
Vector-Enabled Metagenomic (VEM) Surveys Using Whiteflies (Aleyrodidae) Reveal Novel Begomovirus Species in the New and OldWorlds

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Whitefly Collection and Processing for Metagenomic Sequencing
2.2. PCR Assay to Confirm Whitefly Species and Phylogenetic Group
2.3. Metagenomic Data Analysis and Genome Completion
2.4. Genome Sequence Analysis
2.4.1. Pairwise Comparisons
2.4.2. Phylogenetic Analysis
3. Results
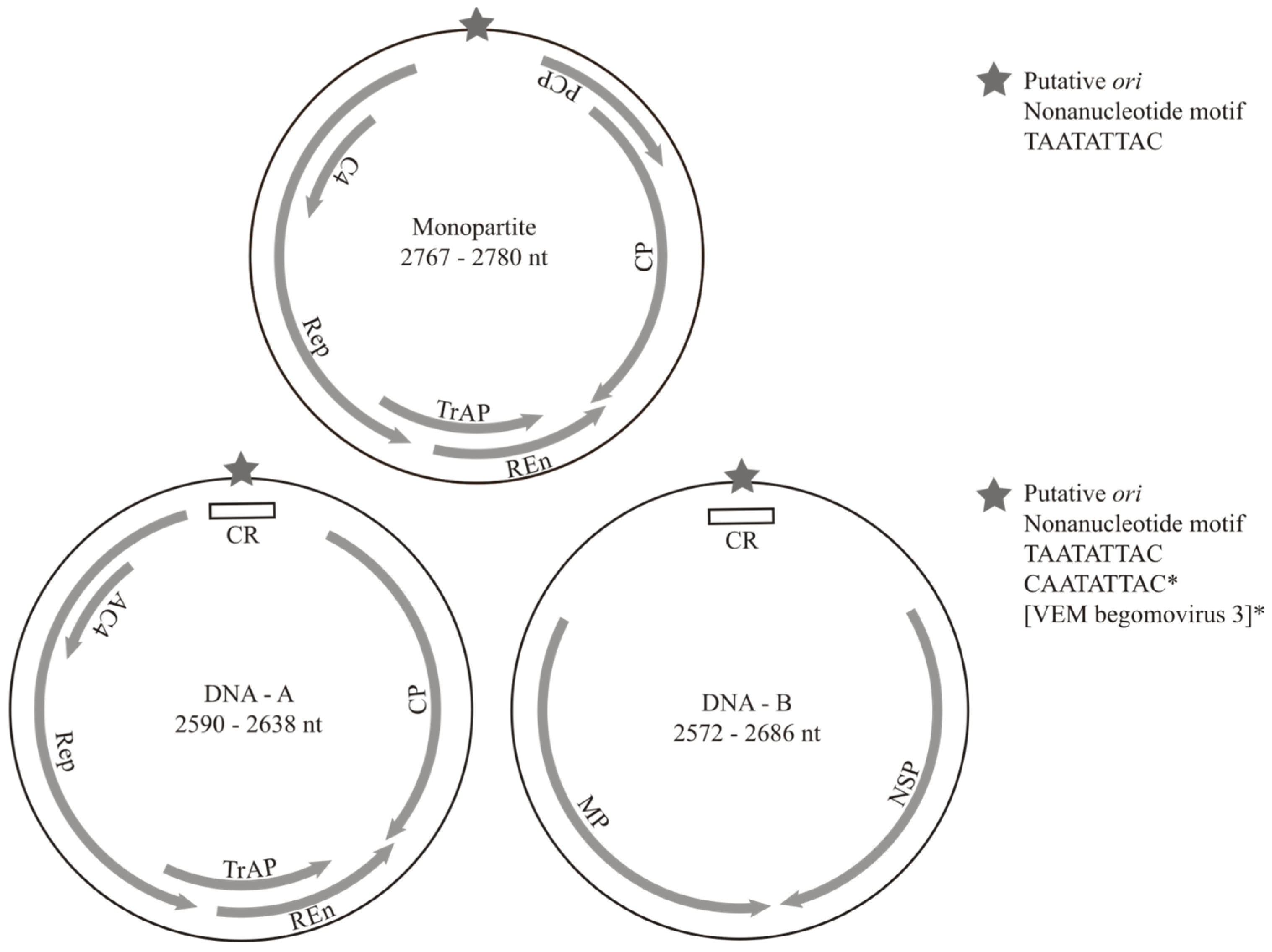
3.1. Overview

{kind=link}
{kind=link}
| Library | Crop/Plant | Location | Collection Date | Whiteflies a |
|---|---|---|---|---|
| CA_S | Squash | Imperial Valley, California, USA | June 2012 | B. tabaci (MEAMI) |
| Guat_T1 | Tomato | Salamá, Baja Verapaz, Guatemala | January 2012 | B. tabaci (NW), T. vaporariorum |
| Guat_T2 | Tomato | El Progreso, Jutiapa, Guatemala | January 2012 | B. tabaci (NW), T. vaporariorum |
| Guat_S | Squash | Teculután, Zacapa, Guatemala | January 2012 | B. tabaci (MEAM1) |
| Spain_B | Bean | Torrox, Málaga, Spain | September 2011 | B. tabaci (MED) |
| Spain_S | Squash | Torrox, Málaga, Spain | September 2011 | B. tabaci (MED) |
| Spain_T | Tomato | Torrox, Málaga, Spain | September 2011 | B. tabaci (MED) |
| Spain_W1 | European black nightshade | Algarrobo-Costa, Málaga, Spain | September 2011 | B. tabaci (MED) |
| Spain_W2 | European black nightshade | Algarrobo-Costa, Málaga, Spain | September 2011 | B. tabaci (MED) |
| Israel_T | Tomato | Bet Dagan, Israel | March 2011 | B. tabaci (MEAM1) |
| Israel_S | Squash | Bet Dagan, Israel | March 2011 | B. tabaci (MEAM1) |
| PR_T1 | Tomato | Santa Isabel, Puerto Rico | November 2010 | B. tabaci (MEAM1) |
| PR_T2 | Tomato | Santa Isabel, Puerto Rico | November 2010 | B. tabaci (MEAM1) |
| PR_P | Pumpkin | Santa Isabel, Puerto Rico | November 2010 | B. tabaci (MEAM1) |
| PR_E | Eggplant | Santa Isabel, Puerto Rico | November 2010 | B. tabaci (MEAM1) |

| Library | Genome a (Accession) | Best match b (% Identity)/Accession Number |
|---|---|---|
| CA_S | VEM Squash leaf curl virus DNA-A * (KT099117) | SLCV, Imperial Valley, CA (99%)/DQ285016 |
| VEM Squash leaf curl virus DNA-B * (KT099159) | SLCV, Imperial Valley, CA (95%)/DQ285017 | |
| Guat_S | VEM Melon chlorotic leaf curl virus DNA-A * (KT099118) | MCLCuV, Guatemala (96%)/AF325497 |
| VEM Melon chlorotic leaf curl virus DNA-B * (KT099160) | MCLCuV, Guatemala (96%)/AF325498 | |
| VEM Tomato severe leaf curl virus DNA-A * (KT099119) | ToSLCV, Guatemala and Mexico (97%)/AF130415/DQ347947 | |
| VEM Sida golden mosaic Honduras virus 1a DNA-A (KT099120) | SiGMHV, Honduras (94%)/Y11097 | |
| VEM Sida golden mosaic Honduras virus 1b DNA-A (KT099121) | SiGMHV, Honduras (94%)/Y11098 | |
| VEM Sida golden mosaic Honduras virus 1 DNA-B (KT099161) | SiGMHV, Honduras (82%)/AJ250731 | |
| VEM begomovirus 1a DNA-A (KT099122) | BICV, Mexico (80%)/JX827487 | |
| VEM begomovirus 1b DNA-A (KT099123) | BICV, Mexico (80%)/JX827488 | |
| VEM begomovirus 1c DNA-A (KT099124) | BICV, Mexico (80%)/JX827489 | |
| VEM begomovirus 2 DNA-A (KT099125) | SiYMV, Cuba (84%)/JN411687 | |
| VEM begomovirus 3a DNA-A (KT099126) | CoYSV, Mexico (83%)/DQ875868 | |
| VEM begomovirus 3b DNA-A (KT099127) | CoYSV, Mexico (83%)/DQ875869 | |
| VEM begomovirus 3a DNA-B (KT099162) | SiGMV/SiYVV, Honduras (82%)/AJ250731/Y11100 | |
| VEM begomovirus 3b DNA-B (KT099163) | SiGMV/SiYVV, Honduras (82%)/AJ250731/Y11101 | |
| VEM begomovirus 4 DNA-A (KT099128) | JMV, Dominican Republic and Florida (85%)/KJ174330/KF998097 | |
| Guat_T2 | VEM Tomato severe leaf curl virus DNA-A * (KT099129) | ToSLCV, Mexico (99%)/DQ347947 |
| VEM Tomato mosaic Havana virus DNA-A * (KT099130) | ToMHV, Nicaragua (99%)/EF088197 | |
| VEM Tomato mosaic Havana virus DNA-B * (KT099164) | ToMHV, Cuba (92%)/Y14875 | |
| Israel_S | VEM Squash leaf curl virus DNA-A (KT099131) | SLCV, Israel (99%)/KM595109 |
| VEM Squash leaf curl v irus DNA-B * (KT099165) | SLCV, Palestine (99%)/KC441466 | |
| VEM Cotton leaf curl Gezira virus DNA-A (KT099132) | CLCuGV, Egypt and Pakistan (94%)/AF155064/FR751146 | |
| PR_T1 | VEM Sweet potato leaf curl virus (KT099133) | SPLCV/SPLCLaV, Brazil and Spain (91%)/FJ969833/EU839579 |
| PR_T2 | VEM begomovirus 5a DNA-A (KT099134) | WfVEM1/SiGMLV, Florida and Jamaica (90%)/HM859902/HQ009522 |
| VEM begomovirus 5b DNA-A (KT099135) | WfVEM1/SiGMLV, Florida and Jamaica (90%)/HM859902/HQ009523 | |
| VEM begomovirus 5c DNA-A (KT099136) | WfVEM1/SiGMLV, Florida and Jamaica (90%)/HM859902/HQ009524 | |
| VEM begomovirus 5d DNA-A (KT099137) | WfVEM1/SiGMLV, Florida and Jamaica (90%)/HM859902/HQ009525 | |
| VEM begomovirus 5e DNA-A (KT099138) | WfVEM1/SiGMLV, Florida and Jamaica (90%)/HM859902/HQ009526 | |
| VEM begomovirus 5 DNA-B (KT099166) | SiYVV, Cuba (79%)/HE806449 | |
| VEM begomovirus 6b DNA-B (KT099167) | SiGMFV, Florida (78%)/HE806443 | |
| PR_P | VEM begomovirus 6 DNA-A (KT099139) | SiYMV, Cuba (88%)/HQ822123 |
| VEM begomovirus 6a DNA-B (KT099168) | SiGMFV, Florida (78%)/HE806443 | |
| VEM Sweet potato leaf curl virus (KT099140) | SPLCV, Puerto Rico (93%)/DQ644563 | |
| VEM Macroptilium mosaic Puerto Rico virus DNA-A* (KT099141) | MaMPRV, Puerto Rico (99%)/AF449192 | |
| VEM Macroptilium mosaic Puerto Rico virus DNA-B* (KT099169) | MaMPRV, Puerto Rico (98%)/AY044134 | |
| PR_E | VEM begomovirus 5f DNA-A (KT099142) | WfVEM1/SiGMLV, Florida and Jamaica (90%)/HM859902/HQ009522 |
| VEM Sweet potato leaf curl virus (KT099143) | SPLCV/SPLCLaV, Brazil, Puerto Rico and Spain (91%)/FJ969833/DQ644563/EU839579 | |
| Spain_B | VEM Sweet potato leaf curl virus 1 (KT099144) | SPLCV, Spain (97%)/EU856364 |
| Spain_S | VEM Sweet potato leaf curl virus 2 (KT099145) | SPLCESV, Spain (94%)/EF456743 |
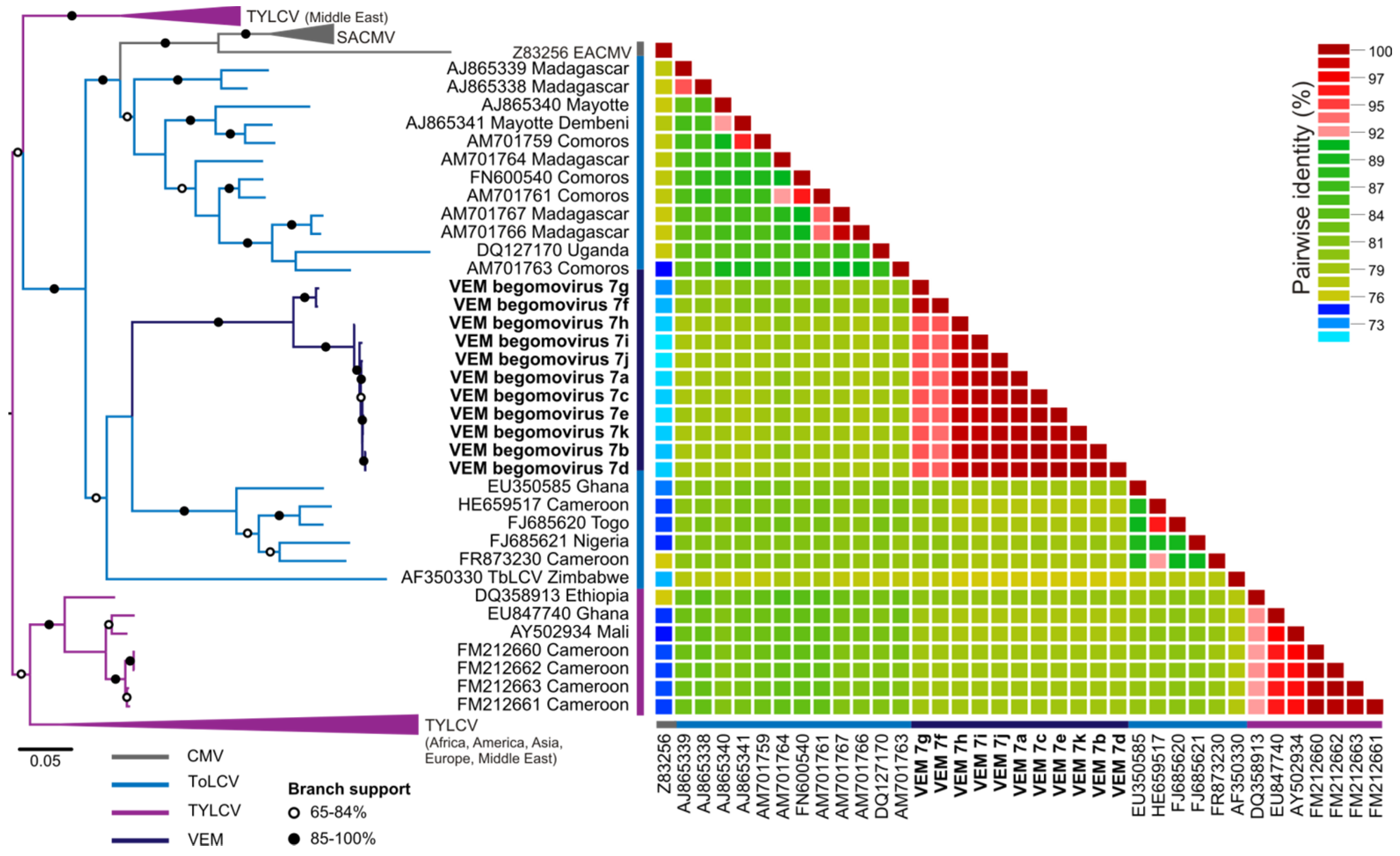
| Spain_T | VEM begomovirus 7a (KT099146) | ToLCNamV, Madagascar (81%)/AM701764 |
| VEM begomovirus 7b (KT099147) | ToLCNamV, Madagascar (81%)/AM701765 | |
| VEM begomovirus 7c (KT099148) | ToLCNamV, Madagascar (81%)/AM701766 | |
| VEM begomovirus 7d (KT099149) | ToLCNamV, Madagascar (81%)/AM701767 | |
| VEM begomovirus 7e (KT099150) | ToLCNamV, Madagascar (81%)/AM701768 | |
| VEM begomovirus 7f (KT099151) | ToLCKMV, Comoros (82%)/AM701759 | |
| VEM begomovirus 7g (KT099152) | ToLCKMV, Comoros (81%)/AM701759 | |
| Spain_W1 | VEM begomovirus 7h (KT099153) | ToLCNamV, Madagascar (81%)/AM701764 |
| VEM begomovirus 7i (KT099154) | ToLCNamV, Madagascar (81%)/AM701765 | |
| VEM begomovirus 7j (KT099155) | ToLCNamV, Madagascar (81%)/AM701766 | |
| VEM begomovirus 7k (KT099156) | ToLCNamV, Madagascar (81%)/AM701767 | |
| VEM Tomato yellow leaf curl virus-[Almeria] 1 * (KT099157) | TYLCV, Spain (99%)/AJ489258 | |
| Spain_W2 | VEM Tomato yellow leaf curl virus-[Almeria] 1a* (KT099158) | TYLCV, Spain (99%)/AJ489258 |
3.2. Bipartite Begomoviruses
3.3. Monopartite Begomoviruses

4. Discussion
4.1. Diversity Revealed by VEM
4.2. Begomovirus Biogeography
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Varsani, A.; Navas-Castillo, J.; Moriones, E.; Hernandez-Zepeda, C.; Idris, A.; Brown, J.K.; Murilo Zerbini, F.; Martin, D.P. Establishment of three new genera in the family Geminiviridae: Becurtovirus, Eragrovirus and Turncurtovirus. Arch. Virol. 2014, 159, 2193–2203. [Google Scholar] [PubMed]
- Mansoor, S.; Briddon, R.W.; Zafar, Y.; Stanley, J. Geminivirus disease complexes: An emerging threat. Trends Plant Sci. 2003, 8, 128–134. [Google Scholar] [CrossRef]
- Polston, J.E.; Anderson, P.K. The emergence of whitefly-transmitted geminiviruses in tomato in the Western Hemisphere. Plant Dis. 1997, 81, 1358–1369. [Google Scholar] [CrossRef]
- Alabi, O.J.; Kumar, P.L.; Naidu, R.A. Cassava mosaic disease: A curse to food security in Sub-Saharan Africa. APSnet Featur. 2011. [Google Scholar] [CrossRef]
- Leke, W.; Mignouna, D.; Brown, J.; Kvarnheden, A. Begomovirus disease complex: Emerging threat to vegetable production systems of West and Central Africa. Agric. Food Secur. 2015, 4, 1. [Google Scholar] [CrossRef]
- Legg, J.P.; Fauquet, C.M. Cassava mosaic geminiviruses in Africa. Plant Mol. Biol. 2004, 56, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.D.; Robinson, D.J. Natural genomic and antigenic variation in whitefly-transmitted geminiviruses (Begomoviruses). Annu. Rev. Phytopathol. 1999, 37, 369–398. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, C. Geminivirus DNA replication. Cell. Mol. Life Sci. 1999, 56, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Briddon, R.W.; Stanley, J. Subviral agents associated with plant single-stranded DNA viruses. Virology 2006, 344, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X. Advances in understanding begomovirus satellites. Annu. Rev. Phytopathol. 2013, 51, 357–381. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.; Coombs, S.; Revill, P.; Harding, R.; Vu, M.; Dale, J. Molecular characterization of begomoviruses and DNA satellites from Vietnam: Additional evidence that the New World geminiviruses were present in the Old World prior to continental separation. J. Gen. Virol. 2008, 89, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Seal, S.E.; VandenBosch, F.; Jeger, M.J. Factors influencing begomovirus evolution and their increasing global significance: Implications for sustainable control. Crit. Rev. Plant Sci. 2006, 25, 23–46. [Google Scholar] [CrossRef]
- Brown, J.K. The Bemisia tabaci complex: Genetic and phenotypic variability drives begomovirus spread and virus diversification. APSnet Featur. 2007. [Google Scholar] [CrossRef]
- Navas-Castillo, J.; Fiallo-Olive, E.; Sanchez-Campos, S. Emerging virus diseases transmitted by whiteflies. Annu. Rev. Phytopathol. 2011, 49, 219–248. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Holmes, E.C. Phylogenetic evidence for rapid rates of molecular evolution in the single-stranded DNA begomovirus Tomato yellow leaf curl virus. J. Virol. 2008, 82, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Holmes, E.C. Validation of high rates of nucleotide substitution in geminiviruses: Phylogenetic evidence from East African cassava mosaic viruses. J. Gen. Virol. 2009, 90, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Lett, J.M.; Varsani, A.; Martin, D.P. Widely conserved recombination patterns among single-stranded DNA viruses. J. Virol. 2009, 83, 2697–2707. [Google Scholar] [CrossRef] [PubMed]
- Jeske, H.; Lutgemeier, M.; Preiss, W. DNA forms indicate rolling circle and recombination-dependent replication of Abutilon mosaic virus. EMBO J. 2001, 20, 6158–6167. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Biagini, P.; Lefeuvre, P.; Golden, M.; Roumagnac, P.; Varsani, A. Recombination in eukaryotic single stranded DNA viruses. Viruses 2011, 3, 1699–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briddon, R.W.; Brown, J.K.; Moriones, E.; Stanley, J.; Zerbini, M.; Zhou, X.; Fauquet, C.M. Recommendations for the classification and nomenclature of the DNA-beta satellites of begomoviruses. Arch. Virol. 2008, 153, 763–781. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, S.; Zafar, Y.; Briddon, R.W. Geminivirus disease complexes: The threat is spreading. Trends Plant Sci. 2006, 11, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.; Lima, A.; Rocha, C.; Castillo-Urquiza, G.; Alves-Junior, M.; Zerbini, F. Recombination and pseudorecombination driving the evolution of the begomoviruses Tomato severe rugose virus (ToSRV) and Tomato rugose mosaic virus (ToRMV): Two recombinant DNA-A components sharing the same DNA-B. Virol. J. 2014, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.T.M.; Sobrinho, R.R.; González-Aguilera, J.; Rocha, C.S.; Silva, S.J.C.; Xavier, C.A.D.; Silva, F.N.; Duffy, S.; Zerbini, F.M. Synonymous site variation due to recombination explains higher genetic variability in begomovirus populations infecting non-cultivated hosts. J. Gen. Virol. 2013, 94, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.M.; Mills-Lujan, K.; Martin, K.; Brown, J.K. Melon chlorotic leaf curl virus: Characterization and differential reassortment with closest relatives reveal adaptive virulence in the Squash leaf curl virus clade and host shifting by the host-restricted Bean calico mosaic virus. J. Virol. 2008, 82, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Andres, S.; Monci, F.; Navas-Castillo, J.; Moriones, E. Begomovirus genetic diversity in the native plant reservoir Solanum nigrum: Evidence for the presence of a new virus species of recombinant nature. Virology 2006, 350, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Martin, D.P.; Hoareau, M.; Naze, F.; Delatte, H.; Thierry, M.; Varsani, A.; Becker, N.; Reynaud, B.; Lett, J.M. Begomovirus “melting pot” in the south-west Indian Ocean islands: Molecular diversity and evolution through recombination. J. Gen. Virol. 2007, 88, 3458–3468. [Google Scholar] [CrossRef] [PubMed]
- Monci, F.; Sánchez-Campos, S.; Navas-Castillo, J.; Moriones, E. A natural recombinant between the geminiviruses Tomato yellow leaf curl Sardinia virus and Tomato yellow leaf curl virus exhibits a novel pathogenic phenotype and is becoming prevalent in spanish populations. Virology 2002, 303, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Moriones, E. Recombination as a motor of host switches and virus emergence: Geminiviruses as case studies. Curr. Opin. Virol. 2015, 10, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Denholm, I.; Cahill, M.; Dennehy, T.J.; Horowitz, A.R. Challenges with managing insecticide resistance in agricultural pests, exemplified by the whitefly Bemisia tabaci. Philos. Trans. R. Soc. Lond. B 1998, 353, 1757–1767. [Google Scholar] [CrossRef]
- De Barro, P.J.; Liu, S.S.; Boykin, L.M.; Dinsdale, A.B. Bemisia tabaci: A statement of species status. Annu. Rev. Entomol. 2011, 56, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Polston, J.E.; de Barro, P.; Boykin, L.M. Transmission specificities of plant viruses with the newly identified species of the Bemisia tabaci species complex. Pest Manag. Sci. 2014, 70, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.R.V.; Henneberry, T.J.; Anderson, P. History, current status, and collaborative research projects for Bemisia tabaci. Crop Prot. 2001, 20, 709–723. [Google Scholar] [CrossRef]
- Silva, S.J.C.; Castillo-Urquiza, G.P.; Hora Júnior, B.T.; Assunção, I.P.; Lima, G.S.A.; Pio-Ribeiro, G.; Mizubuti, E.S.G.; Zerbini, F.M. High genetic variability and recombination in a begomovirus population infecting the ubiquitous weed Cleome affinis in northeastern Brazil. Arch. Virol. 2011, 156, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.J.C.; Castillo-Urquiza, G.P.; Hora-Júnior, B.T.; Assunção, I.P.; Lima, G.S.A.; Pio-Ribeiro, G.; Mizubuti, E.S.G.; Zerbini, F.M. Species diversity, phylogeny and genetic variability of begomovirus populations infecting leguminous weeds in northeastern Brazil. Plant Pathol. 2012, 61, 457–467. [Google Scholar] [CrossRef]
- Rocha, C.S.; Castillo-Urquiza, G.P.; Lima, A.T.M.; Silva, F.N.; Xavier, C.A.D.; Hora-Júnior, B.T.; Beserra-Júnior, J.E.A.; Malta, A.W.O.; Martin, D.P.; Varsani, A.; et al. Brazilian begomovirus populations are highly recombinant, rapidly evolving, and segregated based on geographical location. J. Virol. 2013, 87, 5784–5799. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Shepherd, D.N.; Monjane, A.L.; Owor, B.E.; Erdmann, J.B.; Rybicki, E.P.; Peterschmitt, M.; Briddon, R.W.; Markham, P.G.; Oluwafemi, S.; et al. Recombination, decreased host specificity and increased mobility may have driven the emergence of maize streak virus as an agricultural pathogen. J. Gen. Virol. 2008, 89, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Stobbe, A.H.; Roossinck, M.J. Plant virus metagenomics: What we know and why we need to know more. Front. Plant Sci. 2014, 5, 150. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Zerbini, F.M.; Navas-Castillo, J.; Moriones, E.; Ramos-Sobrinho, R.; Silva, J.F.; Fiallo-Olivé, E.; Briddon, R.; Hernández-Zepeda, C.; Idris, A.; et al. Revision of Begomovirus taxonomy based on pairwise sequence comparisons. Arch. Virol. 2015, 160, 1593–1619. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.; Bamford, D.H.; Breitbart, M.; Davison, A.J.; Ghabrial, S.A.; Gorbalenya, A.E.; Knowles, N.J.; Krell, P.; et al. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2015). Arch. Virol. 2015, 160, 1837–1850. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.R.; Hagen, C.; Lucas, W.J.; Gilbertson, R.L. Exploiting chinks in the plant’s armor: Evolution and emergence of geminiviruses. Annu. Rev. Phytopathol. 2005, 43, 361–394. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.F.; Duffy, S.; Polston, J.E.; Bixby, E.; Vallad, G.E.; Breitbart, M. Exploring the diversity of plant DNA viruses and their satellites using vector-enabled metagenomics on whiteflies. PLoS ONE 2011, 6, e19050. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.F.; Willner, D.L.; Lim, Y.W.; Schmieder, R.; Chau, B.; Nilsson, C.; Anthony, S.; Ruan, Y.J.; Rohwer, F.; Breitbart, M. Broad surveys of DNA viral diversity obtained through viral metagenomics of mosquitoes. PLoS ONE 2011, 6, e20579. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Capobianco, H.; Ng, T.F.; Breitbart, M.; Polston, J.E. RNA viral metagenome of whiteflies leads to the discovery and characterization of a whitefly-transmitted carlavirus in North America. PLoS ONE 2014, 9, e86748. [Google Scholar] [CrossRef] [PubMed]
- Inoue-Nagata, A.K.; Albuquerque, L.C.; Rocha, W.B.; Nagata, T. A simple method for cloning the complete begomovirus genome using the bacteriophage φ29 DNA polymerase. J. Virol. Methods 2004, 116, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Shatters, R.G.; Powell, C.A.; Boykin, L.; Liansheng, H.; McKenzie, C.L.; Shatters, R.G.; Powell, C.A.; Boykin, L.; Liansheng, H.; McKenzie, C.L. Improved DNA barcoding method for Bemisia tabaci and related Aleyrodidae: Development of universal and Bemisia tabaci biotype-specific mitochondrial cytochrome c oxidase I polymerase chain reaction primers. J. Econ. Entomol. 2009, 102, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Fu, L.; Sun, S.; Li, W. Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC Bioinform. 2010, 11, 187. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.S.; Kuchie, J.; Duffy, S. Bioinformatic analysis reveals genome size reduction and the emergence of tyrosine phosphorylation site in the movement protein of New World bipartite begomoviruses. PLoS ONE 2014, 9, e111957. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.D.; Swanson, M.M.; Fargette, D. Begomovirus coat protein: Serology, variation and functions. Physiol. Mol. Plant Pathol. 2002, 60, 257–271. [Google Scholar] [CrossRef]
- Lapidot, M.; Gelbart, D.; Gal-On, A.; Sela, N.; Anfoka, G.; Haj Ahmed, F.; Abou-Jawada, Y.; Sobh, H.; Mazyad, H.; Aboul-Ata, A.-A.; et al. Frequent migration of introduced cucurbit-infecting begomoviruses among Middle Eastern countries. Virol. J. 2014, 11, 181. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Duffy, S.; Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef] [PubMed]
- Laufs, J.; Traut, W.; Heyraud, F.; Matzeit, V.; Rogers, S.G.; Schell, J.; Gronenborn, B. In vitro cleavage and joining at the viral origin of replication by the replication initiator protein of Tomato yellow leaf curl virus. Proc. Natl. Acad. Sci. USA 1995, 92, 3879–3883. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.M.; Hiebert, E.; Bird, J.; Brown, J.K. Two newly described begomoviruses of Macroptilium lathyroides and common bean. Phytopathology 2003, 93, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Delatte, H.; Martin, D.P.; Naze, F.; Goldbach, R.; Reynaud, B.; Peterschmitt, M.; Lett, J.-M. South West Indian Ocean islands tomato begomovirus populations represent a new major monopartite begomovirus group. J. Gen. Virol. 2005, 86, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Thierry, M.; Lefeuvre, P.; Hoareau, M.; Péréfarres, F.; Delatte, H.; Reynaud, B.; Martin, D.P.; Lett, J.M. Differential disease phenotype of begomoviruses associated with tobacco leaf curl disease in Comoros. Arch. Virol. 2012, 157, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.; Al-Saleh, M.; Piatek, M.; Al-Shahwan, I.; Ali, S.; Brown, J. Viral metagenomics: Analysis of begomoviruses by Illumina high-throughput sequencing. Viruses 2014, 6, 1219–1236. [Google Scholar] [CrossRef] [PubMed]
- Bedford, I.D.; Briddon, R.W.; Brown, J.K.; Rosell, R.C.; Markham, P.G. Geminivirus transmission and biological characterisation of Bemisia tabaci (Gennadius) biotypes from different geographic regions. Ann. Appl. Biol. 1994, 125, 311–325. [Google Scholar] [CrossRef]
- Höfer, P.; Bedford, I.D.; Markham, P.G.; Jeske, H.; Frischmuth, T. Coat protein gene replacement results in whitefly transmission of an insect nontransmissible geminivirus isolate. Virology 1997, 236, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.C.; Hu, J.S.; Pollston, J.E.; Ullman, D.E.; Hiebert, E. Complete nucleotide sequence of a non-vector transmissible strain of abutilon mosaic geminivirus in Hawaii. Phytopathology 1996, 86, 608–613. [Google Scholar] [CrossRef]
- Skaljac, M.; Zanic, K.; Ban, S.; Kontsedalov, S.; Ghanim, M. Co-infection and localization of secondary symbionts in two whitefly species. BMC Microbiol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Hidayat, S.H.; Rahmayani, E. Transmission of Tomato leaf curl begomovirus by two different species of whitefly (Hemiptera: Aleyrodidae). Plant Pathol. J. 2007, 23, 57–61. [Google Scholar] [CrossRef]
- Czosnek, H.; Ghanim, M.; Ghanim, M. The circulative pathway of begomoviruses in the whitefly vector Bemisia tabaci—insights from studies with Tomato yellow leaf curl virus. Ann. Appl. Biol. 2002, 140, 215–231. [Google Scholar] [CrossRef]
- Reddy, M.S.; Kanakala, S.; Srinivas, K.P.; Hema, M.; Malathi, V.G.; Sreenivasulu, P. Complete genome sequence of a new begomovirus associated with yellow mosaic disease of Hemidesmus indicus in India. Arch. Virol. 2014, 159, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Berrie, L.C.; Rybicki, E.P.; Rey, M.E.C. Complete nucleotide sequence and host range of South African cassava mosaic virus: Further evidence for recombination amongst begomoviruses. J. Gen. Virol. 2001, 82, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Graham, A.; Martin, D.; Roye, M. Molecular characterization and phylogeny of two begomoviruses infecting Malvastrum americanum in Jamaica: Evidence of the contribution of inter-species recombination to the evolution of malvaceous weed-associated begomoviruses from the Northern Caribbean. Virus Genes 2010, 40, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.M.; Abdel-Salam, A.; Brown, J.K. Introduction of the New World Squash leaf curl virus to squash (Cucurbita pepo) in Egypt: A potential threat to important food crops. Plant Dis 2006, 90, 1262. [Google Scholar] [CrossRef]
- Abudy, A.; Sufrin-Ringwald, T.; Dayan-Glick, C.; Guenoune-Gelbart, D.; Livneh, O.; Zaccai, M.; Lapidot, M. Watermelon chlorotic stunt and Squash leaf curl begomoviruses—New threats to cucurbit crops in the Middle East. Isr. J. Plant Sci. 2010, 58, 33–42. [Google Scholar] [CrossRef]
- Briddon, R.W.; Patil, B.L.; Bagewadi, B.; Nawaz-ul-Rehman, M.S.; Fauquet, C.M. Distinct evolutionary histories of the DNA-A and DNA-B components of bipartite begomoviruses. BMC Evol. Biol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Lozano, G.; Trenado, H.P.; Valverde, R.A.; Navas-Castillo, J. Novel begomovirus species of recombinant nature in sweet potato (Ipomoea batatas) and Ipomoea indica: Taxonomic and phylogenetic implications. J. Gen. Virol. 2009, 90, 2550–2562. [Google Scholar] [CrossRef] [PubMed]
- Seah, Y.M.; Rosario, K.; Breitbart, M.; Duffy, S. No clean sweep for the sweepoviruses: Problems with pairwise percent nucleotide identity as a definitive classification tool. Virus Evol. 2015. in review. [Google Scholar]
- Lefeuvre, P.; Martin, D.P.; Harkins, G.; Lemey, P.; Gray, A.J.A.; Meredith, S.; Lakay, F.; Monjane, A.; Lett, J.-M.; Varsani, A.; et al. The spread of Tomato yellow leaf curl virus from the Middle East to the world. PLoS Pathog. 2010, 6, e1001164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melgarejo, T.A.; Kon, T.; Rojas, M.R.; Paz-Carrasco, L.; Zerbini, F.M.; Gilbertson, R.L. Characterization of a New World monopartite begomovirus causing leaf curl disease of tomato in Ecuador and Peru reveals a new direction in geminivirus evolution. J. Virol. 2013, 87, 5397–5413. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Campos, S.; Martinez-Ayala, A.; Marquez-Martin, B.; Aragon-Caballero, L.; Navas-Castillo, J.; Moriones, E. Fulfilling Koch’s postulates confirms the monopartite nature of tomato leaf deformation virus: A begomovirus native to the New World. Virus Res. 2013, 173, 286–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Liu, Q.; Fan, L.; Cui, X.; Zhou, X. Analysis of synonymous codon usage and evolution of begomoviruses. J. Zhejiang Univ. Sci. B 2008, 9, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Nawaz-ul-Rehman, M.S.; Fauquet, C.M. Evolution of geminiviruses and their satellites. FEBS Lett. 2009, 583, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosario, K.; Seah, Y.M.; Marr, C.; Varsani, A.; Kraberger, S.; Stainton, D.; Moriones, E.; Polston, J.E.; Duffy, S.; Breitbart, M. Vector-Enabled Metagenomic (VEM) Surveys Using Whiteflies (Aleyrodidae) Reveal Novel Begomovirus Species in the New and OldWorlds. Viruses 2015, 7, 5553-5570. https://doi.org/10.3390/v7102895
Rosario K, Seah YM, Marr C, Varsani A, Kraberger S, Stainton D, Moriones E, Polston JE, Duffy S, Breitbart M. Vector-Enabled Metagenomic (VEM) Surveys Using Whiteflies (Aleyrodidae) Reveal Novel Begomovirus Species in the New and OldWorlds. Viruses. 2015; 7(10):5553-5570. https://doi.org/10.3390/v7102895
Chicago/Turabian StyleRosario, Karyna, Yee Mey Seah, Christian Marr, Arvind Varsani, Simona Kraberger, Daisy Stainton, Enrique Moriones, Jane E. Polston, Siobain Duffy, and Mya Breitbart. 2015. "Vector-Enabled Metagenomic (VEM) Surveys Using Whiteflies (Aleyrodidae) Reveal Novel Begomovirus Species in the New and OldWorlds" Viruses 7, no. 10: 5553-5570. https://doi.org/10.3390/v7102895
APA StyleRosario, K., Seah, Y. M., Marr, C., Varsani, A., Kraberger, S., Stainton, D., Moriones, E., Polston, J. E., Duffy, S., & Breitbart, M. (2015). Vector-Enabled Metagenomic (VEM) Surveys Using Whiteflies (Aleyrodidae) Reveal Novel Begomovirus Species in the New and OldWorlds. Viruses, 7(10), 5553-5570. https://doi.org/10.3390/v7102895