
Mutations in MYBPC3 and MYH7 in Association with Brugada Type 1 ECG Pattern: Overlap between Brugada Syndrome and Hypertrophic Cardiomyopathy?

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
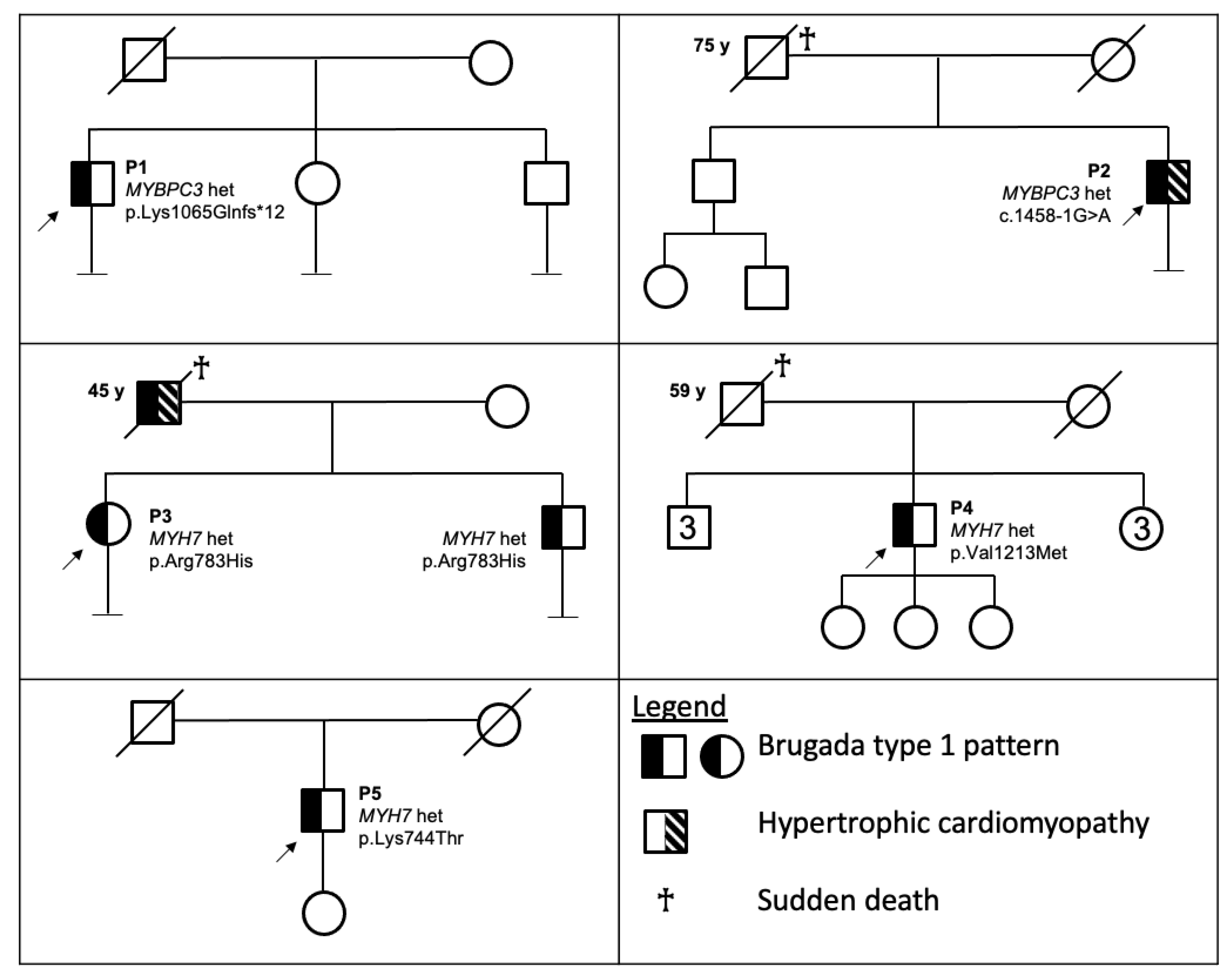
3. Results
3.1. Clinical Evaluation
3.1.1. Patient 1
3.1.2. Patient 2
3.1.3. Patient 3
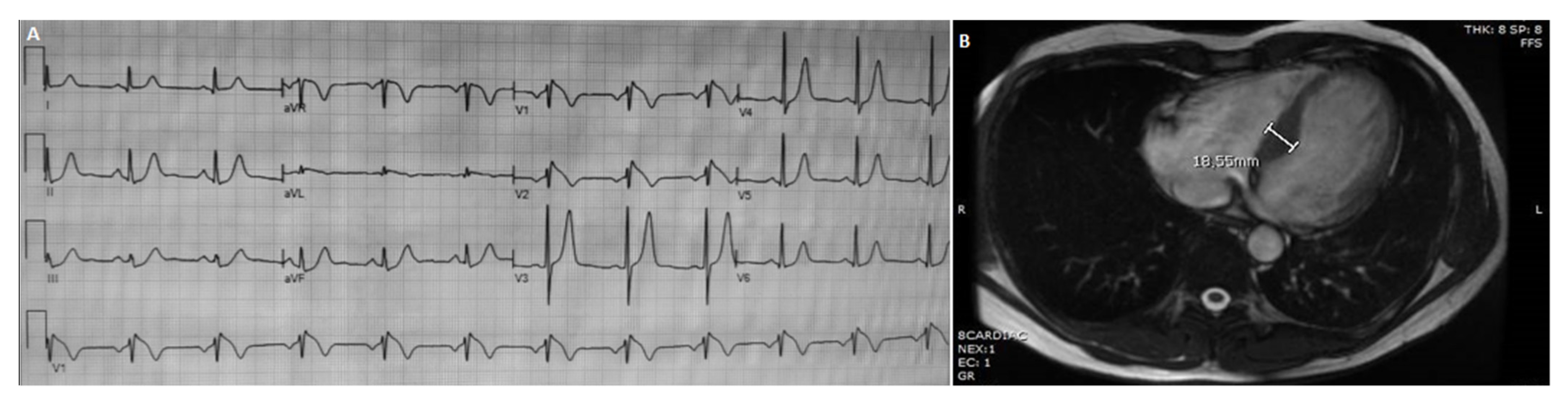
3.1.4. Patient 4
3.1.5. Patient 5
3.2. Genetic Evaluation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilde, A.A.M.; Antzelevitch, C.; Borggrefe, M.; Brugada, J.; Brugada, R.; Brugada, P.; Corrado, D.; Hauer, R.; Kass, R.; Nademanee, K.; et al. Proposed diagnostic criteria for the Brugada syndrome. Eur. Heart J. 2002, 23, 1648–1654. [Google Scholar] [CrossRef]
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef]
- Juang, J.-M.J.; Horie, M. Genetics of Brugada syndrome. J. Arrhythm. 2016, 32, 418–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarquella-Brugada, G.; Campuzano, O.; Arbelo, E.; Brugada, J.; Brugada, R. Brugada syndrome: Clinical and genetic findings. Genet. Med. 2016, 18, 3–12. [Google Scholar] [CrossRef] [Green Version]
- ClinGen. Available online: https://search.clinicalgenome.org/kb/genes/HGNC:10593 (accessed on 15 July 2021).
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Kim, R.; Udupa, S.; Costain, G.; Jobling, R.; Liston, E.; Jamal, S.M.; Szybowska, M.; Morel, C.F.; Bowdin, S.; et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death. Circulation 2018, 138, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Brugada, J.; Brugada, R. Update on Genetic Basis of Brugada Syndrome: Monogenic, Polygenic or Oligogenic? Int. J. Mol. Sci. 2020, 21, 7155. [Google Scholar] [CrossRef] [PubMed]
- Monasky, M.M.; Micaglio, E.; Ciconte, G.; Pappone, C. Brugada Syndrome: Oligogenic or Mendelian Disease? Int. J. Mol. Sci. 2020, 21, 1687. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Remme, C.A.; Tadros, R.; Bezzina, C.R.; Delmar, M. Beyond the One Gene-One Disease Paradigm: Complex Genetics and Pleiotropy in Inheritable Cardiac Disorders. Circulation 2019, 140, 595–610. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Gourraud, J.-B.; Barc, J.; Thollet, A.; Le Scouarnec, S.; Le Marec, H.; Schott, J.-J.; Redon, R.; Probst, V. The Brugada Syndrome: A Rare Arrhythmia Disorder with Complex Inheritance. Front. Cardiovasc. Med. 2016, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Gualandi, F.; Zaraket, F.; Malagù, M.; Parmeggiani, G.; Trabanelli, C.; Fini, S.; Dang, X.; Wei, X.; Fang, M.; Bertini, M.; et al. Mutation Load of Multiple Ion Channel Gene Mutations in Brugada Syndrome. Cardiology 2017, 137, 256–260. [Google Scholar] [CrossRef]
- Moncayo-Arlandi, J.; Brugada, R. Unmasking the molecular link between arrhythmogenic cardiomyopathy and Brugada syndrome. Nat. Rev. Cardiol. 2017, 14, 744–756. [Google Scholar] [CrossRef]
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.V.; et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2019, 12, e002460. [Google Scholar] [CrossRef] [Green Version]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef] [Green Version]
- Girolami, F.; Olivotto, I.; Passerini, I.; Zachara, E.; Nistri, S.; Re, F.; Fantini, S.; Baldini, K.; Torricelli, F.; Cecchi, F. A molecular screening strategy based on beta-myosin heavy chain, cardiac myosin binding protein C and troponin T genes in Italian patients with hypertrophic cardiomyopathy. J. Cardiovasc. Med. 2006, 7, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, M.; Parodi, M.I.; Formisano, F.; Spirito, P.; Autore, C.; Musumeci, M.B.; Favale, S.; Forleo, C.; Rapezzi, C.; Biagini, E.; et al. Targeted next-generation sequencing helps to decipher the genetic and phenotypic heterogeneity of hypertrophic cardiomyopathy. Int. J. Mol. Med. 2016, 38, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldmüller, S.; Müller, M.; Rackebrandt, K.; Binner, P.; Poths, S.; Bonin, M.; Scheffold, T. Array-based resequencing assay for mutations causing hypertrophic cardiomyopathy. Clin. Chem. 2008, 54, 682–687. [Google Scholar] [CrossRef]
- Homburger, J.R.; Green, E.M.; Caleshu, C.; Sunitha, M.S.; Taylor, R.E.; Ruppel, K.M.; Metpally, R.P.R.; Colan, S.D.; Michels, M.; Day, S.M.; et al. Multidimensional structure-function relationships in human β-cardiac myosin from population-scale genetic variation. Proc. Natl. Acad. Sci. USA 2016, 113, 6701–6706. [Google Scholar] [CrossRef] [Green Version]
- Bastiaenen, R.; Cox, A.T.; Castelletti, S.; Wijeyeratne, Y.D.; Colbeck, N.; Pakroo, N.; Ahmed, H.; Bunce, N.; Anderson, L.; Moon, J.C.; et al. Late gadolinium enhancement in Brugada syndrome: A marker for subtle underlying cardiomyopathy? Heart Rhythm 2017, 14, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G. Is it time to include ion channel diseases among cardiomyopathies? J. Electrocardiol. 2005, 38, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, K.; Raju, H.; de Noronha, S.V.; Papadakis, M.; Robinson, L.; Rothery, S.; Makita, N.; Kowase, S.; Boonmee, N.; Vitayakritsirikul, V.; et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J. Am. Coll. Cardiol. 2015, 66, 1976–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behr, E.R.; Ben-Haim, Y.; Ackerman, M.J.; Krahn, A.D.; Wilde, A.A.M. Brugada syndrome and reduced right ventricular outflow tract conduction reserve: A final common pathway? Eur. Heart J. 2021, 42, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, F.; Bertini, M.; Balla, C.; Pestelli, G.; Luisi, A.; Smarrazzo, V.; Farnè, M.; Ferlini, A.; Gualandi, F.; Mele, D. Type 1 Brugada Pattern is associated with echocardiography-detected delayed right ventricular outflow tract contraction. JACC 2021, 77, 2865–2867. [Google Scholar] [CrossRef]
- Di Resta, C.; Pietrelli, A.; Sala, S.; della Bella, P.; De Bellis, G.; Ferrari, M.; Bordoni, R.; Benedetti, S. High-throughput genetic characterization of a cohort of Brugada syndrome patients. Hum. Mol. Genet. 2015, 24, 5828–5835. [Google Scholar] [CrossRef] [Green Version]
- Mango, R.; Luchetti, A.; Sangiuolo, R.; Ferradini, V.; Briglia, N.; Giardina, E.; Ferrè, F.; Citterich, M.H.; Romeo, F.; Novelli, G.; et al. Next Generation Sequencing and Linkage Analysis for the Molecular Diagnosis of a Novel Overlapping Syndrome Characterized by Hypertrophic Cardiomyopathy and Typical Electrical Instability of Brugada Syndrome. Circ. J. 2016, 80, 938–949. [Google Scholar] [CrossRef] [Green Version]
- Pappone, C.; Monasky, M.M.; Ciconte, G. Epicardial ablation in genetic cardiomyopathies: A new frontier. Eur. Heart J. Suppl. 2019, 21, B61–B66. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, P. Brugada syndrome and myocardial histology: Where may the truth Lie? Arch. Cardiovasc. Dis. 2019, 112, 367–369. [Google Scholar] [CrossRef]
- Baudenbacher, F.; Schober, T.; Pinto, J.R.; Sidorov, V.Y.; Hilliard, F.; Solaro, R.J.; Potter, J.D.; Knollmann, B.C. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J. Clin. Investig. 2008, 118, 3893–3903. [Google Scholar] [CrossRef] [Green Version]
- Noyes, A.M.; Zhou, A.; Gao, G.; Gu, L.; Day, S.; Wasserstrom, J.A.; Dudley, S.C. Abnormal sodium channel mRNA splicing in hypertrophic cardiomyopathy. Int. J. Cardiol. 2017, 249, 282–286. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Age of Detection | Type 1 ECG Pattern | Family History | Clinical Evaluation | Gene | c.DNA Change | Protein Change | VarSome | ClinVar | ACMG | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | M | 34 | Spontaneous | - | Asymptomatic except for AVNRT. No signs of HCM. | MYBPC3 | ex 30 c.3192dupC | p.Lys1065Glnfs*12 (K1065Qfs * 12) | P | P | PVS1-PP5-PM2-PP3 (P) | [17] |
| P2 | M | 56 | Spontaneous | + * | Asymptomatic except for sporadic extrasistoles. Instrumental signs of HCM. | MYBPC3 | int 16 c.1458-1G > A | p.? | P | P | PVS1-PP5-PM2-PP3 (P) | [18] |
| P3 | F | 24 | Drug-induced | + ** | History of sincope and Premature Ventricular Contractions. No signs of HCM. | MYH7 | ex 21 c.2348G > A | p.Arg783His (R783H) | LP | VUS/LP | PM1-PM2-PM5-PP2-BP4 (LP) | [19] |
| P4 | M | 61 | Spontaneous | + *** | Asymptomatic. No signs of HCM. | MYH7 | ex 27 c.3637G > A | p.Val1213Met (V1213M) | VUS/LP | VUS | PM2-PP2-PP3 (VUS) | [20] |
| P5 | M | 46 | Spontaneous | - | Asymptomatic. No signs of HCM. | MYH7 | ex 20 c.2231A > C | p.Lys744Thr (K744T) | LP | Not reported | PM1-PM2-PP2-PP3 (LP) | Present paper |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farnè, M.; Balla, C.; Margutti, A.; Selvatici, R.; De Raffele, M.; Di Domenico, A.; Imbrici, P.; De Maria, E.; Biffi, M.; Bertini, M.; et al. Mutations in MYBPC3 and MYH7 in Association with Brugada Type 1 ECG Pattern: Overlap between Brugada Syndrome and Hypertrophic Cardiomyopathy? Cardiogenetics 2021, 11, 139-147. https://doi.org/10.3390/cardiogenetics11030016
Farnè M, Balla C, Margutti A, Selvatici R, De Raffele M, Di Domenico A, Imbrici P, De Maria E, Biffi M, Bertini M, et al. Mutations in MYBPC3 and MYH7 in Association with Brugada Type 1 ECG Pattern: Overlap between Brugada Syndrome and Hypertrophic Cardiomyopathy? Cardiogenetics. 2021; 11(3):139-147. https://doi.org/10.3390/cardiogenetics11030016
Chicago/Turabian StyleFarnè, Marianna, Cristina Balla, Alice Margutti, Rita Selvatici, Martina De Raffele, Assunta Di Domenico, Paola Imbrici, Elia De Maria, Mauro Biffi, Matteo Bertini, and et al. 2021. "Mutations in MYBPC3 and MYH7 in Association with Brugada Type 1 ECG Pattern: Overlap between Brugada Syndrome and Hypertrophic Cardiomyopathy?" Cardiogenetics 11, no. 3: 139-147. https://doi.org/10.3390/cardiogenetics11030016
APA StyleFarnè, M., Balla, C., Margutti, A., Selvatici, R., De Raffele, M., Di Domenico, A., Imbrici, P., De Maria, E., Biffi, M., Bertini, M., Rapezzi, C., Ferlini, A., & Gualandi, F. (2021). Mutations in MYBPC3 and MYH7 in Association with Brugada Type 1 ECG Pattern: Overlap between Brugada Syndrome and Hypertrophic Cardiomyopathy? Cardiogenetics, 11(3), 139-147. https://doi.org/10.3390/cardiogenetics11030016