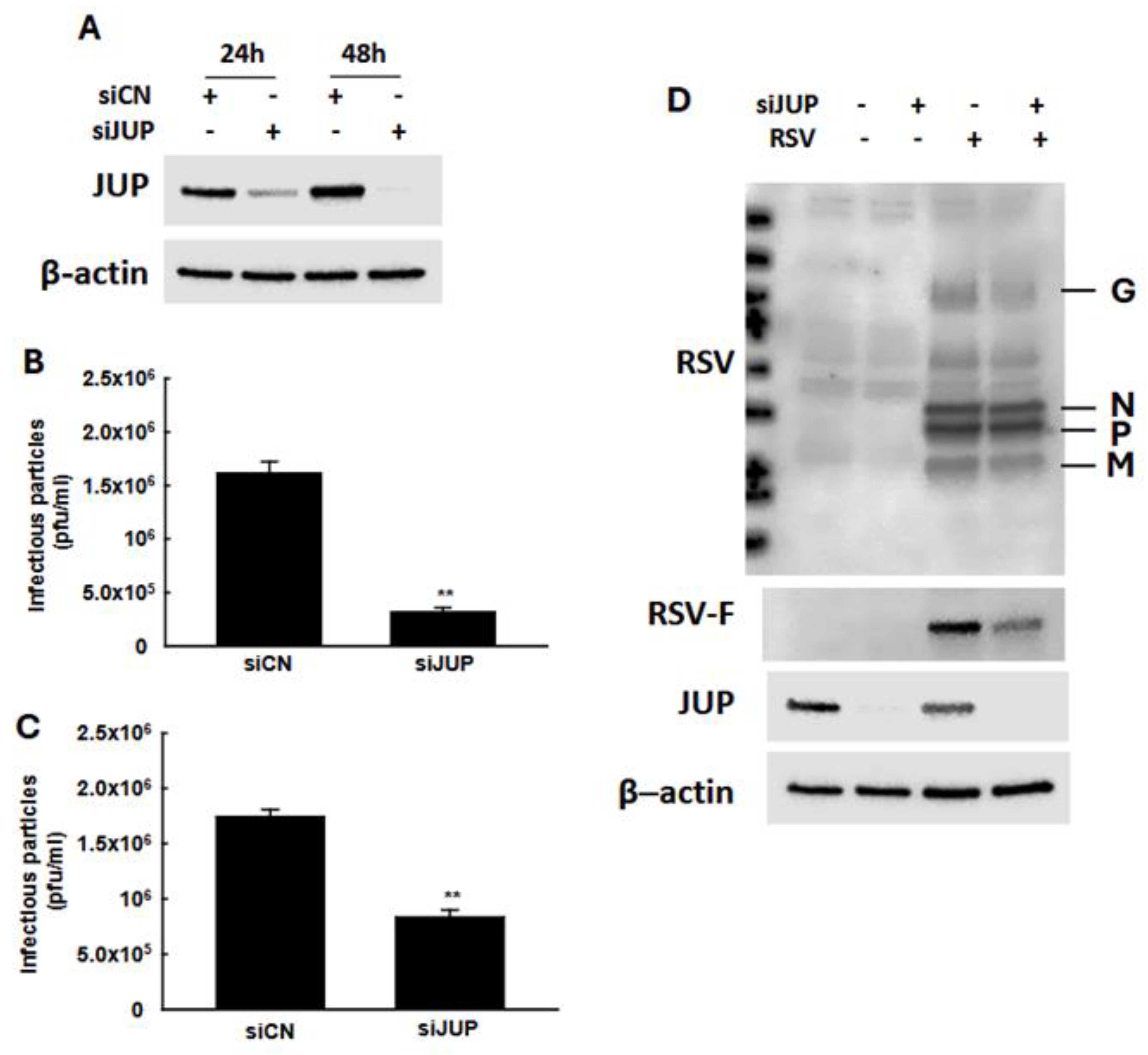
In our previous studies, we demonstrated that EPAC2 facilitates both RSV replication and host inflammatory responses [13,16]. To investigate whether EPAC2-associated JUP contributes to RSV infection, we used JUP-specific siRNA to suppress its expression, followed by an assessment of the impact of JUP silencing on RSV replication. As shown in Figure 3A, treatment with JUP-specific siRNA at a concentration of 100 nM effectively reduced JUP expression, both at 24 h and 48 h post-transfection. We also found that the total number of infectious RSV particles produced by JUP-deficient cells was significantly lower than that of cells treated with scrambled control siRNA (siCN) (Figure 3B). Similarly, the number of infectious particles released into the supernatant was reduced in JUP-siRNA-treated cells compared to siCN-treated cells (Figure 3C). Notably, the reduction in infectious particles released into the supernatant (about 80%) was greater than the reduction observed in the total sample (about 50%), following JUP knockdown. Consistent with these findings, Western blot analysis using anti-RSV antibody confirmed that there was reduced viral protein expression in siJUP-treated cells compared to siCN-treated cells (Figure 3D).
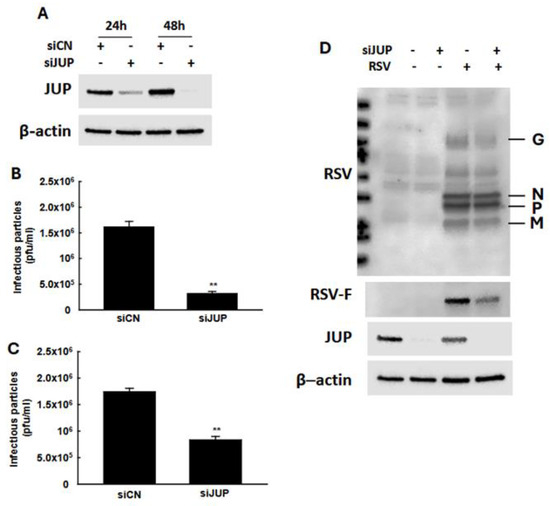
Figure 3.
The impact of JUP on RSV infection. (A) After 24 h or 48 h post-transfection, siCN- or siJUP-transfected A549 cells were subjected to Western blot using anti-JUP antibody to check the efficiency of siJUP in downregulating JUP expression. (B–D), siCN- or siJUP-transfected cells were mock-infected or infected with RSV at an MOI of 1. After 2 h, the cells were washed with PBS twice and then incubated for 15 h. The immune staining using anti-RSV antibodies was used to quantify the infectious particles in the supernatant (B) and total lysed samples (C). Cell pellets were also prepared for Western blot to check the impact of JUP suppression on RSV protein expression. β-actin was used as a protein-loading control (D). The IP experiments were repeated twice. ** denotes p < 0.01, relative to siCN-treated and RSV-infected cells.
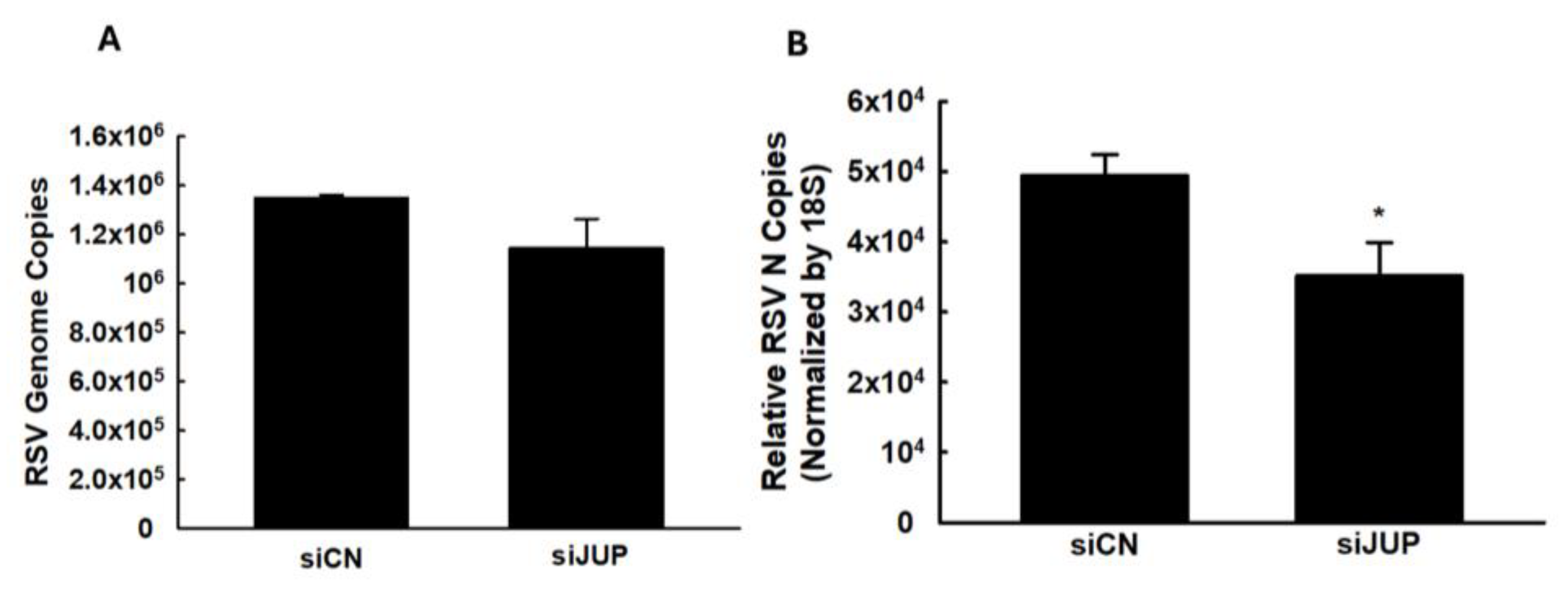
We also investigated whether JUP regulates RSV genome replication and viral gene expression. As shown in Figure 4, silencing JUP did not appear to affect viral genome replication; however, it significantly suppressed the expression of the N gene.
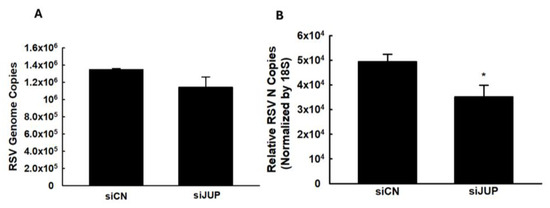
Figure 4.
The effect of JUP on RSV genome replication and gene transcription. siCN- or siJUP-transfected A549 cells were harvested at 24 h p.i., followed by total RNA extraction. The viral genome copies (A) and RSV N gene transcription (B) were assessed by qRT-PCR. * denotes a p value of <0.5, relative to siCN-treated and RSV-infected cells. Four repeats were performed.
3.4. The Impact of JUP on RSV-Induced Cytokines and Chemokines
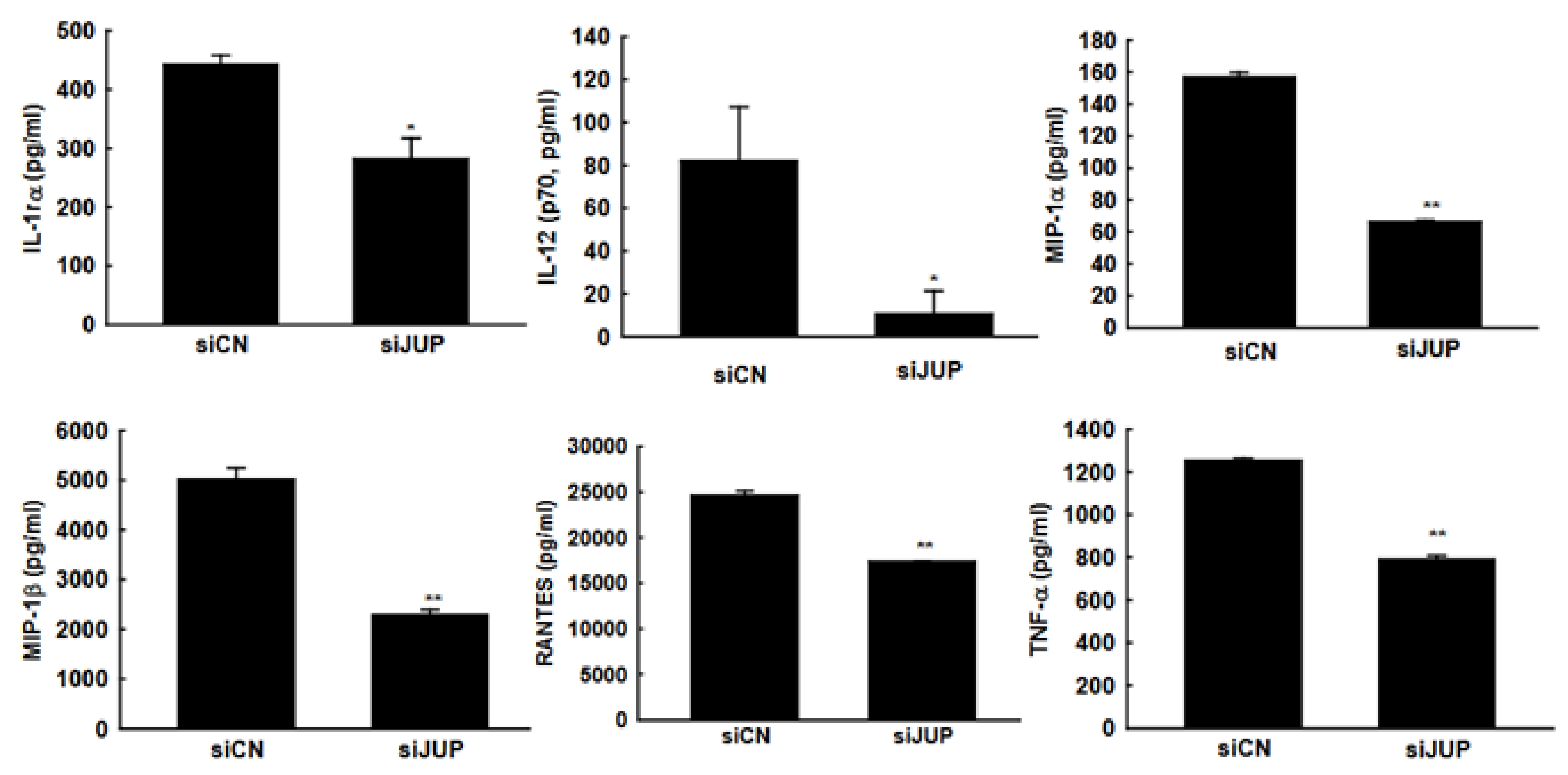
In response to RSV infection, infected cells typically activate cellular responses by producing cytokines and chemokines. Our previous studies demonstrated that EPAC2 promotes inflammation. To evaluate whether JUP suppression has a broader impact on RSV-induced secretion of proinflammatory and immunoregulatory molecules, we analyzed the secretion patterns of chemokines and cytokines in A549 cells with or without JUP silencing (Figure 5). Notably, JUP deficiency resulted in significantly lower levels of IL-1rα, IL-12 (p70), MIP-1α, MIP-1β, RANTES, and TNF-α at 15 h post-infection, compared to siCN-treated cells, supporting the importance of JUP in RSV-induced cellular responses. Unlike EPAC2, JUP did not affect the RSV-induced IP-10 and MCP-1 in A549 cells.
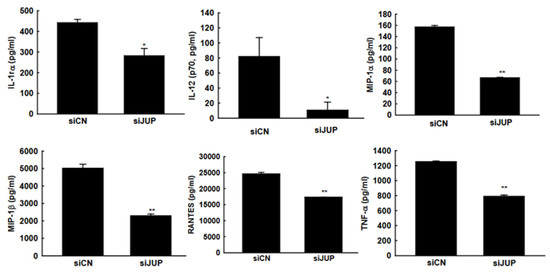
Figure 5.
JUP-regulated cytokine and chemokine induction. siCN- or siJUP-transfected A549 cells were mock infected or infected with RSV, as described in Figure 3. The cell pellets were obtained by centrifugation at 1000 RPM for 5 min, followed by supernatant harvesting. Bio-Plex multiplex assays quantified chemokines and cytokines in the supernatants. * and ** denote p values of <0.5 and <0.01, respectively, relative to siCN-treated and RSV-infected cells. 6 repeats were included.
3.5. Modulation of Cellular Signaling by JUP
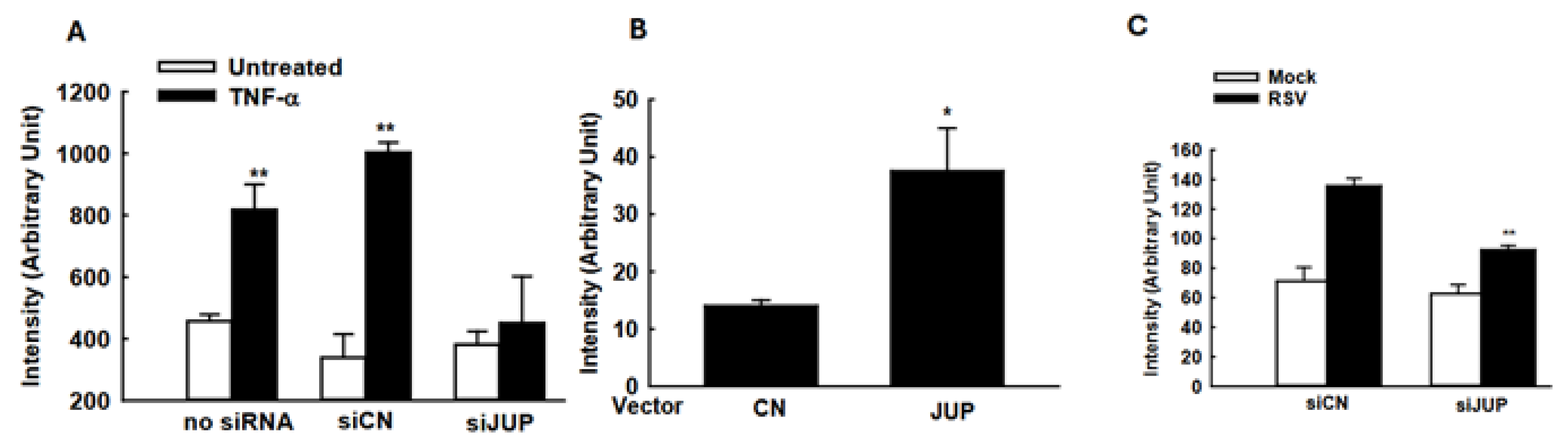
The suppressed cellular responses could be an indirect outcome of the impaired RSV replication by JUP silencing. To investigate whether the JUP knockdown also leads to changes in inflammatory responses independent of viral replication, we used TNF-α to activate NF-κB, a transcription factor that plays an essential role in inducing inflammatory immune mediators in responses to viral infection, and investigated whether silencing JUP impacts NF-κB activation. As shown in Figure 6A, JUP knockdown significantly impaired TNF-α-induced NF-κB activation, in alignment with our previous finding on EPAC2-mediated NF-κB activation and current discovery on EPAC2-JUP interaction.
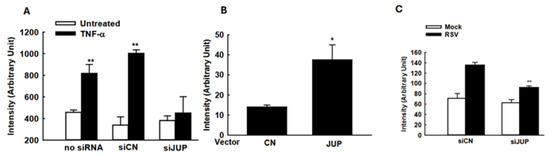
Figure 6.
The impact of JUP on cellular signaling. (A) A549 cells in triplicate were transfected with 0.05 µg/well luciferase reporter gene plasmids containing multiple copies of NF-κB binding sites (NF-κB-Luc) and 100 nM siRNAs, either scrambled or JUP-specific, using lipofectamine 2000. At 24 h post-transfection, the cells were treated with TNF-α at a concentration of 20 ng/mL per well. Following 24 h of treatment, the cells were lysed to measure luciferase reporter activity. Cells without treatment were used as controls. ** denotes p value < 0.01, relative to paired mock-infected samples (white bars). (B) A549 cells were transfected with 0.5 µg/well luciferase reporter plasmids carrying multiple copies of the IRF-3 binding site and 0.5 µg/well JUP-PCDNA3. At 15 h post-transfection, the cells were harvested for luciferase assay. * denotes a p value of <0.05, relative to paired cells with empty vector (CN). (C). A549 cells were transfected with 0.5 µg/well IFN-β luciferase reporter and 100 nM siRNAs specifically against JUP or scrambled siRNAs, by lipofectamine 2000. At 24 h post-transfection, the cells were mock-infected and infected with RSV for 15 hrs, followed by luciferase activity measurement. ** denotes a p value of <0.01, relative to siCN-treated RSV-infected cells; 3–4 repeats were performed.
Many immune mediators also have IRF-3 binding site(s) in addition to the NF-κB binding site. Therefore, we also investigated whether JUP affects cytokine/chemokine expression by regulating IRF-3 activation. As shown in Figure 6B, JUP overexpression led to enhanced IRF-3-mediated luciferase expression. In addition, JUP knockdown significantly suppressed luciferase expression controlled by IFN-β transcription binding sites for NF-κB and IRF-3 (Figure 6C).
4. Discussion
EPAC, a major cellular receptor for cAMP in addition to protein kinase A (PKA), plays major roles in cardiac diseases, cancer, neuronal differentiation, and respiratory inflammation [31,32,33,34]. In non-infectious disease models, EPAC has been reported to promote airway inflammation through the EPAC-activated mitogen-activated protein kinase kinase (MAPKK) pathway [35,36,37,38] or via PLCε [19]. Recently, the significance of EPAC in viral infections was also reported. In MERS-CoV, SARS-CoV-2, or influenza infection, EPAC1 seems to promote viral replication [14,15,18]. Unlike these viruses, our laboratory discovered that it is EPAC2, but not EPAC1, which dominantly affects proinflammatory cellular/pulmonary responses to RSV, human metapneumovirus, and adenovirus [13,17], suggesting that the EPAC isoform evolved in viral infections is pathogen-dependent. However, the molecular mechanisms underlying EPAC-mediated viral infection are not well known, although such information is essential for developing novel therapeutic interventions against viral infections.
By comparing our previous study on EPAC2-mediated and the current study on JUP-mediated cellular responses to RSV infection, we found that EPAC2 deficiency impacts a broader range of cytokines and chemokines than JUP deficiency, supporting the notion that JUP functions as one of the downstream signaling molecules of EPAC2. Unlike EPAC2, JUP did not affect the RSV-induced IP-10 and MCP-1 in A549 cells.
The biological functions of JUP were mostly investigated in the cardiac muscle. In cardiac muscle, JUP is a cytoplasmic component of desmosomes and adherens junctions and is, therefore, essential for its stability [39,40,41]. In cancer, JUP is involved in mediating the cell cytoskeleton actin [20,21]. The role of JUP in viral infection is not well known. We found that JUP deficiency led to a 50% reduction in total infectious particles and an 80% decrease in infectious particles in the supernatant, suggesting that JUP is critical for releasing infectious particles from cells. It has been previously reported that actin controls RSV budding [26]. The importance of actin in mediating virus budding out of cells is also reported for nonlytic rotavirus and SARS-CoV-2 [42,43]. Given the association between JUP and actin, it is not surprising to observe the impact of JUP on viral budding. Combined with our previous finding on viral entry independent of EPAC2 [13], this finding may represent a mechanism by which JUP promotes viral replication via controlling virus budding.
The induction of many cytokines/chemokines is replication-dependent [44,45]. In addition to JUP-enhanced infectious particle production, we also discovered a slight but significant impact of JUP on N gene transcription, possibly leading to increased immune responses to RSV infection. However, we found that JUP could also control cellular responses in a replication-independent manner (Figure 6A, B), similar to our report demonstrating that EPAC2 can control inflammation independent of RSV replication [13] and the report from others showing EPAC2-mediated non-viral respiratory diseases [19].
Based on EAPC2-associated proteins identified by immunoprecipitation/proteomics studies and Cluster analysis by STRING [46], we found that JUP potentially binds to RNA-binding proteins: the nuclear cap-binding protein subunit 1 (NCBP1) and DDX46 (Figure 1C). NCBP1 has been reported to play a crucial role in viral infections by interacting with viral mRNA and influencing antiviral responses [47,48]. DDX46, or DEAD-Box Helicase 46, is a protein that negatively regulates the innate antiviral response to viral infection [49]. Several proteins were not clustered into any groups, including CYP4F11, a cytochrome P450 enzyme. It has been reported previously that the expression of CYP4F11 can be influenced by virus-induced inflammation [50,51]. Some studies have shown that HCV-induced CYP4F12, another important member of P450, is bound to the HCV replication complex to facilitate viral replication [52]. In the future, we will continue to investigate whether these EPAC2-binding proteins are functional in regulating RSV infection.
In summary, this study revealed an important pathway that RSV may use to evade cellular responses, providing a potential therapeutic target for controlling RSV infection.
Author Contributions
Manuscript draft: C.A.T. and X.B.; Experiment performance and data analysis: C.A.T., E.-J.C., E.S.C., D.D., K.K., J.M.C. and K.Z.; Experimental design: S.Y.J., T.W., W.W. and X.B.; Manuscript finalization: X.B. All authors have read and agreed to the published version of the manuscript.
Funding
National Institutes of Health (NIH) R21 AI166543; the American Lung Association ERP-1252718; and NIH T35AI0778878.
Institutional Review Board Statement
Ethical review and approval were waived for this study as it is “Not applicable” for studies not involving humans or animals.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated/analyzed during this study are included in this published article. The datasets and related detailed methods are also available from the corresponding author upon request.
Acknowledgments
This work was supported by grants from the US National Institutes of Health (NIH) R21 AI166543 and ERP-1252718 from the American Lung Association to X.B.; C.T. was supported by NIAID T35 Infectious Diseases & Inflammatory Disorder Training Program (T35AI0778878, PI: T.W.). All individuals included in this section have consented to the acknowledgement.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Esposito, S.; Abu Raya, B.; Baraldi, E.; Flanagan, K.; Martinon Torres, F.; Tsolia, M.; Zielen, S. RSV Prevention in All Infants: Which Is the Most Preferable Strategy? Front. Immunol. 2022, 13, 880368. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.E.; Perez Marc, G.; Zareba, A.M.; Falsey, A.R.; Jiang, Q.; Patton, M.; Polack, F.P.; Llapur, C.; Doreski, P.A.; Ilangovan, K.; et al. Efficacy and Safety of a Bivalent RSV Prefusion F Vaccine in Older Adults. N. Engl. J. Med. 2023, 388, 1465–1477. [Google Scholar] [CrossRef]
- Kampmann, B.; Radley, D.; Munjal, I. Bivalent Prefusion F Vaccine in Pregnancy to Prevent RSV Illness in Infants. Reply. N. Engl. J. Med. 2023, 389, 1053–1055. [Google Scholar] [CrossRef]
- Harris, E. FDA Approves RSV Monoclonal Antibody for Infants and Young Children. JAMA 2023, 330, 586. [Google Scholar] [CrossRef]
- Sugawara, K.; Shibasaki, T.; Takahashi, H.; Seino, S. Structure and functional roles of EPAC2 (Rapgef4). Gene 2016, 575, 577–583. [Google Scholar] [CrossRef]
- Metrich, M.; Lucas, A.; Gastineau, M.; Samuel, J.L.; Heymes, C.; Morel, E.; Lezoualc’h, F. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 2008, 102, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.B.; Santos, D.F.; Laird, D.E.; Doolen, S.; Donahue, R.R.; Wessel, C.R.; Fu, W.; Sinha, G.P.; Wang, P.; Zhou, J.; et al. Methylglyoxal and a spinal TRPA1-AC1-Epac cascade facilitate pain in the db/db mouse model of type 2 diabetes. Neurobiol. Dis. 2019, 127, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kirton, H.; Al-Owais, M.; Thireau, J.; Richard, S.; Steele, D.; Peers, C. EPAC2-Rap1 signaling regulates reactive oxygen species production and susceptibility to cardiac arrhythmias. Antioxid. Redox. Signal 2016, 27, 117–132. [Google Scholar] [CrossRef]
- Singhmar, P.; Huo, X.; Eijkelkamp, N.; Berciano, S.R.; Baameur, F.; Mei, F.C.; Zhu, Y.; Cheng, X.; Hawke, D.; Mayor, F., Jr.; et al. Critical role for Epac1 in inflammatory pain controlled by GRK2-mediated phosphorylation of Epac1. Proc. Natl. Acad. Sci. USA 2016, 113, 3036–3041. [Google Scholar] [CrossRef]
- Shi, G.X.; Rehmann, H.; Andres, D.A. A novel cyclic AMP-dependent Epac-Rit signaling pathway contributes to PACAP38-mediated neuronal differentiation. Mol. Cell Biol. 2006, 26, 9136–9147. [Google Scholar] [CrossRef]
- Sehrawat, S.; Cullere, X.; Patel, S.; Italiano, J., Jr.; Mayadas, T.N. Role of Epac1, an exchange factor for Rap GTPases, in endothelial microtubule dynamics and barrier function. Mol. Biol. Cell 2008, 19, 1261–1270. [Google Scholar] [CrossRef]
- Robichaux, W.G., 3rd; Cheng, X. Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol. Rev. 2018, 98, 919–1053. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Ren, Y.; Chen, Y.; Liu, S.; Wu, W.; Ren, J.; Wang, P.; Garofalo, R.P.; Zhou, J.; Bao, X. Exchange Proteins Directly Activated by cAMP and Their Roles in Respiratory Syncytial Virus Infection. J. Virol. 2018, 92, e01200-18. [Google Scholar] [CrossRef]
- Tao, X.; Mei, F.; Agrawal, A.; Peters, C.J.; Ksiazek, T.G.; Cheng, X.; Tseng, C.T. Blocking of exchange proteins directly activated by cAMP leads to reduced replication of Middle East respiratory syndrome coronavirus. J. Virol. 2014, 88, 3902–3910. [Google Scholar] [CrossRef] [PubMed]
- Drelich, A.; Judy, B.; He, X.; Chang, Q.; Yu, S.; Li, X.; Lu, F.; Wakamiya, M.; Popov, V.; Zhou, J.; et al. Exchange Protein Directly Activated by cAMP Modulates Ebola Virus Uptake into Vascular Endothelial Cells. Viruses 2018, 10, 563. [Google Scholar] [CrossRef]
- Choi, E.J.; Wu, W.; Cong, X.; Zhang, K.; Luo, J.; Ye, S.; Wang, P.; Suresh, A.; Ullah, U.M.; Zhou, J.; et al. Broad Impact of Exchange Protein Directly Activated by cAMP 2 (EPAC2) on Respiratory Viral Infections. Viruses 2021, 13, 1179. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Wu, W.; Zhang, K.; Choi, E.J.; Wang, P.; Ivanciuc, T.; Peniche, A.; Qian, Y.; Garofalo, R.P.; Zhou, J.; et al. Exchange Protein Directly Activated by cAMP 2 Enhances Respiratory Syncytial Virus-Induced Pulmonary Disease in Mice. Front. Immunol. 2021, 12, 757758. [Google Scholar] [CrossRef]
- Foret-Lucas, C.; Figueroa, T.; Bertin, A.; Bessiere, P.; Lucas, A.; Bergonnier, D.; Wasniewski, M.; Servat, A.; Tessier, A.; Lezoualc’h, F.; et al. EPAC1 Pharmacological Inhibition with AM-001 Prevents SARS-CoV-2 and Influenza A Virus Replication in Cells. Viruses 2023, 15, 319. [Google Scholar] [CrossRef]
- Oldenburger, A.; Timens, W.; Bos, S.; Smit, M.; Smrcka, A.V.; Laurent, A.C.; Cao, J.; Hylkema, M.; Meurs, H.; Maarsingh, H.; et al. Epac1 and EPAC2 are differentially involved in inflammatory and remodeling processes induced by cigarette smoke. FASEB J. 2014, 28, 4617–4628. [Google Scholar] [CrossRef]
- Xia, J.; Ma, N.; Shi, Q.; Liu, Q.C.; Zhang, W.; Cao, H.J.; Wang, Y.K.; Zheng, Q.W.; Ni, Q.Z.; Xu, S.; et al. XAF1 promotes colorectal cancer metastasis via VCP-RNF114-JUP axis. J. Cell Biol. 2024, 223, e202303015. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, L.; Qin, Y.; Liu, S.; Qiao, Y.; Wan, X.; Zeng, H.; Tang, X.; Liu, M.; Hou, Y. Effects of differential distributed-JUP on the malignancy of gastric cancer. J. Adv. Res. 2021, 28, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Kloc, M.; Uosef, A.; Wosik, J.; Kubiak, J.Z.; Ghobrial, R.M. Virus interactions with the actin cytoskeleton-what we know and do not know about SARS-CoV-2. Arch. Virol. 2022, 167, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Acharya, D.; Reis, R.; Volcic, M.; Liu, G.; Wang, M.K.; Chia, B.S.; Nchioua, R.; Gross, R.; Munch, J.; Kirchhoff, F.; et al. Actin cytoskeleton remodeling primes RIG-I-like receptor activation. Cell 2022, 185, 3588–3602.e3521. [Google Scholar] [CrossRef]
- Kieser, Q.J.; Granoski, M.J.; McClelland, R.D.; Griffiths, C.; Bilawchuk, L.M.; Stojic, A.; Elawar, F.; Jamieson, K.; Proud, D.; Marchant, D.J. Actin cytoskeleton remodeling disrupts physical barriers to infection and presents entry receptors to respiratory syncytial virus. J. Gen. Virol. 2023, 104, 001923. [Google Scholar] [CrossRef]
- Kay, R.R. Macropinocytosis: Biology and mechanisms. Cells Dev. 2021, 168, 203713. [Google Scholar] [CrossRef] [PubMed]
- Shahriari, S.; Gordon, J.; Ghildyal, R. Host cytoskeleton in respiratory syncytial virus assembly and budding. Virol. J. 2016, 13, 161. [Google Scholar] [CrossRef]
- Deng, J.; Ptashkin, R.N.; Chen, Y.; Cheng, Z.; Liu, G.; Phan, T.; Deng, X.; Zhou, J.; Lee, I.; Lee, Y.S.; et al. Respiratory Syncytial Virus Utilizes a tRNA Fragment to Suppress Antiviral Responses Through a Novel Targeting Mechanism. Mol. Ther. 2015, 23, 1622–1629. [Google Scholar] [CrossRef]
- Choi, E.J.; Ren, J.; Zhang, K.; Wu, W.; Lee, Y.S.; Lee, I.; Bao, X. The Importance of AGO 1 and 4 in Post-Transcriptional Gene Regulatory Function of tRF5-GluCTC, an Respiratory Syncytial Virus-Induced tRNA-Derived RNA Fragment. Int. J. Mol. Sci. 2020, 21, 8766. [Google Scholar] [CrossRef]
- Ren, J.; Liu, T.; Pang, L.; Li, K.; Garofalo, R.P.; Casola, A.; Bao, X. A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J. Gen. Virol. 2011, 92, 2153–2159. [Google Scholar] [CrossRef]
- Saltzman, A.B.; Leng, M.; Bhatt, B.; Singh, P.; Chan, D.W.; Dobrolecki, L.; Chandrasekaran, H.; Choi, J.M.; Jain, A.; Jung, S.Y.; et al. gpGrouper: A Peptide Grouping Algorithm for Gene-Centric Inference and Quantitation of Bottom-Up Proteomics Data. Mol. Cell Proteomics 2018, 17, 2270–2283. [Google Scholar] [CrossRef]
- Almahariq, M.; Tsalkova, T.; Mei, F.C.; Chen, H.; Zhou, J.; Sastry, S.K.; Schwede, F.; Cheng, X. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol. Pharmacol. 2013, 83, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Rodriguez, A.; Ruiz-Hurtado, G.; Sabourin, J.; Gomez, A.M.; Alvarez, J.L.; Benitah, J.P. Proarrhythmic effect of sustained EPAC activation on TRPC3/4 in rat ventricular cardiomyocytes. J. Mol. Cell Cardiol. 2015, 87, 74–78. [Google Scholar] [CrossRef]
- Fujita, T.; Umemura, M.; Yokoyama, U.; Okumura, S.; Ishikawa, Y. The role of Epac in the heart. Cell Mol. Life Sci. 2016, 74, 591–606. [Google Scholar] [CrossRef]
- Holz, G.G. Epac: A new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes 2004, 53, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Roscioni, S.S.; Kistemaker, L.E.; Menzen, M.H.; Elzinga, C.R.; Gosens, R.; Halayko, A.J.; Meurs, H.; Schmidt, M. PKA and Epac cooperate to augment bradykinin-induced interleukin-8 release from human airway smooth muscle cells. Respir. Res. 2009, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Marini, M.; Vittori, E.; Hollemborg, J.; Mattoli, S. Expression of the potent inflammatory cytokines, grnulocyte-macrophage-colony stimulating factor and interleukin-6 and interleukin-8 in bronchial eptihelial cells of patients with asthma. J. Allergy Clin. Immunol. 1992, 89, 1001–1009. [Google Scholar] [CrossRef]
- Shute, J.K.; Vrugt, B.; Lindley, I.J.; Holgate, S.T.; Bron, A.; Aalbers, R.; Djukanovic, R. Free and complexed interleukin-8 in blood and bronchial mucosa in asthma. Am. J. Respir. Crit. Care Med. 1997, 155, 1877–1883. [Google Scholar] [CrossRef]
- Yamamoto, C.; Yoneda, T.; Yoshikawa, M.; Fu, A.; Tokuyama, T.; Tsukaguchi, K.; Narita, N. Airway inflammation in COPD assessed by sputum levels of interleukin-8. Chest 1997, 112, 505–510. [Google Scholar] [CrossRef]
- Ruiz, P.; Birchmeier, W. The plakoglobin knock-out mouse: A paradigm for the molecular analysis of cardiac cell junction formation. Trends Cardiovasc. Med. 1998, 8, 97–101. [Google Scholar] [CrossRef]
- Isac, C.M.; Ruiz, P.; Pfitzmaier, B.; Haase, H.; Birchmeier, W.; Morano, I. Plakoglobin is essential for myocardial compliance but dispensable for myofibril insertion into adherens junctions. J. Cell Biochem. 1999, 72, 8–15. [Google Scholar] [CrossRef]
- Swope, D.; Cheng, L.; Gao, E.; Li, J.; Radice, G.L. Loss of cadherin-binding proteins beta-catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis. Mol. Cell Biol. 2012, 32, 1056–1067. [Google Scholar] [CrossRef]
- Trejo-Cerro, O.; Eichwald, C.; Schraner, E.M.; Silva-Ayala, D.; Lopez, S.; Arias, C.F. Actin-Dependent Nonlytic Rotavirus Exit and Infectious Virus Morphogenetic Pathway in Nonpolarized Cells. J. Virol. 2018, 92, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Swain, J.; Merida, P.; Rubio, K.; Bracquemond, D.; Neyret, A.; Aguilar-Ordonez, I.; Gunther, S.; Barreto, G.; Muriaux, D. F-actin nanostructures rearrangements and regulation are essential for SARS-CoV-2 particle production in host pulmonary cells. iScience 2023, 26, 107384. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Casola, A.; Suarez, G.; Yu, X.; Spetch, L.; Peeples, M.E.; Garofalo, R.P. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2006, 34, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Olszewska-Pazdrak, B.; Casola, A.; Saito, T.; Alam, R.; Crowe, S.E.; Mei, F.; Ogra, P.L.; Garofalo, R.P. Cell-specific expression of RANTES, MCP-1, and MIP-1alpha by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J. Virol. 1998, 72, 4756–4764. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Bier, K.; York, A.; Fodor, E. Cellular cap-binding proteins associate with influenza virus mRNAs. J. Gen. Virol. 2011, 92, 1627–1634. [Google Scholar] [CrossRef]
- Gebhardt, A.; Bergant, V.; Schnepf, D.; Moser, M.; Meiler, A.; Togbe, D.; Mackowiak, C.; Reinert, L.S.; Paludan, S.R.; Ryffel, B.; et al. The alternative cap-binding complex is required for antiviral defense in vivo. PLoS Pathog. 2019, 15, e1008155. [Google Scholar] [CrossRef]
- Zheng, Q.; Hou, J.; Zhou, Y.; Li, Z.; Cao, X. The RNA helicase DDX46 inhibits innate immunity by entrapping m(6)A-demethylated antiviral transcripts in the nucleus. Nat. Immunol. 2017, 18, 1094–1103. [Google Scholar] [CrossRef]
- Wang, G.; Xiao, B.; Deng, J.; Gong, L.; Li, Y.; Li, J.; Zhong, Y. The Role of Cytochrome P450 Enzymes in COVID-19 Pathogenesis and Therapy. Front. Pharmacol. 2022, 13, 791922. [Google Scholar] [CrossRef]
- Stavropoulou, E.; Pircalabioru, G.G.; Bezirtzoglou, E. The Role of Cytochromes P450 in Infection. Front. Immunol. 2018, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.L.; Wang, L.; Cao, Z.Y.; Wang, J.; Jing, M.Z.; Xia, Z.C.; Ao, F.; Ye, L.B.; Liu, S.; Zhu, Y. Inducible CYP4F12 enhances Hepatitis C virus infection via association with viral nonstructural protein 5B. Biochem. Biophys. Res. Commun. 2016, 471, 95–102. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).