
Respiratory Syncytial Virus Persistence in Murine Macrophages Impairs IFN-β Response but Not Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Virus
2.2. Confirmation of Persistent RSV Infection in MΦP
2.3. Western Blot
2.4. Subcellular Fractionation
2.5. Supernatant Collection and mRNA Extraction
2.6. Quantitation of IFN-β by ELISA
2.7. Quantitative RT-PCR
2.8. Macrophage Treatment with Recombinant IFN-β or Supernatants
2.9. Statistics
3. Results
3.1. Persistent RSV Infection is Maintained throughout Passages of MΦP
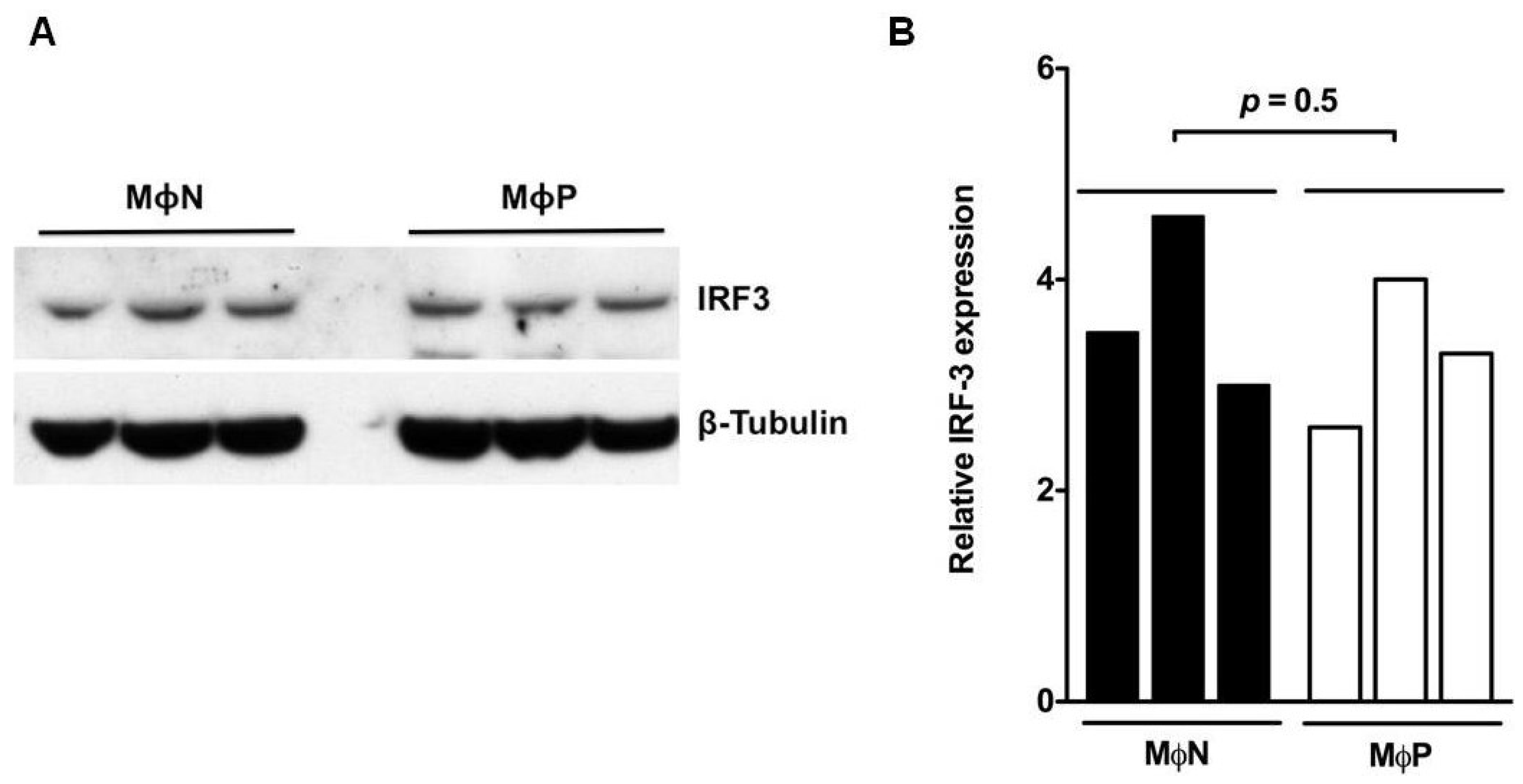
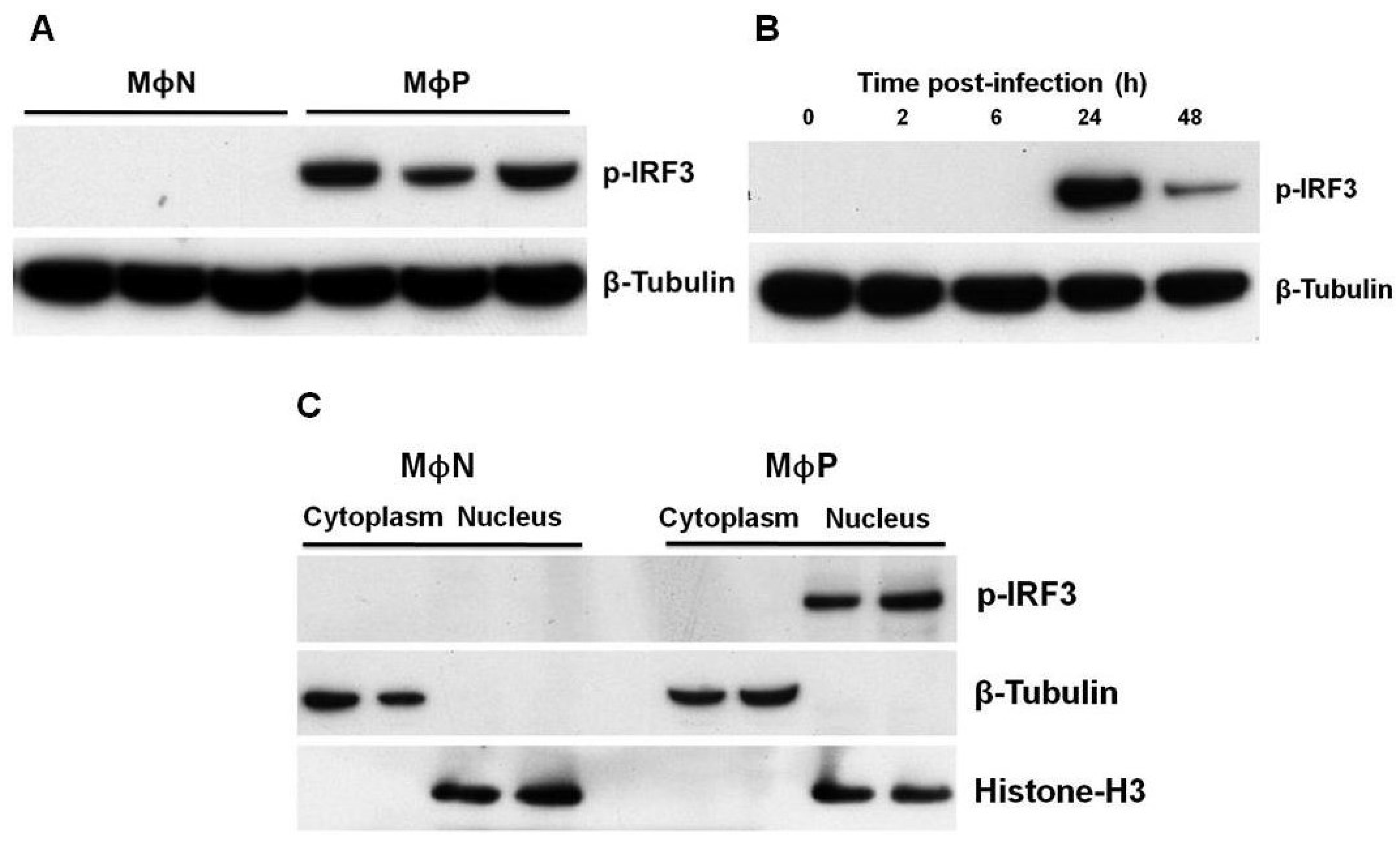
3.2. IRF3 was Constitutively Expressed in Its Phosphorylated Form in MΦP


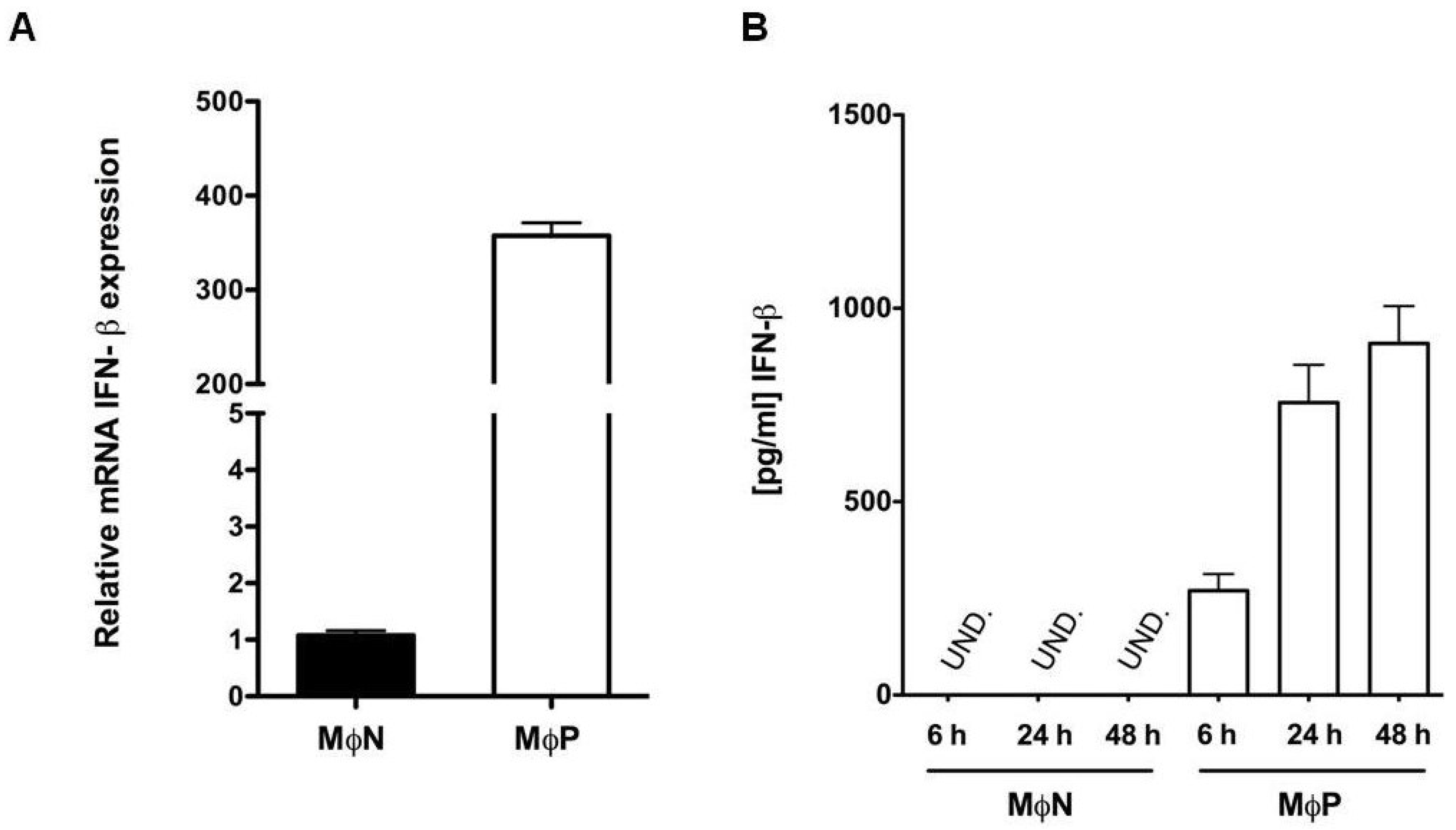
3.3. Persistent RSV Infection Induced Constitutive Expression of IFN-β

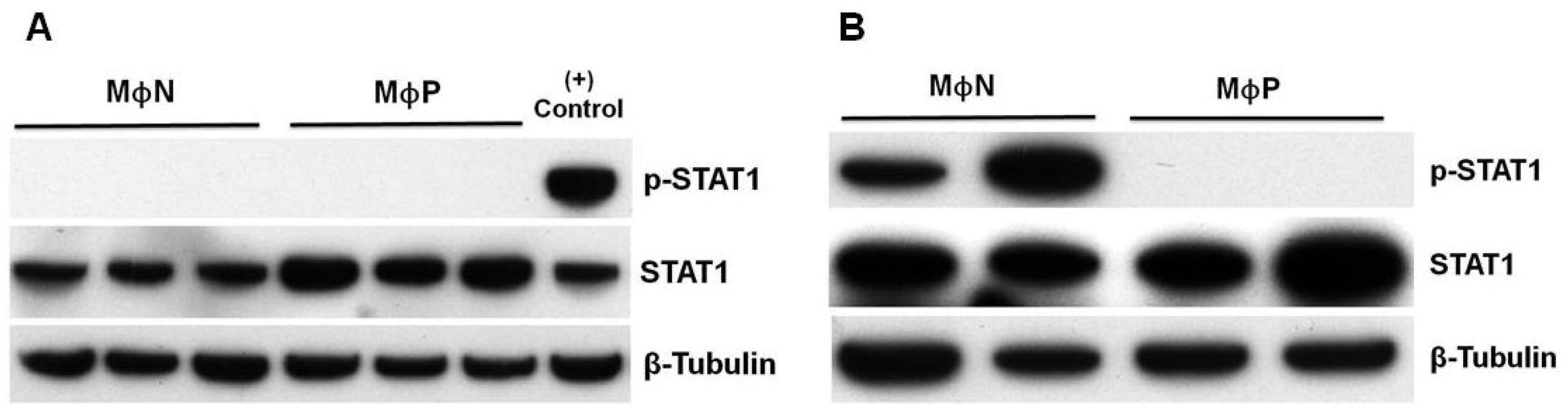
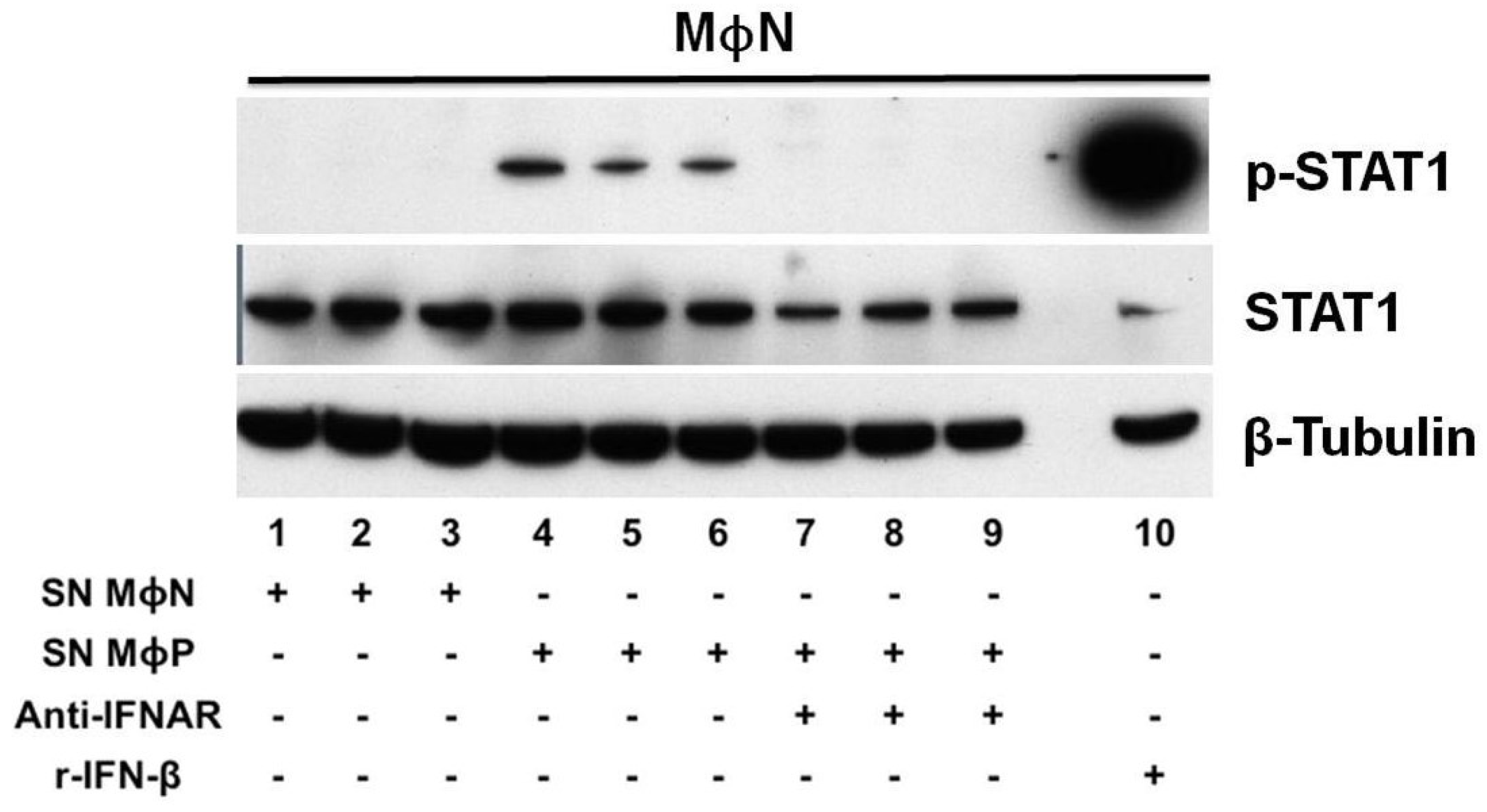
3.4. Persistent RSV Infection Impaired STAT1 Activation and ISGs Expression in Response to IFN-I



4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Hall, C.B.; Simőes, E.A.; Anderson, L.J. Clinical and epidemiologic features of respiratory syncytial virus. Curr. Top. Microbiol. Immunol. 2013, 372, 39–57. [Google Scholar] [PubMed]
- Bardach, A.; Rey-Ares, L.; Cafferata, M.L.; Cormick, G.; Romano, M.; Ruvinsky, S.; Savy, V. Systematic review and meta-analysis of respiratory syncytial virus infection epidemiology in Latin America. Rev. Med. Virol. 2014, 24, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Staat, M.A. Respiratory syncytial virus infections in children. Semin. Respir. Infect. 2002, 17, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Parrott, R.H.; Kim, H.W.; Brandt, C.D.; Chanock, R.M. Respiratory syncytial virus in infants and children. Prev. Med. 1974, 3, 473–480. [Google Scholar] [CrossRef]
- Sigurs, N.; Bjarnason, R.; Sigurbergsson, F.; Kjellman, B. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am. J. Respir. Crit. Care Med. 2000, 16, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Piedimonte, G. Respiratory syncytial virus and asthma: Speed-dating or long-term relationship? Curr. Opin. Pediatr. 2013, 25, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Zomer-Kooijker, K.; van der Ent, C.K.; Ermers, M.J.; Uiterwaal, C.S.; Rovers, M.M.; Bont, L.J.; RSV Corticosteroid Study Group. Increased risk of wheeze and decreased lung function after respiratory syncytial virus infection. PLoS ONE 2014, 9, e87162. [Google Scholar] [PubMed]
- Mejías, A.; Chávez-Bueno, S.; Ramilo, O. Respiratory syncytial virus pneumonia: Mechanisms of inflammation and prolonged airway hyperresponsiveness. Curr. Opin. Infect. Dis. 2005, 18, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Isaia, G.; Teodosiu, O.; Popescu, G.; Athanasiu, P.; Sternberg, I.; Dumitriu, Z. Persistence of viruses in the nasopharynx of apparently healthy children aged 0–5 years. Results of investigations performed in 1982–1983. Virologie 1985, 36, 175–179. [Google Scholar] [PubMed]
- Borg, I.; Rohde, G.; Löseke, S.; Bittscheidt, J.; Schultze-Werninghaus, G.; Stephan, V.; Bufe, A. Evaluation of a quantitative real-time PCR for the detection of respiratory syncytial virus in pulmonary diseases. Eur. Respir. J. 2003, 21, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Cubie, H.A.; Duncan, L.A.; Marshall, L.A.; Smith, N.M. Detection of respiratory syncytial virus nucleic acid in archival postmortem tissue from infants. Pediatr. Pathol. Lab. Med. 1997, 17, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, F.; Gibson, L.F.; Piktel, D.; Othumpangat, S.; Piedimonte, G. Respiratory syncytial virus infection in human bone marrow stromal cells. Am. J. Respir. Cell Mol. Biol. 2011, 45, 277–286. [Google Scholar] [PubMed]
- Sarmiento, R.E.; Tirado, R.; Gómez, B. Characteristics of a respiratory syncytial virus persistently infected macrophage-like culture. Virus Res. 2002, 84, 45–58. [Google Scholar] [CrossRef]
- Martínez, I.; Lombardía, L.; Herranz, C.; García-Barreno, B.; Domínguez, O.; Melero, J.A. Cultures of HEp-2 cells persistently infected by human respiratory syncytial virus differ in chemokine expression and resistance to apoptosis as compared to lytic infections of the same cell type. Virology 2009, 388, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, J.; O’Donnell, D.R.; Rohwedder, A.; Openshaw, P.J. Latency and persistence of respiratory syncytial virus despite T cell immunity. Am. J. Respir. Crit. Care Med. 2004, 169, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Estripeaut, D.; Torres, J.P.; Somers, C.S.; Tagliabue, C.; Khokhar, S.; Bhoj, V.G.; Grube, S.M.; Wozniakowski, A.; Gomez, A.M.; Ramilo, O.; et al. Respiratory syncytial virus persistence in the lungs correlates with airway hyperreactivity in the mouse model. J. Infect. Dis. 2008, 198, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.G.; Hayashi, S.; Bramley, A.M.; Hogg, J.C. Persistence of respiratory syncytial virus genome and protein after acute bronchiolitis in guinea pigs. Chest 1994, 105, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Suhara, W.; Fukuhara, Y.; Fukuda, M.; Nishida, E.; Fujita, T. Direct triggering of the type I interferon system by virus infection: Activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998, 17, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Wathelet, M.G.; Lin, C.H.; Parekh, B.S.; Ronco, L.V.; Howley, P.M.; Maniatis, T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell 1998, 1, 507–518. [Google Scholar] [CrossRef]
- Borrow, P.; Martínez-Sobrido, L.; de la Torre, J.C. Inhibition of the type I interferon antiviral response during arenavirus infection. Viruses 2010, 2, 2443–2480. [Google Scholar] [CrossRef] [PubMed]
- Foy, E.; Li, K.; Wang, C.; Sumpter, R., Jr.; Ikeda, M.; Lemon, S.M.; Gale, M., Jr. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 2003, 300, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Maniatis, T. Negative regulation of interferon-β gene expression during acute and persistent virus infections. PLoS ONE 2011, 6, e20681. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M. Master sensors of pathogenic RNA - RIG-I like receptors. Immunobiology 2013, 218, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Conzelmann, K.K. Transcriptional activation of alpha/beta interferon genes: Interference by nonsegmented negative-strand RNA viruses. J. Virol. 2005, 79, 5241–5248. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ma, G.; Lin, C.H.; Orr, M.; Wathelet, M.G. Mechanism for transcriptional synergy between interferon regulatory factor (IRF)-3 and IRF-7 in activation of the interferon-beta gene promoter. Eur. J. Biochem. 2004, 271, 3693–3703. [Google Scholar] [CrossRef] [PubMed]
- Kraus, T.A.; Lau, J.F.; Parisien, J.P.; Horvath, C.M. A hybrid IRF9-STAT2 protein recapitulates interferon-stimulated gene expression and antiviral response. J. Biol. Chem. 2003, 278, 13033–13038. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef] [PubMed]
- Spann, K.M.; Tran, K.C.; Chi, B.; Rabin, R.L.; Collins, P.L. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J. Virol. 2004, 78, 4363–4369. [Google Scholar] [CrossRef] [PubMed]
- Barik, S. Respiratory syncytial virus mechanisms to interfere with type 1 interferons. Curr. Top. Microbiol. Immunol. 2013, 372, 173–191. [Google Scholar] [PubMed]
- Lo, M.S.; Brazas, R.M.; Holtzman, M.J. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J. Virol. 2005, 79, 9315–9319. [Google Scholar] [CrossRef] [PubMed]
- Spann, K.M.; Tran, K.C.; Collins, P.L. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J. Virol. 2005, 79, 5353–5362. [Google Scholar] [CrossRef] [PubMed]
- Swedan, S.; Musiyenko, A.; Barik, S. Respiratory syncytial virus nonstructural proteins decrease levels of multiple members of the cellular interferon pathways. J. Virol. 2009, 83, 9682–9693. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, T.; Pang, L.; Li, K.; Garofalo, R.P.; Casola, A.; Bao, X. A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J. Gen. Virol. 2011, 92, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Payment, P.; Trudel, M. Methods and Techniques in Virology; Marcel Dekker: New York, NY, USA, 1993; Volume 36–37, pp. 73–74. [Google Scholar]
- Gaona, J.; Santiago-Olivares, C.; Ortega, E.; Gómez, B. Respiratory syncytial virus persistence in macrophages upregulates Fcgamma receptors expression. Viruses 2014, 6, 624–639. [Google Scholar] [CrossRef] [PubMed]
- Senft, A.P.; Taylor, R.H.; Lei, W.; Campbell, S.A.; Tipper, J.L.; Martinez, M.J.; Witt, T.L.; Clay, C.C.; Harrod, K.S. Respiratory syncytial virus impairs macrophage IFN-alpha/beta- and IFN-gamma-stimulated transcription by distinct mechanisms. Am. J. Respir. Cell Mol. Biol. 2010, 42, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Pletneva, L.M.; Haller, O.; Porter, D.D.; Prince, G.A.; Blanco, J.C. Induction of type I interferons and interferon-inducible Mx genes during respiratory syncytial virus infection and reinfection in cotton rats. J. Gen. Virol. 2008, 89, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Zhao, Y.; Kalita, M.; Edeh, C.B.; Paessler, S.; Casola, A.; Teng, M.N.; Garofalo, R.P.; Brasier, A.R. CDK9-dependent transcriptional elongation in the innate interferon-stimulated gene response to respiratory syncytial virus infection in airway epithelial cells. J. Virol. 2013, 87, 7075–7092. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Golovkina, T. Common threads in persistent viral infections. J. Virol. 2010, 84, 4116–4123. [Google Scholar] [CrossRef] [PubMed]
- Chonmaitree, T.; Roberts, N.J., Jr.; Douglas, R.G., Jr.; Hall, C.B.; Simons, R.L. Interferon production by human mononuclear leukocytes: Differences between respiratory syncytial virus and influenza viruses. Infect. Immun. 1981, 32, 300–303. [Google Scholar] [PubMed]
- Guerrero-Plata, A.; Casola, A.; Suarez, G.; Yu, X.; Spetch, L.; Peeples, M.E.; Garofalo, R.P. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2006, 34, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Tran, K.C.; Teng, M.N. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J. Virol. 2009, 83, 3734–3742. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Majumdar, T.; Dhar, J.; Chattopadhyay, S.; Bandyopadhyay, S.K.; Verbovetskaya, V.; Sen, G.C.; Barik, S. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res. 2013, 23, 1025–1042. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Yang, P.; Tang, Y.; Zhao, D. A respiratory syncytial virus persistent-infected cell line system reveals the involvement of SOCS1 in the innate antiviral response. Virol. Sin. 2015, 30, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Toledo, E.; Gómez, B. National Autonomous University of Mexico (UNAM); Ciudad Universitaria: D.F., Mexico, 2014. [Google Scholar]
- Génin, P.; Algarté, M.; Roof, P.; Lin, R.; Hiscott, J. Regulation of RANTES chemokine gene expression requires cooperativity between NF-kappa B and IFN-regulatory factor transcription factors. J. Immunol. 2000, 164, 5352–5361. [Google Scholar] [CrossRef] [PubMed]
- Jie, Z.; Dinwiddie, D.L.; Senft, A.P.; Harrod, K.S. Regulation of STAT signaling in mouse bone marrow derived dendritic cells by respiratory syncytial virus. Virus Res. 2011, 156, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, M.; Shi, L.; Varga, S.M.; Barik, S.; Behlke, M.A.; Look, D.C. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology 2006, 344, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Midulla, F.; Villani, A.; Panuska, J.R.; Dab, I.; Kolls, J.K.; Merolla, R.; Ronchetti, R. Respiratory syncytial virus lung infection in infants: Immunoregulatory role of infected alveolar macrophages. J. Infect. Dis. 1993, 168, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Panuska, J.R.; Cirino, N.M.; Midulla, F.; Despot, J.E.; McFadden, E.R., Jr.; Huang, Y.T. Productive infection of isolated human alveolar macrophages by respiratory syncytial virus. J. Clin. Investig. 1990, 83, 113–119. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera-Toledo, E.; Torres-González, L.; Gómez, B. Respiratory Syncytial Virus Persistence in Murine Macrophages Impairs IFN-β Response but Not Synthesis. Viruses 2015, 7, 5361-5374. https://doi.org/10.3390/v7102879
Rivera-Toledo E, Torres-González L, Gómez B. Respiratory Syncytial Virus Persistence in Murine Macrophages Impairs IFN-β Response but Not Synthesis. Viruses. 2015; 7(10):5361-5374. https://doi.org/10.3390/v7102879
Chicago/Turabian StyleRivera-Toledo, Evelyn, Laura Torres-González, and Beatriz Gómez. 2015. "Respiratory Syncytial Virus Persistence in Murine Macrophages Impairs IFN-β Response but Not Synthesis" Viruses 7, no. 10: 5361-5374. https://doi.org/10.3390/v7102879
APA StyleRivera-Toledo, E., Torres-González, L., & Gómez, B. (2015). Respiratory Syncytial Virus Persistence in Murine Macrophages Impairs IFN-β Response but Not Synthesis. Viruses, 7(10), 5361-5374. https://doi.org/10.3390/v7102879