- Review

The Genetic Architecture of Sudden Cardiac Death: A State-of-the-Art Review
- Sabrina Montuoro,
- Emanuele Monda and
- Giuseppe Limongelli
- + 14 authors
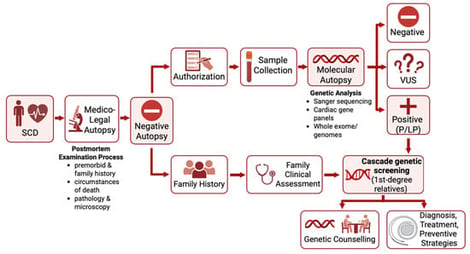
Sudden cardiac death (SCD) is a major global health issue, defined as sudden natural death presumed to be of cardiac cause. While in the elderly SCD is commonly associated with coronary artery disease, in the younger population it is linked to inherited cardiomyopathies or channelopathies, even though SCD can remain unexplained even after a comprehensive autopsy in a substantial proportion of cases. In this context, genetic testing has gained importance, supported by the widespread availability of techniques such as next-generation and whole-exome/genome sequencing and their reduced costs. This state-of-the-art review summarizes the genetic bases of sudden cardiac death among cardiomyopathies, channelopathies and in sudden unexplained death presumed to be of arrhythmic cause. Among the structural causes, inherited cardiomyopathies such as hypertrophic, dilated, non-dilated left ventricular, arrhythmogenic right ventricular and restrictive ones represent major substrates for malignant ventricular arrhythmias mostly arising from variants in sarcomeric or desmosomal genes. Channelopathies (long or short QT syndrome, Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia) are caused by variants in genes encoding cardiac ion channels and/or regulatory proteins, which equally predispose to high risk of life-threatening ventricular arrhythmias. In sudden arrhythmic death syndrome, with a structurally normal heart, post-mortem genetic testing (molecular autopsy) can uncover an underlying inherited condition. However, variants of uncertain significance are detected in more than half of the cases, underscoring the need for a multidisciplinary approach. Genetic testing also plays a key role in cascade screening of first-degree relatives. While monogenic variants drive risk in inherited cardiac disorders, emerging evidence suggests that polygenic contributions may modulate SCD susceptibility, highlighting future roles for polygenic risk scores in risk stratification.
19 March 2026