
Exploring Avian Influenza Viruses in Yakutia—The Largest Breeding Habitat of Wild Migratory Birds in Northeastern Siberia

 , , , , , , , , , , , , , and
, , , , , , , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Issues
2.2. Sample Collection
2.3. Avian Influenza Virus Isolation Using Chicken Embryos
2.4. RNA Extraction, Reverse Transcription, and PCR
2.5. Sequencing of AIVs
2.6. Search for the Nearest Identical Sequences for AIVs
2.7. Phylogenetic and Cluster Analyses of AIVs
2.8. Search for Amino Acid Substitutions in Internal Genes of AIVs
3. Results
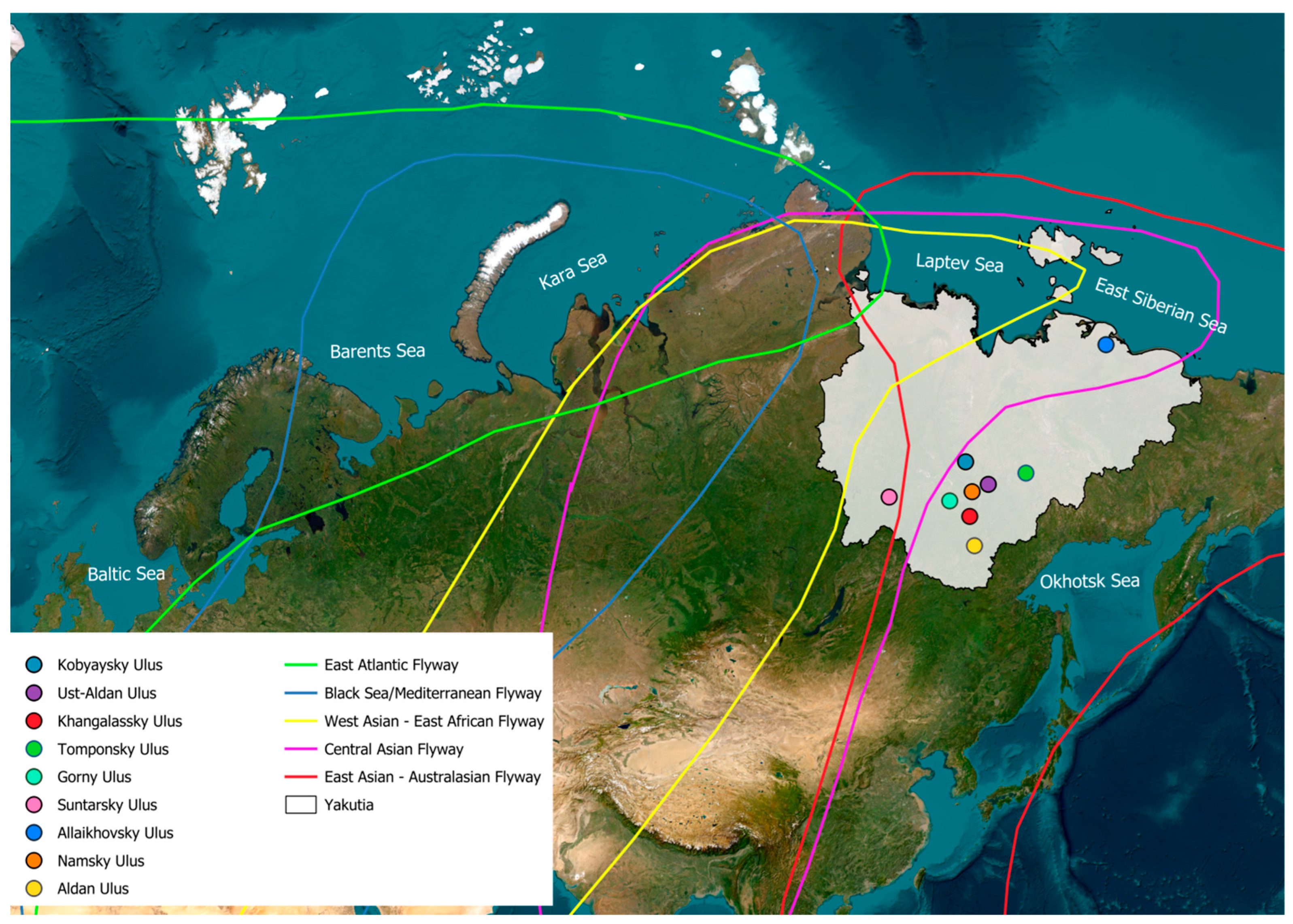
3.1. Ecological Features of Study Area and Samples
3.2. AIVs in Wild Birds of Yakutia
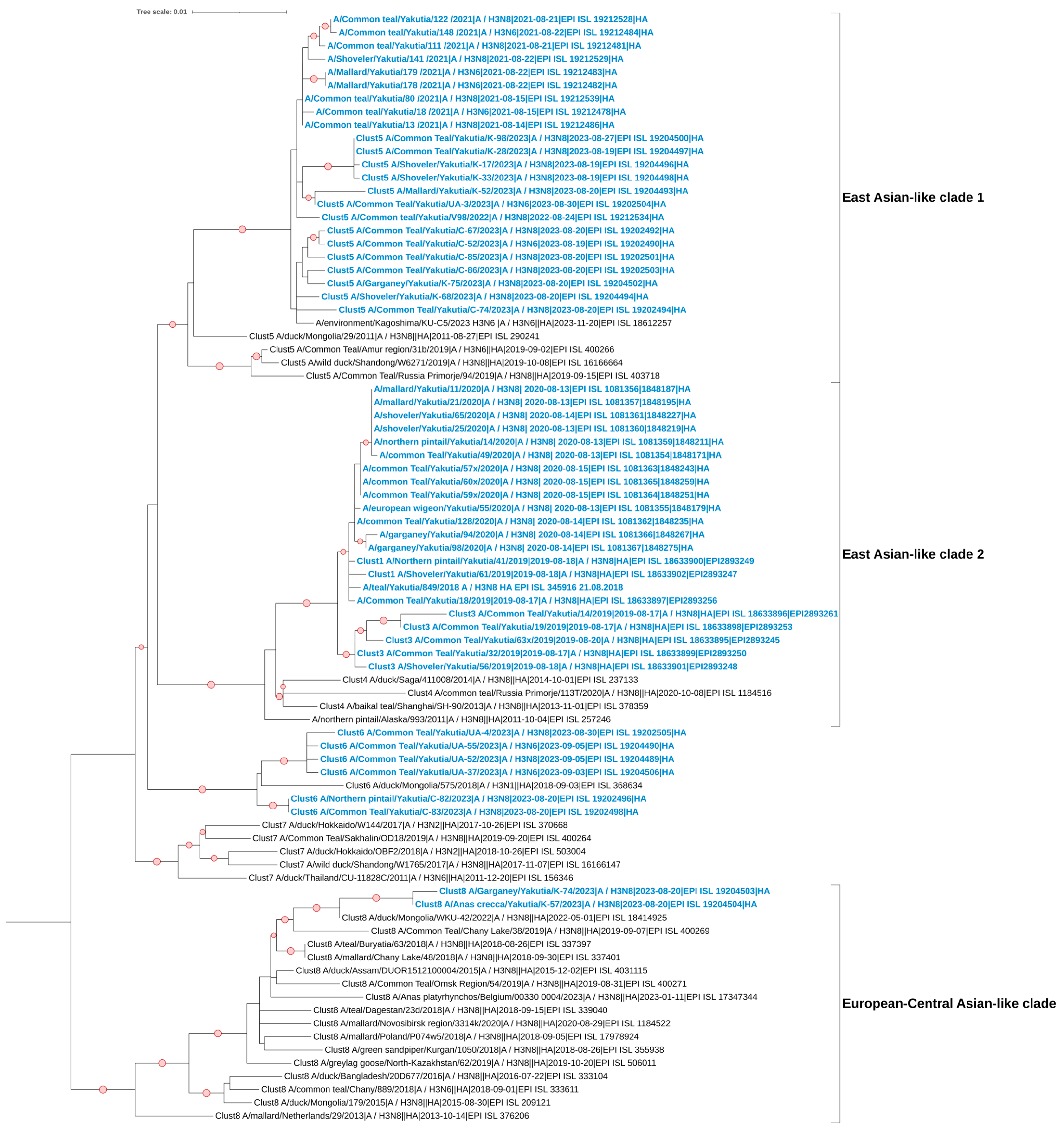
3.3. Cluster Analysis and the Nearest Sequences from the GISAID Database for the Internal Genes of AIVs
3.4. Amino Acid Substitutions in the Proteins of the Internal Genes of AIVs
3.5. Phylogenetic and Cluster Analyses of AIVs
4. Discussion
5. Limitations of the Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Committee on Taxonomy of Viruses. ICTV Taxonomy History. International Committee on Taxonomy of Viruses. Available online: https://ictv.global/taxonomy (accessed on 1 March 2025).
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and Ecology of Influenza A Viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef]
- Burrough, E.R.; Magstadt, D.R.; Petersen, B.; Timmermans, S.J.; Gauger, P.C.; Zhang, J.; Siepker, C.; Mainenti, M.; Li, G.; Thompson, A.C.; et al. Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4b Virus Infection in Domestic Dairy Cattle and Cats, United States, 2024. Emerg. Infect. Dis. 2024, 30, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasan, C.C.; Li, F.; Wang, D. Emerging Threats of Highly Pathogenic Avian Influenza A (H5N1) in US Dairy Cattle: Understanding Cross-Species Transmission Dynamics in Mammalian Hosts. Viruses 2024, 16, 1703. [Google Scholar] [CrossRef]
- Blagodatski, A.; Trutneva, K.; Glazova, O.; Mityaeva, O.; Shevkova, L.; Kegeles, E.; Onyanov, N.; Fede, K.; Maznina, A.; Khavina, E.; et al. Avian Influenza in Wild Birds and Poultry: Dissemination Pathways, Monitoring Methods, and Virus Ecology. Pathogens 2021, 10, 630. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Peiris, M.; Chen, H.; Guan, Y. H5N1 Outbreaks and Enzootic Influenza. Emerg. Infect. Dis. 2006, 12, 3–8. [Google Scholar] [CrossRef]
- Mv, V.; Ni, G. Birds of Yakutia: Fauna Diversity, Ecology, Role in Ecosystems and Human Life. J. Biodivers. Endanger. Species 2013, 1, 114. [Google Scholar] [CrossRef]
- Okazaki, K.; Takada, A.; Ito, T.; Imai, M.; Takakuwa, H.; Hatta, M.; Ozaki, H.; Tanizaki, T.; Nagano, T.; Ninomiya, A.; et al. Precursor Genes of Future Pandemic Influenza Viruses Are Perpetuated in Ducks Nesting in Siberia. Arch. Virol. 2000, 145, 885–893. [Google Scholar] [CrossRef]
- Marchenko, V.Y.; Susloparov, I.M.; Kolosova, N.P.; Goncharova, N.I.; Shipovalov, A.V.; Durymanov, A.G.; Ilyicheva, T.N.; Budatsirenova, L.V.; Ivanova, V.K.; Ignatyev, G.A.; et al. Influenza A(H5N8) Virus Isolation in Russia, 2014. Arch. Virol. 2015, 160, 2857–2860. [Google Scholar] [CrossRef]
- WHO. World Health Organization Collecting, Preserving and Shipping Specimens for the Diagnosis of Avian Influenza A(H5N1) Virus Infection: Guide for Field Operations; World Health Organization: Geneva, Switzerland, 2006; Available online: https://iris.who.int/bitstream/handle/10665/69392/WHO_CDS_EPR_ARO_2006.1_eng.pdf (accessed on 1 March 2025).
- Avian Influenza-WOAH-World Organisation for Animal Health. Available online: https://www.woah.org/en/disease/avian-influenza/ (accessed on 14 November 2023).
- Spackman, E. Animal Influenza Virus, 2nd ed.; Humana Press: New York, NY, USA, 2014. [Google Scholar]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St. George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-Reaction Genomic Amplification Accelerates Sequencing and Vaccine Production for Classical and Swine Origin Human Influenza A Viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef]
- CFIA-NCFAD/Nf-Flu. Available online: https://github.com/CFIA-NCFAD/nf-flu/tree/3.2.1?tab=readme-ov-file (accessed on 1 March 2025).
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow Enables Reproducible Computational Workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; the UGENE team. Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Ragonnet-Cronin, M.; Hodcroft, E.; Hué, S.; Fearnhill, E.; Delpech, V.; Brown, A.J.L.; Lycett, S. Automated Analysis of Phylogenetic Clusters. BMC Bioinform. 2013, 14, 317. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Suttie, A.; Deng, Y.-M.; Greenhill, A.R.; Dussart, P.; Horwood, P.F.; Karlsson, E.A. Inventory of Molecular Markers Affecting Biological Characteristics of Avian Influenza A Viruses. Virus Genes 2019, 55, 739–768. [Google Scholar] [CrossRef]
- Gao, W.; Zu, Z.; Liu, J.; Song, J.; Wang, X.; Wang, C.; Liu, L.; Tong, Q.; Wang, M.; Sun, H.; et al. Prevailing I292V PB2 Mutation in Avian Influenza H9N2 Virus Increases Viral Polymerase Function and Attenuates IFN-β Induction in Human Cells. J. Gen. Virol. 2019, 100, 1273–1281. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Kiso, M.; Yasuhara, A.; Ito, M.; Shu, Y.; Kawaoka, Y. Enhanced Replication of Highly Pathogenic Influenza A(H7N9) Virus in Humans. Emerg. Infect. Dis. 2018, 24, 746–750. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Yamada, S.; Fukuyama, S.; Murakami, S.; Zhao, D.; Uraki, R.; Watanabe, T.; Tomita, Y.; Macken, C.; Neumann, G.; et al. Virulence-Affecting Amino Acid Changes in the PA Protein of H7N9 Influenza A Viruses. J. Virol. 2014, 88, 3127–3134. [Google Scholar] [CrossRef]
- Xiao, C.; Ma, W.; Sun, N.; Huang, L.; Li, Y.; Zeng, Z.; Wen, Y.; Zhang, Z.; Li, H.; Li, Q.; et al. PB2-588 V Promotes the Mammalian Adaptation of H10N8, H7N9 and H9N2 Avian Influenza Viruses. Sci. Rep. 2016, 6, 19474. [Google Scholar] [CrossRef]
- Schmolke, M.; Manicassamy, B.; Pena, L.; Sutton, T.; Hai, R.; Varga, Z.T.; Hale, B.G.; Steel, J.; Pérez, D.R.; García-Sastre, A. Differential Contribution of PB1-F2 to the Virulence of Highly Pathogenic H5N1 Influenza A Virus in Mammalian and Avian Species. PLoS Pathog. 2011, 7, e1002186. [Google Scholar] [CrossRef]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A Single Mutation in the PB1-F2 of H5N1 (HK/97) and 1918 Influenza A Viruses Contributes to Increased Virulence. PLoS Pathog. 2007, 3, e141. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, X.; Gao, W.; Wang, C.; Wang, J.; Sun, H.; Sun, Y.; Guo, L.; Zhang, R.; Chang, K.-C.; et al. Prevailing PA Mutation K356R in Avian Influenza H9N2 Virus Increases Mammalian Replication and Pathogenicity. J. Virol. 2016, 90, 8105–8114. [Google Scholar] [CrossRef]
- Song, J.; Feng, H.; Xu, J.; Zhao, D.; Shi, J.; Li, Y.; Deng, G.; Jiang, Y.; Li, X.; Zhu, P.; et al. The PA Protein Directly Contributes to the Virulence of H5N1 Avian Influenza Viruses in Domestic Ducks. J. Virol. 2011, 85, 2180–2188. [Google Scholar] [CrossRef]
- Song, J.; Xu, J.; Shi, J.; Li, Y.; Chen, H. Synergistic Effect of S224P and N383D Substitutions in the PA of H5N1 Avian Influenza Virus Contributes to Mammalian Adaptation. Sci. Rep. 2015, 5, 10510. [Google Scholar] [CrossRef]
- Tada, T.; Suzuki, K.; Sakurai, Y.; Kubo, M.; Okada, H.; Itoh, T.; Tsukamoto, K. Emergence of Avian Influenza Viruses with Enhanced Transcription Activity by a Single Amino Acid Substitution in the Nucleoprotein during Replication in Chicken Brains. J. Virol. 2011, 85, 10354–10363. [Google Scholar] [CrossRef]
- Tada, T.; Suzuki, K.; Sakurai, Y.; Kubo, M.; Okada, H.; Itoh, T.; Tsukamoto, K. NP Body Domain and PB2 Contribute to Increased Virulence of H5N1 Highly Pathogenic Avian Influenza Viruses in Chickens. J. Virol. 2011, 85, 1834–1846. [Google Scholar] [CrossRef] [PubMed]
- Korzhuev, S.S. Yakutiya; Nauka: Moscow, Russia, 1965. [Google Scholar]
- Degtyarev, A.G. Hunting and Game Birds of the Republic of Sakha (Yakutia); Yakutsk Branch of State Publishing House of SB RAS: Yakutsk, Russia, 2004. [Google Scholar]
- Lee, K.; Yu, D.; Martínez-López, B.; Yoon, H.; Kang, S.-I.; Hong, S.-K.; Lee, I.; Kang, Y.; Jeong, W.; Lee, E. Fine-Scale Tracking of Wild Waterfowl and Their Impact on Highly Pathogenic Avian Influenza Outbreaks in the Republic of Korea, 2014–2015. Sci. Rep. 2020, 10, 18631. [Google Scholar] [CrossRef] [PubMed]
- Doko, T.; Chen, W.; Hijikata, N.; Yamaguchi, N.; Hiraoka, E.; Fujita, M.; Uchida, K.; Shimada, T.; Higuchi, H. Migration Patterns and Characteristics of Eurasian Wigeons (Mareca Penelope) Wintering in Southwestern Japan Based on Satellite Tracking. Zool. Sci. 2019, 36, 490. [Google Scholar] [CrossRef]
- Cho, H.-J.; Kim, D.-H.; Kim, I.-K.; Kang, T.-H.; Park, C.-Y.; Shin, Y.-U.; Han, S.-W.; Hur, W.-H.; Moon, O.-K.; Yu, J.-P. Report on Bird-Banding in Korea—About Ducks. J. Asia-Pac. Biodivers. 2013, 6, 383–390. [Google Scholar] [CrossRef]
- Si, Y.; Xu, Y.; Xu, F.; Li, X.; Zhang, W.; Wielstra, B.; Wei, J.; Liu, G.; Luo, H.; Takekawa, J.; et al. Spring Migration Patterns, Habitat Use, and Stopover Site Protection Status for Two Declining Waterfowl Species Wintering in China as Revealed by Satellite Tracking. Ecol. Evol. 2018, 8, 6280–6289. [Google Scholar] [CrossRef]
- Finlayson, C.M.; Rastogi, G.; Mishra, D.R.; Pattnaik, A.K. (Eds.) Ecology, Conservation, and Restoration of Chilika Lagoon, India; Wetlands: Ecology, Conservation and Management; Springer International Publishing: Cham, Switzerland, 2020; Volume 6. [Google Scholar] [CrossRef]
- Shemyakin, E.V.; Degtyarev, V.G.; Egorov, N.N.; Kirillin, R.A.; Huang, K.Y.; Dubovitskiy, N.A.; Sharshov, K.A.; Kulikova, O.Y. New Summertime Records of the Bar-Headed Goose (Anser indicus) Far Outside Its Known Breeding Range and the Associated Risk of H5N1 Virus Introduction. J. Asian Ornithol. 2023, 39, 12–16. [Google Scholar]
- Fedorov, A.N.; Vasilyev, N.F.; Torgovkin, Y.I.; Shestakova, A.A.; Varlamov, S.P.; Zheleznyak, M.N.; Shepelev, V.V.; Konstantinov, P.Y.; Kalinicheva, S.S.; Basharin, N.I.; et al. Permafrost-Landscape Map of the Republic of Sakha (Yakutia) on a Scale 1:1,500,000. Geosciences 2018, 8, 465. [Google Scholar] [CrossRef]
- Iijima, Y.; Fedorov, A.N.; Park, H.; Suzuki, K.; Yabuki, H.; Maximov, T.C.; Ohata, T. Abrupt Increases in Soil Temperatures Following Increased Precipitation in a Permafrost Region, Central Lena River Basin, Russia. Permafr. Periglac. 2010, 21, 30–41. [Google Scholar] [CrossRef]
- Mine, J.; Tsunekuni, R.; Tanikawa, T.; Uchida, Y.; Dubovitskiy, N.; Derko, A.; Sobolev, I.; Shestopalov, A.; Sharshov, K.; Saito, T. Genetics of Japanese H5N8 High Pathogenicity Avian Influenza Viruses Isolated in Winter 2020–2021 and Their Genetic Relationship with Avian Influenza Viruses in Siberia. Transbounding Emerg. Dis. 2022, 69, e2195–e2213. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.; Zhang, H.; Zhao, C.; Zhang, Y.; Shen, J.; Sun, X.; Xu, H.; Xie, Y.; Gao, X.; et al. Prevalence, Evolution, Replication and Transmission of H3N8 Avian Influenza Viruses Isolated from Migratory Birds in Eastern China from 2017 to 2021. Emerg. Microbes Infect. 2023, 12, 2184178. [Google Scholar] [CrossRef]
- He, J.; Gong, L.; Chen, X.; Cheng, D.; Hou, S.; Kong, M.; Wei, X.; Yu, J.; Zhu, Q.; Li, W.; et al. A Retrospective Investigation of a Case of Dual Infection by Avian-Origin Influenza A (H10N5) and Seasonal Influenza A (H3N2) Viruses—Anhui Province, China, December 2023-January 2024. China CDC Wkly. 2024, 6, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, M.; Zhang, H.; Zhao, C.; Zhang, Y.; He, G.; Deng, G.; Cui, P.; Li, Y.; Liu, W.; et al. Emergence, Evolution, and Biological Characteristics of H10N4 and H10N8 Avian Influenza Viruses in Migratory Wild Birds Detected in Eastern China in 2020. Microbiol. Spectr. 2022, 10, e0080722. [Google Scholar] [CrossRef] [PubMed]
- Veen, J.; Yurlov, A.K.; Delany, S.N.; Mihantiev, A.I.; Selivanova, M.A.; Boere, G.C. An Atlas of Movements of Southwest Siberian Waterbirds; Wetlands International: Wageningen, The Netherlands, 2005. [Google Scholar]
- Shen, J.; Zhang, H.; Sun, X.; Zhang, Y.; Wang, M.; Guan, M.; Liu, L.; Li, W.; Xu, H.; Xie, Y.; et al. Evolution and Biological Characteristics of H11 Avian Influenza Viruses Isolated from Migratory Birds and Pigeons. Emerg. Microbes Infect. 2024, 13, 2398641. [Google Scholar] [CrossRef]
- Zhao, C.; Guo, J.; Zeng, X.; Shi, J.; Deng, G.; Zhang, Y.; Wang, Y.; Ma, Q.; Gao, X.; Cui, P.; et al. Novel H7N7 Avian Influenza Viruses Detected in Migratory Wild Birds in Eastern China between 2018 and 2020. Microbes Infect. 2022, 24, 105013. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Species | Total Number | Number of AIVs | Positivity Rate (%) |
|---|---|---|---|---|
| Accipitriformes (n = 4) | black kite (Milvus migrans) | 4 | 0 | 0 |
| Anseriformes (n = 1851) | greater white-fronted goose (Anser albifrons) | 13 | 0 | 0 |
| northern pintail (Anas acuta) | 61 | 3 | 4.92 | |
| northern shoveler (Anas clypeata) | 174 | 16 | 9.2 | |
| common teal (Anas crecca) | 516 | 36 | 6.98 | |
| falcated duck (Anas falcata) | 31 | 1 | 3.23 | |
| baikal teal (Anas formosa) | 20 | 1 | 5 | |
| European wigeon (Anas penelope) | 113 | 1 | 0.9 | |
| Mallard (Anas platyrhynchos) | 220 | 8 | 3.63 | |
| garganey (Anas querquedula) | 58 | 6 | 10.34 | |
| snow goose (Anser caerulescens) | 24 | 0 | 0 | |
| bean goose (Anser fabalis) | 2 | 0 | 0 | |
| bar-headed goose (Anser indicus) | 4 | 0 | 0 | |
| common pochard (Aythya ferina) | 1 | 0 | 0 | |
| tufted duck (Aythya fuligula) | 250 | 1 | 0.4 | |
| brant (Branta bernicla) | 8 | 0 | 0 | |
| goldeneye (Bucephala clangula) | 10 | 0 | 0 | |
| long-tailed duck (Clangula hyemalis) | 9 | 0 | 0 | |
| tundra swan (Cygnus bewickii) | 218 | 0 | 0 | |
| harlequin duck (Histrionicus histrionicus) | 4 | 0 | 0 | |
| Eurasian wigeon (Mareca penelope) | 57 | 0 | 0 | |
| velvet scoter (Melanitta fusca) | 1 | 0 | 0 | |
| smew (Mergellus albellus) | 6 | 0 | 0 | |
| common merganser (Mergus merganser) | 2 | 0 | 0 | |
| red-breasted merganser (Mergus serrator) | 8 | 0 | 0 | |
| spectacled eider (Somateria fischeri) | 41 | 0 | 0 | |
| Charadriiformes (n = 73) | sharp-tailed sandpiper (Calidris acuminata) | 2 | 0 | 0 |
| pectoral sandpiper (Calidris melanotos) | 1 | 0 | 0 | |
| temminck’s stint (Calidris temminckii) | 2 | 0 | 0 | |
| white-winged tern (Chlidonias leucopterus) | 4 | 0 | 0 | |
| common snipe (Gallinago gallinago) | 1 | 0 | 0 | |
| pintailed snipe (Gallinago stenura) | 1 | 0 | 0 | |
| European herring gull (Larus argentatus) | 4 | 0 | 0 | |
| common gull (Larus canus) | 10 | 0 | 0 | |
| glaucous gull (Larus hyperboreus) | 1 | 0 | 0 | |
| black-headed gull (Larus ridibundus) | 13 | 0 | 0 | |
| vega gull (Larus vegae) | 4 | 0 | 0 | |
| red phalarope (Phalaropus fulicarius) | 1 | 0 | 0 | |
| gray plover (Pluvialis squatarola) | 1 | 0 | 0 | |
| common tern (Sterna hirundo) | 14 | 0 | 0 | |
| wood sandpiper (Tringa glareola) | 1 | 0 | 0 | |
| green shank (Tringa nebularia) | 1 | 0 | 0 | |
| green sandpiper (Tringa ochropus) | 1 | 0 | 0 | |
| marsh sandpiper (Tringa stagnatilis) | 10 | 0 | 0 | |
| northern lapwing (Vanellus vanellus) | 1 | 0 | 0 | |
| Galliformes (n = 17) | hazel grouse (Bonasa bonasia) | 7 | 0 | 0 |
| willow ptarmigan (Lagopus lagopus) | 8 | 0 | 0 | |
| black grouse (Lyrurus tetrix) | 2 | 0 | 0 | |
| Gaviiformes (n = 1) | black-throated loon (Gavia arctica) | 1 | 0 | 0 |
| Gruiformes (n = 4) | white-naped crane (Antigone vipio) | 1 | 0 | 0 |
| coot (Fulica atra) | 1 | 0 | 0 | |
| water rail (Rallus aquaticus) | 1 | 0 | 0 | |
| brown-cheeked rail (Rallus indicus) | 1 | 0 | 0 | |
| Passeriformes (n = 1) | carrion crow (Corvus corone) | 1 | 0 | 0 |
| Pelecaniformes (n = 2) | gray heron (Ardea cinerea) | 2 | 0 | 0 |
| Podicipediformes (n = 17) | red-necked grebe (Podiceps grisegena) | 17 | 0 | 0 |
| 9 orders | 56 species | 1970 | 73 | 3.71 |
| Name | Subtype | Host | Date | ID |
|---|---|---|---|---|
| A/Teal/Yakutia/802/2018 | H4N6 | common teal (Anas crecca) | 20 August 2018 | EPI ISL 345915 |
| A/Teal/Yakutia/849/2018 | H3N8 | common teal (Anas crecca) | 21 August 2018 | EPI ISL 345916 |
| A/Common Teal/Yakutia/14/2019 | H3N8 | common teal (Anas crecca) | 17 August 2019 | EPI ISL 18633896 |
| A/Common Teal/Yakutia/18/2019 | H3N8 | common teal (Anas crecca) | 17 August 2019 | EPI ISL 18633897 |
| A/Common Teal/Yakutia/19/2019 | H3N8 | common teal (Anas crecca) | 17 August 2019 | EPI ISL 18633898 |
| A/Common Teal/Yakutia/32/2019 | H3N8 | common teal (Anas crecca) | 17 August 2019 | EPI ISL 18633899 |
| A/Northern pintail/Yakutia/41/2019 | H3N8 | northern pintail (Anas acuta) | 18 August 2019 | EPI ISL 18633900 |
| A/Shoveler/Yakutia/56/2019 | H3N8 | northern shoveler (Anas clypeata) | 18 August 2019 | EPI ISL 18633901 |
| A/Shoveler/Yakutia/61/2019 | H3N8 | northern shoveler (Anas clypeata) | 18 August 2019 | EPI ISL 18633902 |
| A/Common Teal/Yakutia/2x/2019 | H10N3 | common teal (Anas crecca) | 19 August 2019 | EPI ISL 18633890 |
| A/Common Teal/Yakutia/6x/2019 | H4N6 | common teal (Anas crecca) | 19 August 2019 | EPI ISL 18633892 |
| A/Shoveler/Yakutia/28x/2019 | H4N6 | northern shoveler (Anas clypeata) | 19 August 2019 | EPI ISL 18633893 |
| A/Common Teal/Yakutia/63x/2019 | H3N8 | common teal (Anas crecca) | 20 August 2019 | EPI ISL 18633895 |
| A/Shoveler/Yakutia/68x/2019 | H4N6 | northern shoveler (Anas clypeata) | 20 August 2019 | EPI ISL 18633894 |
| A/Shoveler/Yakutia/57/2019 | H4N6 | northern shoveler (Anas clypeata) | 8 September 2018 | EPI ISL 18633935 |
| A/Common Teal/Yakutia/49/2020 | H3N8 | common teal (Anas crecca) | 13 August 2020 | EPI ISL 1081354 |
| A/European Wigeon/Yakutia/55/2020 | H3N8 | European wigeon (Anas penelope) | 13 August 2020 | EPI ISL 1081355 |
| A/Mallard/Yakutia/11/2020 | H3N8 | mallard (Anas platyrhynchos) | 13 August 2020 | EPI ISL 1081356 |
| A/Mallard/Yakutia/21/2020 | H3N8 | mallard (Anas platyrhynchos) | 13 August 2020 | EPI ISL 1081357 |
| A/Mallard/Yakutia/47/2020 | H7N7 | mallard (Anas platyrhynchos) | 13 August 2020 | EPI ISL 1081358 |
| A/Northern Pintail/Yakutia/14/2020 | H3N8 | northern pintail (Anas acuta) | 13 August 2020 | EPI ISL 1081359 |
| A/Shoveler/Yakutia/25/2020 | H3N8 | northern shoveler (Anas clypeata) | 13 August 2020 | EPI ISL 1081360 |
| A/Common Teal/Yakutia/128/2020 | H3N8 | common teal (Anas crecca) | 14 August 2020 | EPI ISL 1081362 |
| A/Garganey/Yakutia/94/2020 | H3N8 | garganey (Anas querquedula) | 14 August 2020 | EPI ISL 1081366 |
| A/Garganey/Yakutia/98/2020 | H3N8 | garganey (Anas querquedula) | 14 August 2020 | EPI ISL 1081367 |
| A/Shoveler/Yakutia/65/2020 | H3N8 | northern shoveler (Anas clypeata) | 14 August 2020 | EPI ISL 1081361 |
| A/Common Teal/Yakutia/57x/2020 | H3N8 | common teal (Anas crecca) | 15 August 2020 | EPI ISL 1081363 |
| A/Common Teal/Yakutia/59x/2020 | H3N8 | common teal (Anas crecca) | 15 August 2020 | EPI ISL 1081364 |
| A/Common Teal/Yakutia/60x/2020 | H3N8 | common teal (Anas crecca) | 15 August 2020 | EPI ISL 1081365 |
| A/Common Teal/Yakutia/13/2021 | H3N8 | common teal (Anas crecca) | 14 August 2021 | EPI ISL 19212486 |
| A/Common Teal/Yakutia/18/2021 | H3N6 | common teal (Anas crecca) | 15 August 2021 | EPI ISL 19212478 |
| A/Common Teal/Yakutia/80/2021 | H3N8 | common teal (Anas crecca) | 15 August 2021 | EPI ISL 19212539 |
| A/Common Teal/Yakutia/111/2021 | H3N8 | common teal (Anas crecca) | 21 August 2021 | EPI ISL 19212481 |
| A/Common Teal/Yakutia/122/2021 | H3N8 | common teal (Anas crecca) | 21 August 2021 | EPI ISL 19212528 |
| A/Common Teal/Yakutia/146/2021 | H4N6 | common teal (Anas crecca) | 22 August 2021 | EPI ISL 19212487 |
| A/Common Teal/Yakutia/148/2021 | H3N6 | common teal (Anas crecca) | 22 August 2021 | EPI ISL 19212484 |
| A/Mallard/Yakutia/178/2021 | H3N6 | mallard (Anas platyrhynchos) | 22 August 2021 | EPI ISL 19212482 |
| A/Mallard/Yakutia/179/2021 | H3N6 | mallard (Anas platyrhynchos) | 22 August 2021 | EPI ISL 19212483 |
| A/Shoveler/Yakutia/141/2021 | H3N8 | northern shoveler (Anas clypeata) | 22 August 2021 | EPI ISL 19212529 |
| A/MallardxNorthern Pintail Hybrid/Yakutia/V46/2022 | H4N6 | hybrid between garganey (Anas querquedula) and northern pintail (Anas acuta) | 15 August 2022 | EPI ISL 19212538 |
| A/Shoveler/Yakutia/Bo17/2022 | H4N6 | northern shoveler (Anas clypeata) | 20 August 2022 | EPI ISL 19212537 |
| A/Shoveler/Yakutia/Bo29/2022 | H4N6 | northern shoveler (Anas clypeata) | 20 August 2022 | EPI ISL 19212536 |
| A/Shoveler/Yakutia/Bo30/2022 | H4N6 | northern shoveler (Anas clypeata) | 20 August 2022 | EPI ISL 19212535 |
| A/Common Teal/Yakutia/V98/2022 | H3N8 | common teal (Anas crecca) | 24 August 2022 | EPI ISL 19212534 |
| A/Common Teal/Yakutia/V109/2022 | H4N6 | common teal (Anas crecca) | 5 September 2022 | EPI ISL 19212533 |
| A/Mallard/Yakutia/C-3/2023 | H5N3 | mallard (Anas platyrhynchos) | 18 August 2023 | EPI ISL 19202486 |
| A/Mallard/Yakutia/C-9/2023 | H4N6 | mallard (Anas platyrhynchos) | 18 August 2023 | EPI ISL 19202488 |
| A/Common Teal/Yakutia/C-52/2023 | H3N6 | common teal (Anas crecca) | 19 August 2023 | EPI ISL 19202490 |
| A/Common Teal/Yakutia/K-28/2023 | H3N8 | common teal (Anas crecca) | 19 August 2023 | EPI ISL 19204497 |
| A/Shoveler/Yakutia/K-17/2023 | H3N8 | northern shoveler (Anas clypeata) | 19 August 2023 | EPI ISL 19204496 |
| A/Shoveler/Yakutia/K-33/2023 | H3N8 | northern shoveler (Anas clypeata) | 19 August 2023 | EPI ISL 19204498 |
| A/Anas crecca/Yakutia/K-57/2023 | H3N8 | common teal (Anas crecca) | 20 August 2023 | EPI ISL 19204504 |
| A/Common Teal/Yakutia/C-67/2023 | H3N8 | common teal (Anas crecca) | 20 August 2023 | EPI ISL 19202492 |
| A/Common Teal/Yakutia/C-74/2023 | H3N8 | common teal (Anas crecca) | 20 August 2023 | EPI ISL 19202494 |
| A/Common Teal/Yakutia/C-83/2023 | H3N8 | common teal (Anas crecca) | 20 August 2023 | EPI ISL 19202498 |
| A/Common Teal/Yakutia/C-85/2023 | H3N8 | common teal (Anas crecca) | 20 August 2023 | EPI ISL 19202501 |
| A/Common Teal/Yakutia/C-86/2023 | H3N8 | common teal (Anas crecca) | 20 August 2023 | EPI ISL 19202503 |
| A/Garganey/Yakutia/K-74/2023 | H3N8 | garganey (Anas querquedula) | 20 August 2023 | EPI ISL 19204503 |
| A/Garganey/Yakutia/K-75/2023 | H3N8 | garganey (Anas querquedula) | 20 August 2023 | EPI ISL 19204502 |
| A/Mallard/Yakutia/K-52/2023 | H3N8 | mallard (Anas platyrhynchos) | 20 August 2023 | EPI ISL 19204493 |
| A/Northern Pintail/Yakutia/C-82/2023 | H3N8 | northern pintail (Anas acuta) | 20 August 2023 | EPI ISL 19202496 |
| A/Northern Shoveler/Yakutia/K-56/2023 | H4N6 | northern shoveler (Anas clypeata) | 20 August 2023 | EPI ISL 19204505 |
| A/Shoveler/Yakutia/K-68/2023 | H3N8 | northern shoveler (Anas clypeata) | 20 August 2023 | EPI ISL 19204494 |
| A/Shoveler/Yakutia/K-72/2023 | H4N6 | northern shoveler (Anas clypeata) | 20 August 2023 | EPI ISL 19204495 |
| A/Tufted Duck/Yakutia/K-37/2023 | H11N9 | tufted duck (Aythya fuligula) | 20 August 2023 | EPI ISL 19204499 |
| A/Common Teal/Yakutia/K-96/2023 | H4N6 | common teal (Anas crecca) | 27 August 2023 | EPI ISL 19204501 |
| A/Common Teal/Yakutia/K-98/2023 | H3N8 | common teal (Anas crecca) | 27 August 2023 | EPI ISL 19204500 |
| A/Common Teal/Yakutia/UA-3/2023 | H3N6 | common teal (Anas crecca) | 30 August 2023 | EPI ISL 19202504 |
| A/Common Teal/Yakutia/UA-4/2023 | H3N8 | common teal (Anas crecca) | 30 August 2023 | EPI ISL 19202505 |
| A/Common Teal/Yakutia/UA-37/2023 | H3N6 | common teal (Anas crecca) | 3 September 2023 | EPI ISL 19204506 |
| A/Common Teal/Yakutia/UA-52/2023 | H3N8 | common teal (Anas crecca) | 5 September 2023 | EPI ISL 19204489 |
| A/Common Teal/Yakutia/UA-55/2023 | H3N6 | common teal (Anas crecca) | 5 September 2023 | EPI ISL 19204490 |
| A/Baikal Teal/Yakutia/OP-16/2023 | H11N9 | baikal teal (Anas formosa) | 6 September 2023 | EPI ISL 19204492 |
| A/Falcated Duck/Yakutia/OP-62/2023 | H10N3 | falcated duck (Anas falcata) | 27 September 2023 | EPI ISL 19204491 |
| Virus | Subtype | PB2 | PB1 | PA | NP | MP | NS | Genotype |
|---|---|---|---|---|---|---|---|---|
| 802/2018 | H4N6 | Mongolia | Shanghai | Mongolia | Korea | Bangladesh | Shandong | |
| 148/2021 | H3N6 | Vietnam | South_Korea | Chany | Jiangxi | Hunan | Mongolia | |
| 47/2020 | H7N7 | Buryatia | South_Korea | Korea | Kagoshima | |||
| UA-4/2023 | H3N8 | Hubei | Amur_region | Shandong | Hunan | Novosibirsk_region | ||
| C-82/2023 | H3N8 | Russia_Primorje | Russia_Primorje | Amur_region | Mongolia | Mongolia | Russia_Primorje | |
| C-83/2023 | H3N8 | Mongolia | ||||||
| 2x/2019 | H10N3 | Omsk_Region | Ningxia | Chany | Tomsk | Korea | Germany-HE | |
| C-3/2023 | H5N3 | Bangladesh | Saga | North_Kazakhstan | Korea | Bangladesh | Novosibirsk_region | |
| UA-3/2023 | H3N6 | Russia_Primorje | Amur_region | Amur_region | Yamaguchi | Shanxi | Jiangxi | |
| UA-52/2023 | H3N8 | Chany | Mongolia | Mongolia | ||||
| V98/2022 | H3N8 | Toyama | Kagoshima | Alaska | South_Africa | |||
| K-52/2023 | H3N8 | Kagoshima | Jiangxi | Mongolia | ||||
| 179/2021 | H3N6 | Shandong | Saga | Amur_region | Hokkaido | Amur_region | ||
| 18/2019 | H3N8 | Mongolia | Mongolia | Korea | Ibaraki | Korea | Hunan | |
| 122/2021 | H3N8 | Shandong | Korea | South_Korea | Jiangxi | Georgia | Mongolia | |
| V109/2022 | H4N6 | South_Korea | Japan | Wakayama | Zhejiang | South_Korea | ||
| OP-62/2023 | H10N3 | Bangladesh | South_Korea | Bangladesh | Jiangxi | |||
| V46/2022 | H4N6 | Shandong | Hokkaido | Hubei | ||||
| 13/2021 | H3N8 | Toyama | Chiba | Jiangxi | Bangladesh | Mongolia | ||
| C-52/2023 | H3N6 | Kagoshima | Bangladesh | South_Korea | Bangladesh | Mongolia | ||
| 111/2021 | H3N8 | Toyama | South_Korea | Jiangxi | Hokkaido | South_Africa | ||
| UA-55/2023 | H3N6 | Bangladesh | Amur_region | Amur_region | ||||
| Bo30/2022 | H4N6 | Mongolia | Sakhalin | South_Korea | South_Korea | Bangladesh | Mongolia | |
| UA-37/2023 | H3N6 | Amur_region | Amur_region | Jiangxi | Hokkaido | Amur_region | ||
| 18/2021 | H3N6 | Shandong | Korea | Bangladesh | Jiangxi | Mongolia | Kagoshima | |
| 63x/2019 | H3N8 | Mongolia | Mongolia | Amur_region | Kyoto | Korea | Hunan | |
| 41/2019 | H3N8 | Mongolia | Korea | Ibaraki | ||||
| 178/2021 | H3N6 | Toyama | Amur_region | Jiangxi | Hokkaido | Amur_region | ||
| 14/2019 | H3N8 | Egypt | Mongolia | Korea | Ibaraki | Hunan | Shandong | g1 |
| 32/2019 | H3N8 | |||||||
| 56/2019 | H3N8 | |||||||
| 61/2019 | H3N8 | |||||||
| 849/2018 | H3N8 | Novosibirsk_region | ||||||
| K-57/2023 | H3N8 | Bangladesh | Shandong | Chany | Mongolia | Shanxi | Buryatia | g2 |
| K-74/2023 | H3N8 | |||||||
| K-33/2023 | H3N8 | Bangladesh | Bangladesh | Kagoshima | ||||
| 146/2021 | H4N6 | Shandong | Toyama | Bangladesh | Jiangxi | Mongolia | Egypt | |
| C-67/2023 | H3N8 | South_Korea | Bangladesh | Mongolia | ||||
| C-85/2023 | H3N8 | Kagoshima | Okayama | Bangladesh | Mongolia | |||
| C-9/2023 | H4N6 | Shandong | Toyama | Jiangxi | Mongolia | Omsk_region | ||
| K-28/2023 | H3N8 | South_Korea | Kagoshima | Bangladesh | Yamaguchi | Bangladesh | Kagoshima | g3 |
| K-96/2023 | H4N6 | Shandong | Kagoshima | Jiangxi | Mongolia | |||
| K-98/2023 | H3N8 | South_Korea | Bangladesh | Bangladesh | South_Korea | |||
| K-56/2023 | H4N6 | Shandong | Toyama | Kagoshima | ||||
| Bo17/2022 | H4N6 | Mongolia | Shanghai | |||||
| Bo29/2022 | H4N6 | Bangladesh | Kagoshima | |||||
| K-17/2023 | H3N8 | South_Korea | Kagoshima | Bangladesh | South_Korea | |||
| K-68/2023 | H3N8 | Shandong | Bangladesh | Okayama | Kagoshima | |||
| K-72/2023 | H4N6 | Toyama | Kagoshima | Mongolia | Buryatia | |||
| OP-16/2023 | H11N9 | Kagoshima | Kagoshima | Hokkaido | South_Korea | Tottori | South_Korea | g4 |
| K-37/2023 | H11N9 | |||||||
| 80/2021 | H3N8 | Shandong | Toyama | South_Korea | Jiangxi | Hokkaido | Alaska | g5 |
| 141/2021 | H3N8 | Mongolia | ||||||
| C-74/2023 | H3N8 | Shandong | Bangladesh | Mongolia | Shandong | Hokkaido | Mongolia | g6 |
| C-86/2023 | H3N8 | Kagoshima | Bangladesh | |||||
| K-75/2023 | H3N8 | Kagoshima | Bangladesh | Jiangxi | Shimane | |||
| 128/2020 | H3N8 | Mongolia | Mongolia | Amur_region | Ibaraki | Korea | Shandong | g7 |
| 49/2020 | H3N8 | |||||||
| 57x/2020 | H3N8 | |||||||
| 59x/2020 | H3N8 | |||||||
| 60x/2020 | H3N8 | |||||||
| 55/2020 | H3N8 | |||||||
| 94/2020 | H3N8 | |||||||
| 98/2020 | H3N8 | |||||||
| 11/2020 | H3N8 | |||||||
| 21/2020 | H3N8 | |||||||
| 14/2020 | H3N8 | |||||||
| 25/2020 | H3N8 | |||||||
| 65/2020 | H3N8 | |||||||
| 19/2019 | H3N8 | Mongolia | Mongolia | Amur_region | Eastern_China | Korea | Hunan | g8 |
| 57/2019 | H4N6 | |||||||
| 68x/2019 | H4N6 | |||||||
| 6x/2019 | H4N6 | Mongolia | Mongolia | Amur_region | Eastern_China | Korea | Hunan | g9 |
| 28x/2019 | H4N6 |
| Segment/Protein | Mutation | Effects | Strains | References |
|---|---|---|---|---|
| PB2 | I292V | Increased polymerase activity in mammalian cell line, increased virulence in mice Increased polymerase activity in mammalian cell line | A/Common Teal/Yakutia/2x/2019 (H10N3) A/Mallard/Yakutia/C-3/2023 (H5N3) A/Common Teal/Yakutia/148/2021 (H3N6) A/Garganey/Yakutia/K-74/2023 (H3N8) A/Shoveler/Yakutia/K-33/2023 (H3N8) A/Anas crecca/Yakutia/K-57/2023 (H3N8) | [26] |
| PB2 | K482R | Increased polymerase activity in mammalian cell line | A/Common Teal/Yakutia/UA-37/2023 (H3N6) | [27,28] |
| PB2 | A588V | Increased polymerase activity and replication in mammalian and avian cell lines, increased virulence in mice | A/Common Teal/Yakutia/148*/2021 (H3N6) | [29] |
| PB1-F2 | N66S | Enhanced replication, virulence, and antiviral response in mice | A/Teal/Yakutia/849/2018 (H3N8) A/Mallard/Yakutia/47/2020 (H7N7) A/Shoveler/Yakutia/56/2019 (H3N8) A/Shoveler/Yakutia/61/2019 (H3N8) A/Northern Pintail/Yakutia/14/2020(H3N8) A/Common Teal/Yakutia/18/2019 (H3N8) A/Common Teal/Yakutia/32/2019 (H3N8) A/Common Teal/Yakutia/63x/2019 (H3N8) A/Common Teal/Yakutia/2x/2019 (H10N3) A/Common Teal/Yakutia/V109/2022(H4N6) A/Common Teal/Yakutia/122/2021 (H3N8) A/Common Teal/Yakutia/18/2021 (H3N6) A/MallardxNorthern Pintail Hybrid/Yakutia/V46/2022 (H4N6) A/Common Teal/Yakutia/148/2021 (H3N6) A/Falcated Duck/Yakutia/OP-62/2023 (H10N3) A/Garganey/Yakutia/K-74/2023 (H3N8) A/Shoveler/Yakutia/K-33/2023 (H3N8) A/Anas crecca/Yakutia/K-57/2023 (H3N8) | [30,31] |
| PA | K356R | Increase polymerase activity and enhanced replication in mammalian cell line, increased virulence in mice | A/Common Teal/Yakutia/C-67/2023(H3N8) A/Common Teal/Yakutia/C-52/2023(H3N6) A/Common Teal/Yakutia/C-85/2023(H3N8) | [32] |
| PA | S224P, N383D | Increased polymerase activity and enhanced viral replication in duck and mouse cell lines, increased virulence in mice and ducks | A/Common Teal/Yakutia/80/2021 (H3N8) | [33,34] |
| NP | M105V | Increased virulence in chickens | A/Mallard/Yakutia/47/2020 (H7N7) A/Common Teal/Yakutia/2x/2019 (H10N3) A/Shoveler/Yakutia/Bo30/2022 (H4N6) A/Common Teal/Yakutia/C-52/2023(H3N6) A/Common Teal/Yakutia/UA-4/2023 (H3N8) A/Falcated Duck/Yakutia/OP-62/2023 (H10N3) | [35,36] |
| NP | I109T | Increased polymerase activity and viral replication in chickens (but not ducks), increased virulence in chickens | A/Common Teal/Yakutia/UA-4/2023 (H3N8) | [35,36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasianov, N.; Sharshov, K.; Derko, A.; Sobolev, I.; Dubovitskiy, N.; Loginova, A.; Shemyakin, E.; Vladimirtseva, M.; Egorov, N.; Gabyshev, V.; et al. Exploring Avian Influenza Viruses in Yakutia—The Largest Breeding Habitat of Wild Migratory Birds in Northeastern Siberia. Viruses 2025, 17, 632. https://doi.org/10.3390/v17050632
Kasianov N, Sharshov K, Derko A, Sobolev I, Dubovitskiy N, Loginova A, Shemyakin E, Vladimirtseva M, Egorov N, Gabyshev V, et al. Exploring Avian Influenza Viruses in Yakutia—The Largest Breeding Habitat of Wild Migratory Birds in Northeastern Siberia. Viruses. 2025; 17(5):632. https://doi.org/10.3390/v17050632
Chicago/Turabian StyleKasianov, Nikita, Kirill Sharshov, Anastasiya Derko, Ivan Sobolev, Nikita Dubovitskiy, Arina Loginova, Evgeniy Shemyakin, Maria Vladimirtseva, Nikolay Egorov, Viacheslav Gabyshev, and et al. 2025. "Exploring Avian Influenza Viruses in Yakutia—The Largest Breeding Habitat of Wild Migratory Birds in Northeastern Siberia" Viruses 17, no. 5: 632. https://doi.org/10.3390/v17050632
APA StyleKasianov, N., Sharshov, K., Derko, A., Sobolev, I., Dubovitskiy, N., Loginova, A., Shemyakin, E., Vladimirtseva, M., Egorov, N., Gabyshev, V., Kim, Y., Lee, S.-H., Cho, A. Y., Kim, D.-H., Kim, T.-H., Song, C.-S., Jeong, H., Jheong, W., Hong, Y., ... Shestopalov, A. (2025). Exploring Avian Influenza Viruses in Yakutia—The Largest Breeding Habitat of Wild Migratory Birds in Northeastern Siberia. Viruses, 17(5), 632. https://doi.org/10.3390/v17050632