Integration Site and Clonal Expansion in Human Chronic Retroviral Infection and Gene Therapy

{kind=link}
{kind=link}
{kind=link}
Abstract
:2. HTLV-1
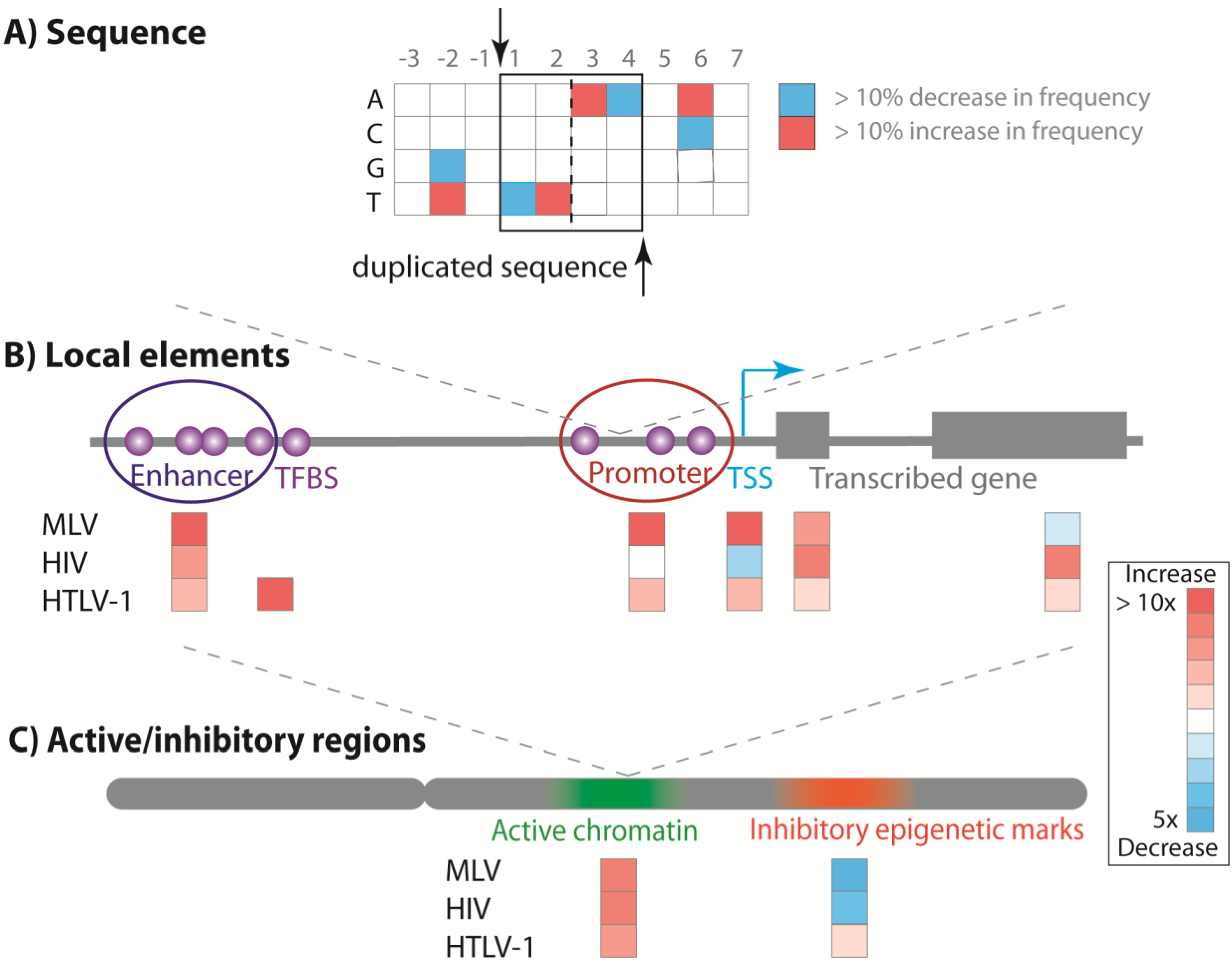
5. Retroviral Integration Site Preferences
5.1. Primary DNA Target Sequence
5.2. Local Genomic Features
5.3. Transcriptionally Active Chromatin

5.4. Host Factor Interaction with the Viral Integrase
6. Integration Site Clonal Expansion in Vivo
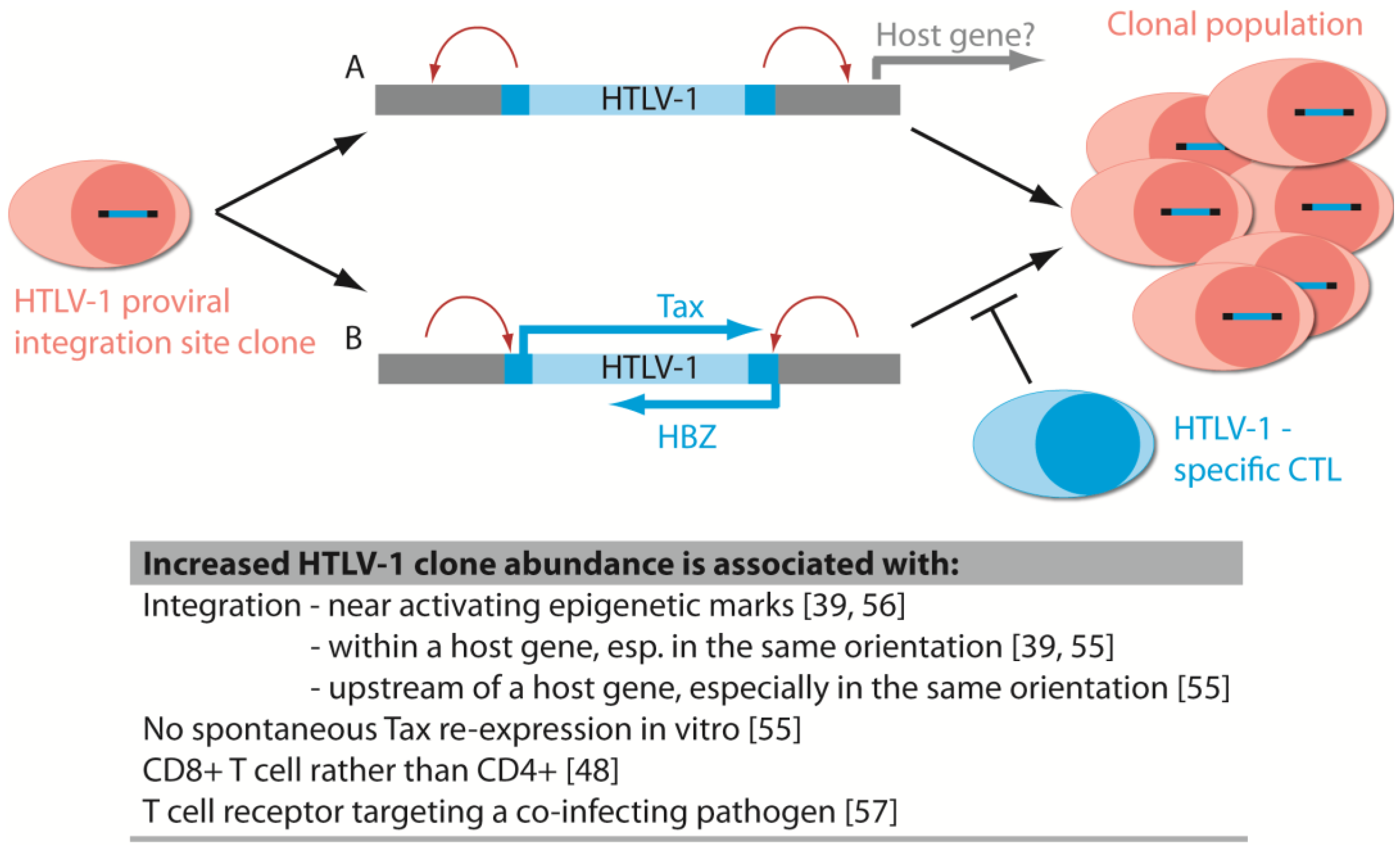
6.1. Disease-Associated Clonal Expansion Is Observed in Vivo
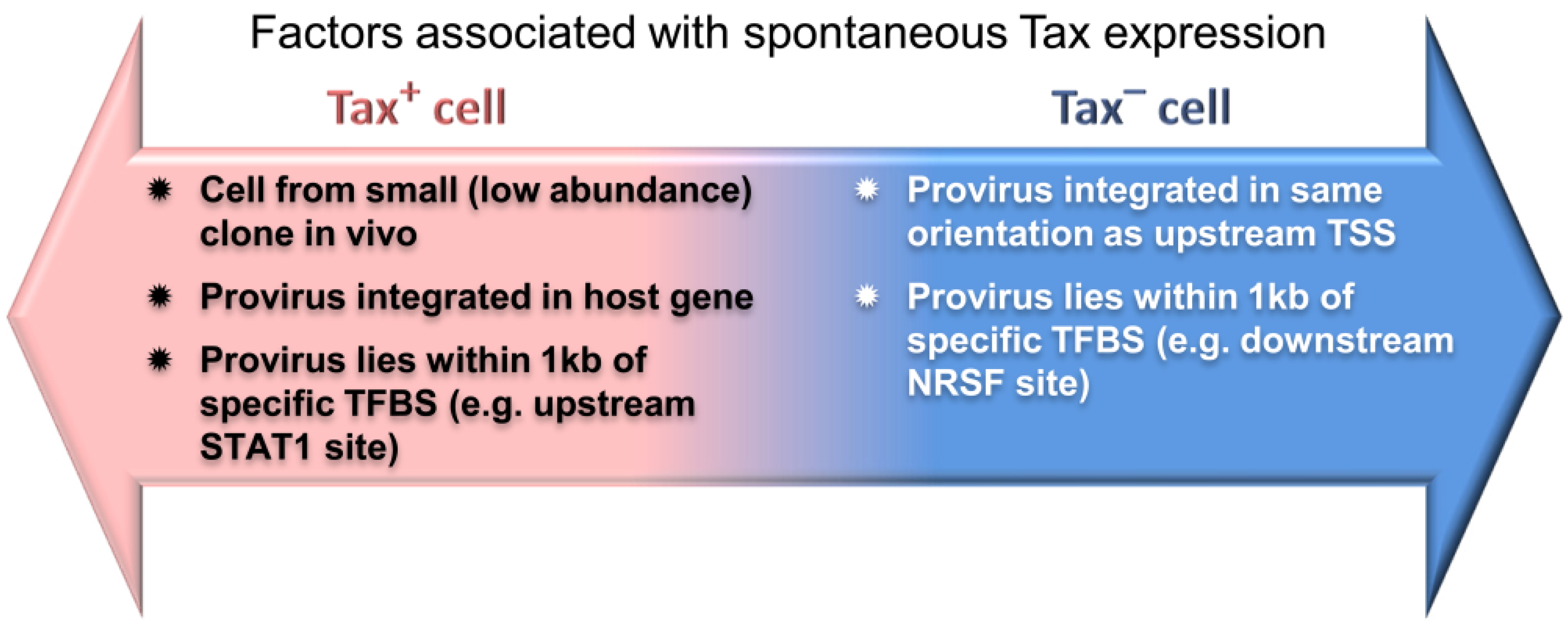
6.2. The Host Immune Response Affects HTLV-1 Integration Site Clone Survival and Abundance

6.3. The Role of Integration Site and Transgene Expression in Gene Therapy Clone Expansion
6.4. The Viral Promoter/Enhancer Alters Host Gene Expression and Oligoclonal Proliferation
6.5. Integration Bias Influences Integration Site Profile in Vivo
6.6. Other Causes of Asymmetrical Expansion of Retrovirus-Infected Clones in Vivo
7. Concluding Thoughts

Author Contributions
Conflicts of Interest
References and Notes
- Blikstad, V.; Benachenhou, F.; Sperber, G.O.; Blomberg, J. Evolution of human endogenous retroviral sequences: A conceptual account. Cell. Mol. Life Sci. 2008, 65, 3348–3365. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Lock, W.M.; Mager, D.L. Endogenous retroviral LTRs as promoters for human genes: A critical assessment. Gene 2009, 448, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Jeang, K.-T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.C.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R.M. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Manel, N.; Kim, F.J.; Kinet, S.; Taylor, N.; Sitbon, M.; Battini, J.-L. The ubiquitous glucose transporter GLUT-1 is a receptor for HTLV. Cell 2003, 115, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.B.; Rowan, A.G.; Melamed, A.; Taylor, G.P.; Bangham, C.R.M. HTLV-1-infected T cells contain a single integrated provirus in natural infection. Blood 2012, 120, 3488–3490. [Google Scholar] [CrossRef] [PubMed]
- Boxus, M.; Willems, L. Mechanisms of HTLV-1 persistence and transformation. Br. J. Cancer 2009, 101, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Jeang, K.T. Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: viral infectivity, Tax, HBZ and therapy. Oncogene 2011, 30, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.H.; Edwards, A.J.; Cruickshank, J.K.; Rudge, P.; Dalgleish, A.G. In vivo cellular tropism of human T-cell leukemia virus type 1. J. Virol. 1990, 64, 5682–5687. [Google Scholar] [PubMed]
- Ijichi, S.; Ramundo, M.B.; Takahashi, H.; Hall, W.W. In vivo cellular tropism of human T cell leukemia virus type II (HTLV-II). J. Exp. Med. 1992, 176, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Kannian, P.; Yin, H.; Doueiri, R.; Lairmore, M.D.; Fernandez, S.; Green, P.L. Distinct transformation tropism exhibited by human T lymphotropic virus type 1 (HTLV-1) and HTLV-2 is the result of postinfection T cell clonal expansion. J. Virol. 2012, 86, 3757–3766. [Google Scholar] [CrossRef] [PubMed]
- Moritoyo, T.; Reinhart, T.A.; Moritoyo, H.; Sato, E.; Izumo, S.; Osame, M.; Haase, A.T. Human T-lymphotropic virus type I-associated myelopathy and tax gene expression in CD4+ T lymphocytes. Ann. Neurol. 1996, 40, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Hanon, E.; Hall, S.; Taylor, G.P.; Saito, M.; Davis, R.; Tanaka, Y.; Usuku, K.; Osame, M.; Weber, J.N.; Bangham, C.R.M. Abundant Tax protein expression in CD4+ T cells infected with human T-cell lymphotropic virus type I (HTLV-I) is prevented by cytotoxic T lymphocytes. Blood 2000, 95, 1386–1392. [Google Scholar] [PubMed]
- Kannagi, M.; Harada, S.; Maruyama, I.; Inoko, H.; Igarashi, H.; Kuwashima, G.; Sato, S.; Morita, M.; Kidokoro, M.; Sugimoto, M. Predominant recognition of human T cell leukemia virus type I (HTLV-I) pX gene products by human CD8+ cytotoxic T cells directed against HTLV-I-infected cells. Int. Immunol. 1991, 3, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Goon, P.K.C.; Biancardi, A.; Fast, N.; Igakura, T.; Hanon, E.; Mosley, A.J.; Asquith, B.; Gould, K.G.; Marshall, S.; Taylor, G.P.; et al. Human T cell lymphotropic virus (HTLV) type-1-specific CD8+ T cells: frequency and immunodominance hierarchy. J. Infect. Dis. 2004, 189, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
- Satou, Y.; Yasunaga, J.-I.; Yoshida, M.; Matsuoka, M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc. Natl. Acad. Sci. USA 2006, 103, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Nosaka, K.; Yasunaga, J.-I.; Maeda, M.; Mueller, N.; Okayama, A.; Matsuoka, M. Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology 2005, 2, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilburn, S.; Rowan, A.; Demontis, M.-A.; MacNamara, A.; Asquith, B.; Bangham, C.R.M.; Taylor, G.P. In vivo expression of human T-lymphotropic virus type 1 basic leucine-zipper protein generates specific CD8+ and CD4+ T-lymphocyte responses that correlate with clinical outcome. J. Infect. Dis. 2011, 203, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Macnamara, A.; Rowan, A.; Hilburn, S.; Kadolsky, U.; Fujiwara, H.; Suemori, K.; Yasukawa, M.; Taylor, G.; Bangham, C.R.M.; Asquith, B. HLA class I binding of HBZ determines outcome in HTLV-1 infection. PLoS Pathog. 2010, 6, e1001117. [Google Scholar]
- Qasim, W.; Gaspar, H.B.; Thrasher, A.J. Progress and prospects: Gene therapy for inherited immunodeficiencies. Gene Ther. 2009, 16, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Antoine, C.; Müller, S.; Cant, A.; Cavazzana-Calvo, M.; Veys, P.; Vossen, J.; Fasth, A.; Heilmann, C.; Wulffraat, N.; Seger, R.; et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968–99. Lancet 2003, 361, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.-L.; et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.B.; Parsley, K.L.; Howe, S.; King, D.; Gilmour, K.C.; Sinclair, J.; Brouns, G.; Schmidt, M.; von Kalle, C.; Barington, T.; et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet 2004, 364, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Maetzig, T.; Galla, M.; Baum, C.; Schambach, A. Gammaretroviral vectors: Biology, technology and application. Viruses 2011, 3, 677–713. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human [bgr]-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef]
- Sakuma, T.; Barry, M.A.; Ikeda, Y. Lentiviral vectors: Basic to translational. Biochem. J. 2012, 443, 603–618. [Google Scholar] [CrossRef] [PubMed]
- Matreyek, K.A.; Engelman, A. Viral and cellular requirements for the nuclear entry of retroviral preintegration nucleoprotein complexes. Viruses 2013, 5, 2483–2511. [Google Scholar] [CrossRef] [PubMed]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol 1998, 72, 8463–8471. [Google Scholar] [PubMed]
- Mueller, P.R.; Wold, B. In vivo footprinting of a muscle specific enhancer by ligation mediated PCR. Science 1989, 246, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Schwarzwaelder, K.; Bartholomae, C.; Zaoui, K.; Ball, C.; Pilz, I.; Braun, S.; Glimm, H.; Kalle, C.V. High-resolution insertion-site analysis by linear amplification—Mediated PCR (LAM-PCR). Nat. Methods 2007, 4, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, R.; Eckenberg, R.; Paruzynski, A.; Bartholomae, C.C.; Nowrouzi, A.; Arens, A.; Howe, S.J.; Recchia, A.; Cattoglio, C.; Wang, W.; et al. Comprehensive genomic access to vector integration in clinical gene therapy. Nat. Med. 2009, 15, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Brugman, M.H.; Suerth, J.D.; Rothe, M.; Suerbaum, S.; Schambach, A.; Modlich, U.; Kustikova, O.; Baum, C. Evaluating a ligation-mediated PCR and pyrosequencing method for the detection of clonal contribution in polyclonal retrovirally transduced samples. Hum. Gene Ther. Methods 2013, 24, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Paruzynski, A.; Arens, A.; Gabriel, R.; Bartholomae, C.C.; Scholz, S.; Wang, W.; Wolf, S.; Glimm, H.; Schmidt, M.; von Kalle, C. Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing. Nat. Protoc. 2010, 5, 1379–1395. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.A.; Malani, N.; Melamed, A.; Gormley, N.; Carter, R.; Bentley, D.; Berry, C.; Bushman, F.D.; Taylor, G.P.; Bangham, C.R.M. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T cell clones. Blood 2011, 117, 3113–3122. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.C.; Gillet, N.A.; Melamed, A.; Gormley, N.; Bangham, C.R.M.; Bushman, F. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics 2012, 28, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Firouzi, S.; Lopez, Y.; Suzuki, Y.; Nakai, K.; Sugano, S.; Yamochi, T.; Watanabe, T. Development and validation of a new high-throughput method to investigate the clonality of HTLV-1-infected cells based on provirus integration sites. Genome Med. 2014, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Brady, T.; Roth, S.L.; Malani, N.; Wang, G.P.; Berry, C.C.; Leboulch, P.; Hacein-Bey-Abina, S.; Cavazzana-Calvo, M.; Papapetrou, E.P.; Sadelain, M.; et al. A method to sequence and quantify DNA integration for monitoring outcome in gene therapy. Nucl. Acids Res. 2011, 39, e72. [Google Scholar] [CrossRef]
- Holman, A.G.; Coffin, J.M. Symmetrical base preferences surrounding HIV-1, avian sarcoma/leukosis virus, and murine leukemia virus integration sites. Proc. Natl. Acad. Sci. USA 2005, 102, 6103–6107. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M.; Munroe, D.J. Weak palindromic consensus sequences are a common feature found at the integration target sites of many retroviruses. J. Virol. 2005, 79, 5211–5214. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Craigie, R. Integration of mini-retroviral DNA: A cell-free reaction for biochemical analysis of retroviral integration. Proc. Natl. Acad. Sci. USA 1989, 86, 3065–3069. [Google Scholar] [CrossRef] [PubMed]
- Derse, D.; Crise, B.; Li, Y.; Princler, G.; Lum, N.; Stewart, C.; McGrath, C.F.; Hughes, S.H.; Munroe, D.J.; Wu, X. Human T-cell leukemia virus type 1 integration target sites in the human genome: Comparison with those of other retroviruses. J. Virol. 2007, 81, 6731–6741. [Google Scholar] [CrossRef] [PubMed]
- Meekings, K.N.; Leipzig, J.; Bushman, F.D.; Taylor, G.P.; Bangham, C.R.M. HTLV-1 integration into transcriptionally active genomic regions is associated with proviral expression and with HAM/TSP. PLoS Pathog. 2008, 4, e1000027. [Google Scholar]
- Melamed, A.; Witkover, A.D.; Laydon, D.J.; Brown, R.; Ladell, K.; Miners, K.; Rowan, A.G.; Gormley, N.; Price, D.A.; Taylor, G.P.; et al. Clonality of HTLV-2 in natural infection. PLoS Pathog. 2014, 10, e1004006. [Google Scholar]
- De Ravin, S.S.; Su, L.; Theobald, N.; Choi, U.; Macpherson, J.L.; Poidinger, M.; Symonds, G.; Pond, S.M.; Ferris, A.L.; Hughes, S.H.; et al. Enhancers are major targets for murine leukemia virus vector integration. J. Virol. 2014, 88, 4504–4513. [Google Scholar] [CrossRef] [PubMed]
- LaFave, M.C.; Varshney, G.K.; Gildea, D.E.; Wolfsberg, T.G.; Baxevanis, A.D.; Burgess, S.M. MLV integration site selection is driven by strong enhancers and active promoters. Nucl. Acids Res. 2014, 42, 4257–4269. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Cattoglio, C.; Pellin, D.; Rizzi, E.; Maruggi, G.; Corti, G.; Miselli, F.; Sartori, D.; Guffanti, A.; di Serio, C.; Ambrosi, A.; et al. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood 2010, 116, 5507–5517. [Google Scholar] [CrossRef] [PubMed]
- Schröder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, e234. [Google Scholar] [CrossRef] [Green Version]
- Melamed, A.; Laydon, D.J.; Gillet, N.A.; Tanaka, Y.; Taylor, G.P.; Bangham, C.R.M. Genome-wide determinants of proviral targeting, clonal abundance and expression in natural HTLV-1 infection. PLoS Pathogens 2013, 9, e1003271. [Google Scholar] [CrossRef]
- Cook, L.B.; Melamed, A.; Niederer, H.; Valganon, M.; Laydon, D.; Foroni, L.; Taylor, G.P.; Matsuoka, M.; Bangham, C.R.M. The role of HTLV-1 clonality, proviral structure and genomic integration site in adult T cell leukemia/lymphoma. Blood 2014, 123, 3925–3931. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.A.; Cook, L.; Laydon, D.J.; Hlela, C.; Verdonck, K.; Alvarez, C.; Gotuzzo, E.; Clark, D.; Farre, L.; Bittencourt, A.; et al. Strongyloidiasis and infective dermatitis alter human T lymphotropic virus-1 clonality in vivo. PLoS Pathog. 2013, 9, e1003263. [Google Scholar]
- Niederer, H.A.; Laydon, D.J.; Melamed, A.; Elemans, M.; Asquith, B.; Matsuoka, M.; Bangham, C.R. HTLV-1 proviral integration sites differ between asymptomatic carriers and patients with HAM/TSP. Virol. J. 2014, 11, 172. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Brady, T.; Agosto, L.M.; Malani, N.; Berry, C.C.; OʼDoherty, U.; Bushman, F. HIV integration site distributions in resting and activated CD4+ T cells infected in culture. AIDS 2009, 23, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Bartholomae, C.C.; Arens, A.; Balaggan, K.S.; Yanez-Munoz, R.J.; Montini, E.; Howe, S.J.; Paruzynski, A.; Korn, B.; Appelt, J.U.; Macneil, A.; et al. Lentiviral vector integration profiles differ in rodent postmitotic tissues. Mol. Ther. 2011, 19, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, M.K.; Yamashita, M.; Emerman, M.; Ciuffi, A.; Marshall, H.; Crawford, G.; Collins, F.; Shinn, P.; Leipzig, J.; Hannenhalli, S.; et al. Retroviral DNA integration: Viral and cellular determinants of target-site selection. PLoS Pathog. 2006, 2, e60. [Google Scholar] [CrossRef]
- Engelman, A.; Cherepanov, P. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 2008, 4, e1000046. [Google Scholar]
- Eidahl, J.O.; Crowe, B.L.; North, J.A.; McKee, C.J.; Shkriabai, N.; Feng, L.; Plumb, M.; Graham, R.L.; Gorelick, R.J.; Hess, S.; et al. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucl. Acids Res. 2013, 41, 3924–3936. [Google Scholar] [CrossRef] [PubMed]
- Van Nuland, R.; van Schaik, F.; Simonis, M.; van Heesch, S.; Cuppen, E.; Boelens, R.; Timmers, H.; van Ingen, H. Nucleosomal DNA binding drives the recognition of H3K36-methylated nucleosomes by the PSIP1-PWWP domain. Epigenetics Chromatin 2013, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Kolasinska-Zwierz, P.; Down, T.; Latorre, I.; Liu, T.; Liu, X.S.; Ahringer, J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genetics 2009, 41, 376–381. [Google Scholar] [CrossRef]
- Ocwieja, K.E.; Brady, T.L.; Ronen, K.; Huegel, A.; Roth, S.L.; Schaller, T.; James, L.C.; Towers, G.J.; Young, J.A.T.; Chanda, S.K.; et al. HIV integration targeting: A pathway involving transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog. 2011, 7, e1001313. [Google Scholar]
- Ferris, A.L.; Wu, X.; Hughes, C.M.; Stewart, C.; Smith, S.J.; Milne, T.A.; Wang, G.G.; Shun, M.C.; Allis, C.D.; Engelman, A.; et al. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl. Acad. Sci. USA 2010, 107, 3135–3140. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Shun, M.C.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog. 2009, 5, e1000259. [Google Scholar]
- De Rijck, J.; de Kogel, C.; Demeulemeester, J.; Vets, S.; El Ashkar, S.; Malani, N.; Bushman, F.D.; Landuyt, B.; Husson, S.J.; Busschots, K.; et al. The BET family of proteins targets moloney murine leukemia virus integration near transcription start sites. Cell. Rep. 2013, 5, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.S.; Maetzig, T.; Maertens, G.N.; Sharif, A.; Rothe, M.; Weidner-Glunde, M.; Galla, M.; Schambach, A.; Cherepanov, P.; Schulz, T.F. Bromo- and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J. Virol. 2013, 87, 12721–12736. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; Siegel, R.M.; Jang, M.K.; Lenardo, M.J.; Ozato, K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol. Cell. 2004, 13, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Umehara, T.; Nakano, K.; Jang, M.K.; Shirouzu, M.; Morita, S.; Uda-Tochio, H.; Hamana, H.; Terada, T.; Adachi, N.; et al. Crystal structure of the human BRD2 bromodomain: insights into dimerization and recognition of acetylated histone H4. J. Biol. Chem. 2007, 282, 4193–4201. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Laydon, D.J.; Melamed, A.; Sim, A.; Gillet, N.A.; Sim, K.; Darko, S.; Kroll, J.S.; Douek, D.C.; Price, D.A.; Bangham, C.R.M.; et al. Quantification of HTLV-1 clonality and TCR diversity. PLoS Computat. Biol. 2014, 10, e1003646. [Google Scholar] [CrossRef]
- Bangham, C.R.M. CTL quality and the control of human retroviral infections. Eur. J. Immunol. 2009, 39, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Boztug, K.; Schmidt, M.; Schwarzer, A.; Banerjee, P.P.; Díez, I.A.; Dewey, R.A.; Böhm, M.; Nowrouzi, A.; Ball, C.R.; Glimm, H.; et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 2010, 363, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Braun, C.J.; Boztug, K.; Paruzynski, A.; Witzel, M.; Schwarzer, A.; Rothe, M.; Modlich, U.; Beier, R.; Gohring, G.; Steinemann, D.; et al. Gene therapy for Wiskott-Aldrich syndrome--long-term efficacy and genotoxicity. Sci. Transl. Med. 2014, 6, 227ra33. [Google Scholar] [CrossRef]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Kramer, A.; Schwable, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [PubMed]
- Hacein-Bey-Abina, S.; Hauer, J.; Lim, A.; Picard, C.; Wang, G.P.; Berry, C.C.; Martinache, C.; Rieux-Laucat, F.; Latour, S.; Belohradsky, B.H.; et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2010, 363, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Kustikova, O.; Fehse, B.; Modlich, U.; Yang, M.; Düllmann, J.; Kamino, K.; von Neuhoff, N.; Schlegelberger, B.; Li, Z.; Baum, C. Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking. Science 2005, 308, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Thrasher, A.J. Gene therapy for PIDs: Progress, pitfalls and prospects. Gene 2013, 525, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Cassani, B.; Andolfi, G.; Mirolo, M.; Biasco, L.; Recchia, A.; Urbinati, F.; Valacca, C.; Scaramuzza, S.; Aker, M.; et al. Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J. Clin. Investig. 2007, 117, 2233–2240. [Google Scholar] [CrossRef] [PubMed]
- Van Dooren, S.; Pybus, O.G.; Salemi, M.; Liu, H.-F.; Goubau, P.; Remondegui, C.; Talarmin, A.; Gotuzzo, E.; Alcantara, L.C.J.; Galvão-Castro, B.; et al. The low evolutionary rate of human T-cell lymphotropic virus type-1 confirmed by analysis of vertical transmission chains. Mol. Biol. Evol. 2004, 21, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.A.; Gutiérrez, G.; Rodriguez, S.M.; de Brogniez, A.; Renotte, N.; Alvarez, I.; Trono, K.; Willems, L. Massive depletion of bovine leukemia virus proviral clones located in genomic transcriptionally active sites during primary infection. PLoS Pathog. 2013, 9, e1003687. [Google Scholar]
- Florins, A.; de Brogniez, A.; Elemans, M.; Bouzar, A.-B.; François, C.; Reichert, M.; Asquith, B.; Willems, L. Viral expression directs the fate of B cells in bovine leukemia virus-infected sheep. J. Virol. 2012, 86, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.F.; Huynh, K.D.; Lee, J.T. Perinucleolar targeting of the inactive X during S phase: evidence for a role in the maintenance of silencing. Cell 2007, 129, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.B.; Cooray, S.; Gilmour, K.C.; Parsley, K.L.; Zhang, F.; Adams, S.; Bjorkegren, E.; Bayford, J.; Brown, L.; Davies, E.G.; et al. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci. Transl. Med. 2011, 3, 97ra80. [Google Scholar]
- Candotti, F.; Shaw, K.L.; Muul, L.; Carbonaro, D.; Sokolic, R.; Choi, C.; Schurman, S.H.; Garabedian, E.; Kesserwan, C.; Jagadeesh, G.J.; et al. Gene therapy for adenosine deaminase–deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood 2012, 120, 3635–3646. [Google Scholar] [CrossRef] [PubMed]
- Davé, U.P.; Akagi, K.; Tripathi, R.; Cleveland, S.M.; Thompson, M.A.; Yi, M.; Stephens, R.; Downing, J.R.; Jenkins, N.A.; Copeland, N.G. Murine leukemias with retroviral insertions at LMO2 are predictive of the leukemias induced in SCID-X1 patients following retroviral gene therapy. PLoS Genetics 2009, 5, e1000491. [Google Scholar] [CrossRef]
- Bessis, N.; GarciaCozar, F.J.; Boissier, M.C. Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther. 2004, 11, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Willemsen, R.; van Elzakker, P.; van Steenbergen-Langeveld, S.; Broertjes, M.; Oosterwijk-Wakka, J.; Oosterwijk, E.; Sleijfer, S.; Debets, R.; Gratama, J.W. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011, 117, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Mauro, V.P.; Chappell, S.A. A critical analysis of codon optimization in human therapeutics. Trends Mol. Med. 2014, in press. [Google Scholar]
- Sherrill-Mix, S.; Lewinski, M.; Famiglietti, M.; Bosque, A.; Malani, N.; Ocwieja, K.; Berry, C.; Looney, D.; Shan, L.; Agosto, L.; et al. HIV latency and integration site placement in five cell-based models. Retrovirology 2013, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Dahabieh, M.; Ooms, M.; Brumme, C.; Taylor, J.; Harrigan, P.R.; Simon, V.; Sadowski, I. Direct non-productive HIV-1 infection in a T-cell line is driven by cellular activation state and NFkappaB. Retrovirology 2014, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Uren, A.G.; Kool, J.; Berns, A.; van Lohuizen, M. Retroviral insertional mutagenesis: past, present and future. Oncogene 2005, 24, 7656–7672. [Google Scholar] [CrossRef] [PubMed]
- Ranzani, M.; Annunziato, S.; Adams, D.J.; Montini, E. Cancer gene discovery: Exploiting insertional mutagenesis. Mol. Cancer Res. 2013, 11, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- McCormack, M.P.; Young, L.F.; Vasudevan, S.; de Graaf, C.A.; Codrington, R.; Rabbitts, T.H.; Jane, S.M.; Curtis, D.J. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science 2010, 327, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kuhlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.M.; Choi, U.; Theobald, N.; Linton, G.; Long Priel, D.A.; Kuhns, D.; Malech, H.L. Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils. Blood 2010, 115, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Recchia, A.; Bonini, C.; Magnani, Z.; Urbinati, F.; Sartori, D.; Muraro, S.; Tagliafico, E.; Bondanza, A.; Stanghellini, M.T.; Bernardi, M.; et al. Retroviral vector integration deregulates gene expression but has no consequence on the biology and function of transplanted T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Cassani, B.; Montini, E.; Maruggi, G.; Ambrosi, A.; Mirolo, M.; Selleri, S.; Biral, E.; Frugnoli, I.; Hernandez-Trujillo, V.; di Serio, C.; et al. Integration of retroviral vectors induces minor changes in the transcriptional activity of T cells from ADA-SCID patients treated with gene therapy. Blood 2009, 114, 3546–3556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Markus, J.; Bies, J.; Paul, T.; Wolff, L. Three murine leukemia virus integration regions within 100 kilobases upstream of c-myb are proximal to the 5ʼ regulatory region of the gene through DNA looping. J. Virol. 2012, 86, 10524–10532. [Google Scholar] [CrossRef] [PubMed]
- Cesana, D.; Sgualdino, J.; Rudilosso, L.; Merella, S.; Naldini, L.; Montini, E. Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J. Clin. Investig. 2012, 122, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Moiani, A.; Paleari, Y.; Sartori, D.; Mezzadra, R.; Miccio, A.; Cattoglio, C.; Cocchiarella, F.; Lidonnici, M.R.; Ferrari, G.; Mavilio, F. Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J. Clin. Investig. 2012, 122, 1653–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Nagel, J.; Gross, B.; Meggendorfer, M.; Preiss, C.; Grez, M.; Brack-Werner, R.; Dietzel, S. Stably integrated and expressed retroviral sequences can influence nuclear location and chromatin condensation of the integration locus. Chromosoma 2012, 121, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Zychlinski, D.; Schambach, A.; Modlich, U.; Maetzig, T.; Meyer, J.; Grassman, E.; Mishra, A.; Baum, C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol. Ther. 2008, 16, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Cesana, D.; Ranzani, M.; Volpin, M.; Bartholomae, C.; Duros, C.; Artus, A.; Merella, S.; Benedicenti, F.; Sergi Sergi, L.; Sanvito, F.; et al. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol. Ther. 2014, 22, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Thornhill, S.I.; Schambach, A.; Howe, S.J.; Ulaganathan, M.; Grassman, E.; Williams, D.; Schiedlmeier, B.; Sebire, N.J.; Gaspar, H.B.; Kinnon, C.; et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol. Ther. 2008, 16, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Bangham, C.R.; Cook, L.B.; Melamed, A. HTLV-1 clonality in adult T-cell leukaemia and non-malignant HTLV-1 infection. Semin Cancer Biol. 2014, 26, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Bartolomae, C.C.; Cesana, D.; Cartier, N.; Aubourg, P.; Ranzani, M.; Cesani, M.; Benedicenti, F.; Plati, T.; Rubagotti, E.; et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood 2011, 117, 5332–5339. [Google Scholar] [CrossRef] [PubMed]
- Cattoglio, C.; Facchini, G.; Sartori, D.; Antonelli, A.; Miccio, A.; Cassani, B.; Schmidt, M.; von Kalle, C.; Howe, S.; Thrasher, A.J.; et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 2007, 110, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Biasco, L.; Ambrosi, A.; Pellin, D.; Bartholomae, C.; Brigida, I.; Roncarolo, M.G.; di Serio, C.; von Kalle, C.; Schmidt, M.; Aiuti, A. Integration profile of retroviral vector in gene therapy treated patients is cell‐specific according to gene expression and chromatin conformation of target cell. EMBO Mol. Med. 2011, 3, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Kustikova, O.S.; Geiger, H.; Li, Z.; Brugman, M.H.; Chambers, S.M.; Shaw, C.A.; Pike-Overzet, K.; Ridder, D.D.; Staal, F.J.T.; Keudell, G.V.; et al. Retroviral vector insertion sites associated with dominant hematopoietic clones mark “stemness” pathways. Blood 2007, 109, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Martins, V.C.; Busch, K.; Juraeva, D.; Blum, C.; Ludwig, C.; Rasche, V.; Lasitschka, F.; Mastitsky, S.E.; Brors, B.; Hielscher, T.; et al. Cell competition is a tumour suppressor mechanism in the thymus. Nature 2014, 509, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Fields, P.A.; Taylor, G.P. “Antivirals” in the treatment of adult T cell leukaemia-lymphoma (ATLL). Curr. Hematol. Malig. Rep. 2012, 7, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Yonekura, K.; Kanzaki, T.; Gunshin, K.; Kawakami, N.; Takatsuka, Y.; Nakano, N.; Tokunaga, M.; Kubota, A.; Takeuchi, S.; Kanekura, T.; et al. Effect of anti-CCR4 monoclonal antibody (mogamulizumab) on adult T-cell leukemia-lymphoma: Cutaneous adverse reactions may predict the prognosis. J. Dermatol. 2014, 41, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Di, W.L.; Mellerio, J.E.; Bernadis, C.; Harper, J.; Abdul-Wahab, A.; Ghani, S.; Chan, L.; Martinez-Queipo, M.; Hara, H.; McNicol, A.M.; et al. Phase I study protocol for ex vivo lentiviral gene therapy for the inherited skin disease, Netherton syndrome. Hum. Gene ther. Clin. Dev. 2014, 24, 182–190. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niederer, H.A.; Bangham, C.R.M. Integration Site and Clonal Expansion in Human Chronic Retroviral Infection and Gene Therapy. Viruses 2014, 6, 4140-4164. https://doi.org/10.3390/v6114140
Niederer HA, Bangham CRM. Integration Site and Clonal Expansion in Human Chronic Retroviral Infection and Gene Therapy. Viruses. 2014; 6(11):4140-4164. https://doi.org/10.3390/v6114140
Chicago/Turabian StyleNiederer, Heather A., and Charles R. M. Bangham. 2014. "Integration Site and Clonal Expansion in Human Chronic Retroviral Infection and Gene Therapy" Viruses 6, no. 11: 4140-4164. https://doi.org/10.3390/v6114140
APA StyleNiederer, H. A., & Bangham, C. R. M. (2014). Integration Site and Clonal Expansion in Human Chronic Retroviral Infection and Gene Therapy. Viruses, 6(11), 4140-4164. https://doi.org/10.3390/v6114140