RNA Virus Evolution via a Quasispecies-Based Model Reveals a Drug Target with a High Barrier to Resistance


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
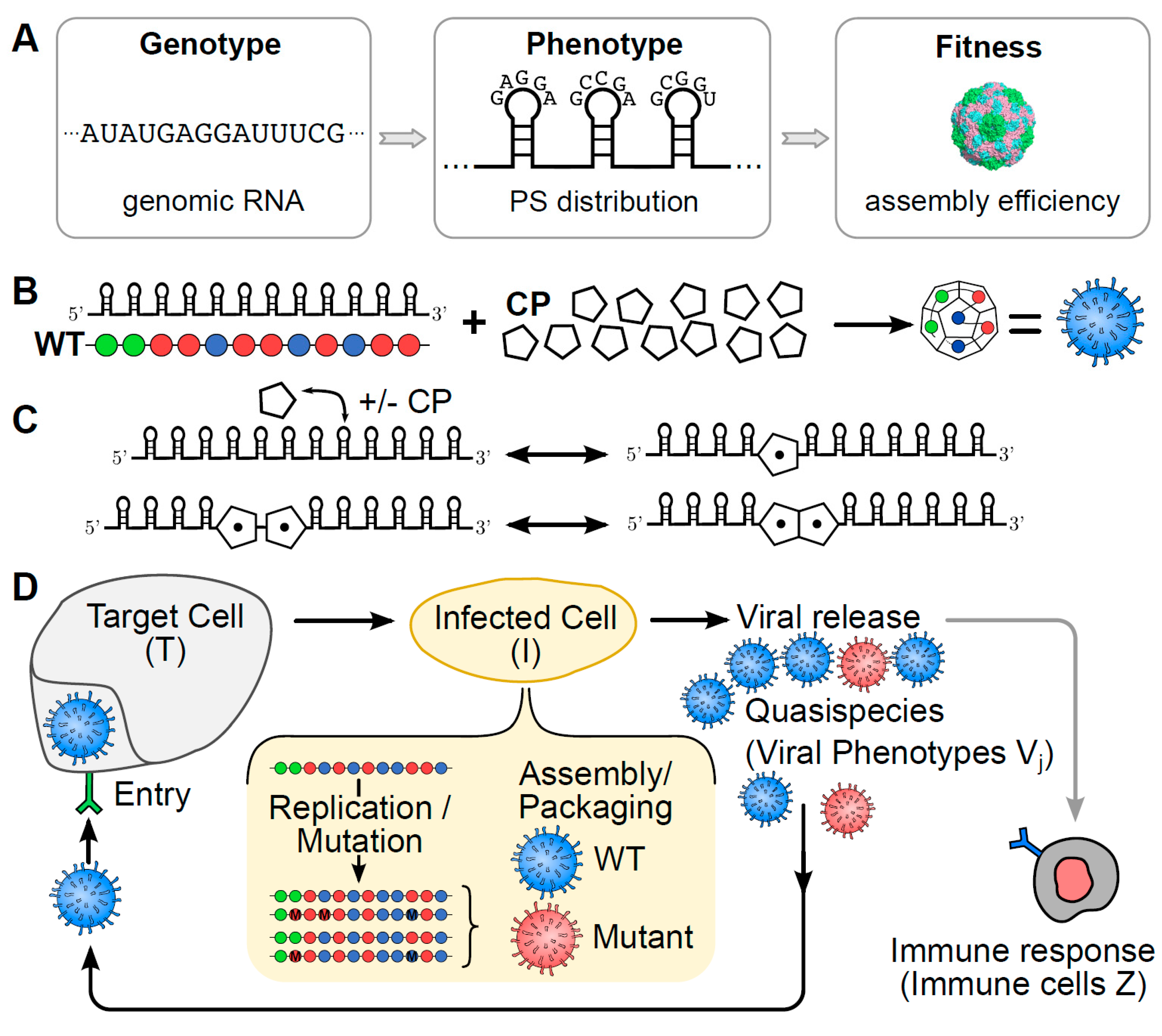
2. Materials and Methods
- The replication step is simulated by creating positive-sense copies from negative-sense templates and vice versa. Starting with the parent phenotype , polymerase randomly copies any of the positive-sense, or in later rounds also negative-sense templates present in the infected cell, until 2000 positive-sense vRNAs are accumulated. Copying errors result in mutations that are assumed to occur with a fixed per-nucleotide mutation rate of per nucleotide, which equates to, on average, one nucleotide error per genome-copying event, as is typical of Picornaviruses [20]. Since we are working with phenotypes instead of genotypes, PSs are mutated at a rate of per PS per genome, reflecting the situation where approximately 5% of the genome contains sequence motifs important for PS function.
- The assembly step is simulated by giving each positive-sense vRNA created in the replication step the chance to package based on its phenotype and the associated probability of packaging obtained from our pre-computed phenotype-fitness map. Mimicking in vivo scenarios, our PS-mediated assembly model simulates ssRNA virus assembly against a backdrop of cRNAs. The latter are associated with a uniformly small chance of packaging. Successfully encapsidated cRNAs act as immunogens in our model, stimulating the immune response, as they are indistinguishable from viral particles at the particle exterior. However, although cRNAs are allowed to enter target cells, they do not result in the production of additional viral particles. Following the assembly step, progeny vRNAs and misencapsidated cRNAs that are fully encapsidated are released into the extracellular environment and are added to the total viral load.
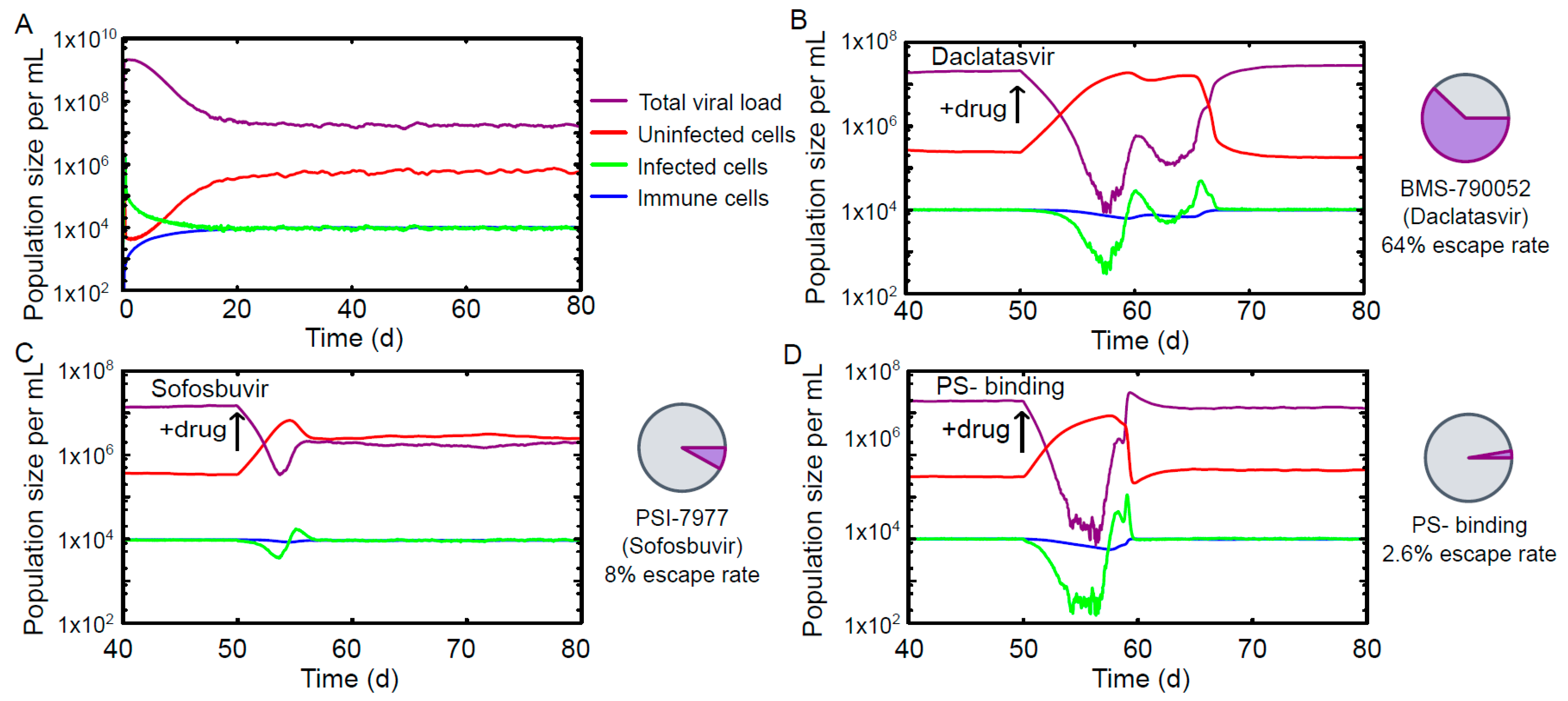
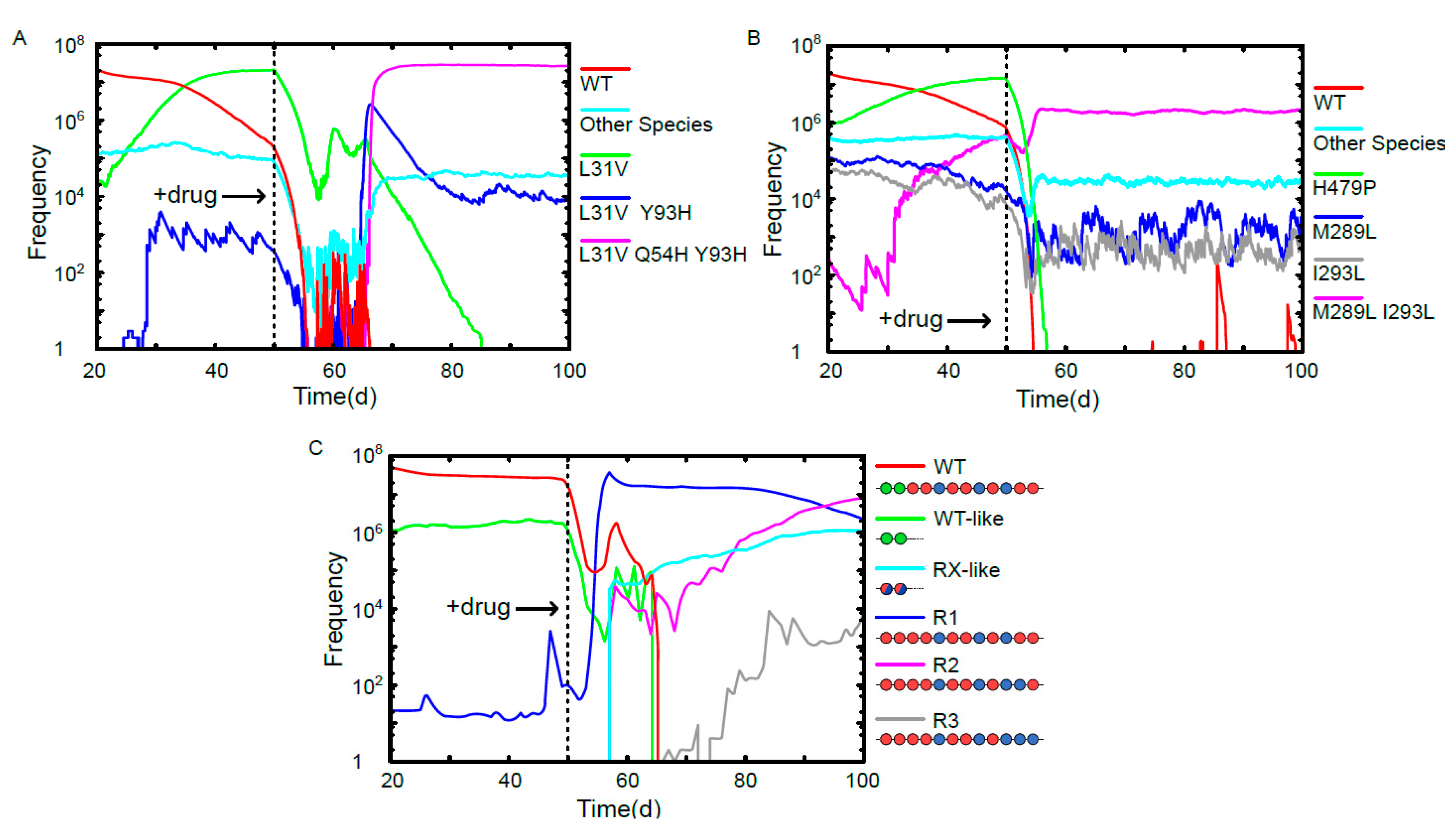
3. Results
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eigen, M.; McCaskill, J.; Schuster, P. Molecular Quasi-Species. J. Phys. Chem. 1998, 92, 6881–6891. [Google Scholar] [CrossRef]
- Lauring, A.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Fontana, W.; Schuster, P. Shaping space: The possible and the attainable in RNA genotype-phenotype mapping. J. Theor. Biol. 1998, 194, 491–515. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Andino, R. Closing the gap: The challenges in converging theoretical, computational, experimental and real-life studies in virus evolution. Curr. Opin. Virol. 2012, 2, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.-J.; Reifman, J. A quantitative quasispecies theory-based model of virus escape mutation under immune selection. Proc. Natl. Acad. Sci. USA 2012, 109, 12980–12985. [Google Scholar] [CrossRef] [PubMed]
- Stockley, P.G.; Twarock, R.; Bakker, S.E.; Barker, A.M.; Borodavka, A.; Dykeman, E.; Ford, R.J.; Pearson, A.R.; Phillips, S.E.V.; Ranson, N.A.; et al. Packaging signals in single-stranded RNA viruses: Nature’s alternative to a purely electrostatic assembly mechanism. J. Biol. Phys. 2013, 39, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Dykeman, E.C.; Stockley, P.G.; Twarock, R. Building a viral capsid in the presence of genomic RNA. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2013, 87, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dykeman, E.C.; Stockley, P.G.; Twarock, R. Solving a Levinthal’s paradox for virus assembly identifies a unique antiviral strategy. Proc. Natl. Acad. Sci. USA 2014, 111, 5361–5366. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Dykeman, E.C.; Coutts, R.H.A.; Lomonossoff, G.P.; Rowlands, D.J.; Phillips, S.E.V.; Ranson, N.; Twarock, R.; Tuma, R.; Stockley, P.G. Revealing the density of encoded functions in a viral RNA. Proc. Natl. Acad. Sci. USA 2015, 112, 201420812. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.; Buldu, J.M.; Stich, M.; Manrubia, S.C. Topological structure of the space of phenotypes: The case of RNA neutral networks. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Shakeel, S.; Dykeman, E.C.; White, S.J.; Ora, A.; Cockburn, J.J.B.; Butcher, S.J.; Stockley, P.G.; Twarock, R. Genomic RNA folding mediates assembly of human parechovirus. Nat. Commun. 2017, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; White, S.J.; Thompson, R.F.; Bingham, R.; Weiß, E.U.; Maskell, D.P.; Zlotnick, A.; Dykeman, E.C.; Tuma, R.; Twarock, R.; et al. HBV RNA pre-genome encodes specific motifs that mediate interactions with the viral core protein that promote nucleocapsid assembly. Nat. Microbiol. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Stewart, H.; Bingham, R.J.; White, S.J.; Dykeman, E.C.; Zothner, C.; Tuplin, A.K.; Stockley, P.G.; Twarock, R.; Harris, M. Identification of novel RNA secondary structures within the hepatitis C virus genome reveals a cooperative involvement in genome packaging. Sci. Rep. 2016, 6, 22952. [Google Scholar] [CrossRef] [PubMed]
- Dykeman, E.C.; Stockley, P.G.; Twarock, R. Packaging signals in two single-stranded RNA viruses imply a conserved assembly mechanism and geometry of the packaged genome. J. Mol. Biol. 2013, 425, 3235–3249. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Wroblewski, E.; Leonov, G.; Phillips, S.E.V.; Tuma, R.; Twarock, R.; Stockley, P.G. Rewriting nature’s assembly manual for an ssRNA virus. Proc. Natl. Acad. Sci. USA 2017, 114, 1114. [Google Scholar] [CrossRef] [PubMed]
- Berger, B.; Shor, P.W.; Tucker-Kellogg, L.; King, J. Local rule-based theory of virus shell assembly. Proc. Natl. Acad. Sci. USA 1994, 91, 7732–7736. [Google Scholar] [CrossRef] [PubMed]
- Zlotnick, A. To build a Virus Capsid. J. Mol. Biol. 1994, 241, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Lago, H.; Parrott, A.M.; Moss, T.; Stonehouse, N.J.; Stockley, P.G. Probing the kinetics of formation of the bacteriophage MS2 translational operator complex: Identification of a protein conformer unable to bind RNA. J. Mol. Biol. 2001, 305, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Drake, J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; May, R.M. Virus Dynamics: Mathematical Principles of Immunology and Virology; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- De Boer, R.J. Which of our modeling predictions are robust? PLoS Comput. Biol. 2012, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.U. Hepatitis C Viral Dynamics in Vivo and the Antiviral Efficacy of Interferon-Therapy. Science 1998, 282, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M. Hepatitis C Virus: From Laboratory to Clinic; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Fridell, R.A.; Wang, C.; Sun, J.H.; O’Boyle, D.R., II; Nower, P.; Valera, L.; Qiu, D.; Roberts, S.; Huang, X.; Kienzle, B.; et al. Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in Humans: In Vitro and In Vivo correlations. Hepatology 2011, 54, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Espiritu, C.; Bansal, S.; Micolochick Steuer, H.M.; Niu, C.; Zennou, V.; Keilman, M.; Zhu, Y.; Lan, S.; Otto, M.J.; et al. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob. Agents Chemother. 2012, 56, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, A.; Brodsky, L.; Andino, R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 2013, 505, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Andino, R. Exploring the fitness landscapes of an RNA virus using a universal barcode microarray. J. Virol. 2011, 85, 3780–3791. [Google Scholar] [CrossRef] [PubMed]
- Borderia, A.V.; Isakov, O.; Moratorio, G.; Henningsson, R.; Agüera-González, S.; Organtini, L.; Gnädig, N.F.; Blanc, H.; Alcover, A.; Hafenstein, S.; et al. Group selection and contribution of minority variants during virus adaptation determines virus fitness and phenotype. PLoS Pathog. 2015, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Bonhoeffer, S.; Chappey, C.; Parkin, N.T.; Whitcomb, J.M.; Petropoulos, C.J. Evidence for positive epistasis in HIV-1. Science 2004, 306, 1547–1550. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, Z.; Lai, M.; Shu, S.; Du, Y.; Zhou, Z.H.; Sun, R. In situ structures of the genome and genome-delivery apparatus in a single-stranded RNA virus. Nature 2016, 541, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Koning, R.I.; Gomez-Blanco, J.; Akopjana, I.; Vargas, J.; Kazaks, A.; Tars, K.; Carazo, J.M.; Koster, A.J. Asymmetric cryo-EM reconstruction of phage MS2 reveals genome structure in situ. Nat. Commun. 2016, 7, 12524. [Google Scholar] [CrossRef] [PubMed]
- Gorzelnik, K.V.; Cui, Z.; Reed, C.A.; Jakana, J.; Young, R.; Zhang, J. Asymmetric cryo-EM structure of the canonical Allolevivirus Qβ reveals a single maturation protein and the genomic ssRNA in situ. Proc. Natl. Acad. Sci. USA 2016, 113, 11519–11524. [Google Scholar] [CrossRef] [PubMed]
- Tanner, E.J.; Liu, H.; Oberste, M.S.; Pallansch, M.; Collett, M.S.; Kirkegaard, K. Dominant drug targets suppress the emergence of antiviral resistance. eLIFE 2014, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Crowder, S.; Kirkegaard, K. Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nat. Genet. 2005, 37, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Wendt, E.; Andino, R. Engineering attenuated virus vaccines by controlling replication fidelity. Nat. Med. 2008, 14, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Bonhoeffer, S.; May, R.M.; Shaw, G.M.; Nowak, M. A Virus dynamics and drug therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 6971–6976. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bingham, R.J.; Dykeman, E.C.; Twarock, R. RNA Virus Evolution via a Quasispecies-Based Model Reveals a Drug Target with a High Barrier to Resistance. Viruses 2017, 9, 347. https://doi.org/10.3390/v9110347
Bingham RJ, Dykeman EC, Twarock R. RNA Virus Evolution via a Quasispecies-Based Model Reveals a Drug Target with a High Barrier to Resistance. Viruses. 2017; 9(11):347. https://doi.org/10.3390/v9110347
Chicago/Turabian StyleBingham, Richard J., Eric C. Dykeman, and Reidun Twarock. 2017. "RNA Virus Evolution via a Quasispecies-Based Model Reveals a Drug Target with a High Barrier to Resistance" Viruses 9, no. 11: 347. https://doi.org/10.3390/v9110347
APA StyleBingham, R. J., Dykeman, E. C., & Twarock, R. (2017). RNA Virus Evolution via a Quasispecies-Based Model Reveals a Drug Target with a High Barrier to Resistance. Viruses, 9(11), 347. https://doi.org/10.3390/v9110347