
MYH7 Genotype–Phenotype Correlation in a Cohort of Finnish Patients


 and
and
Abstract
:1. Introduction
2. Materials and Methods
Ethics
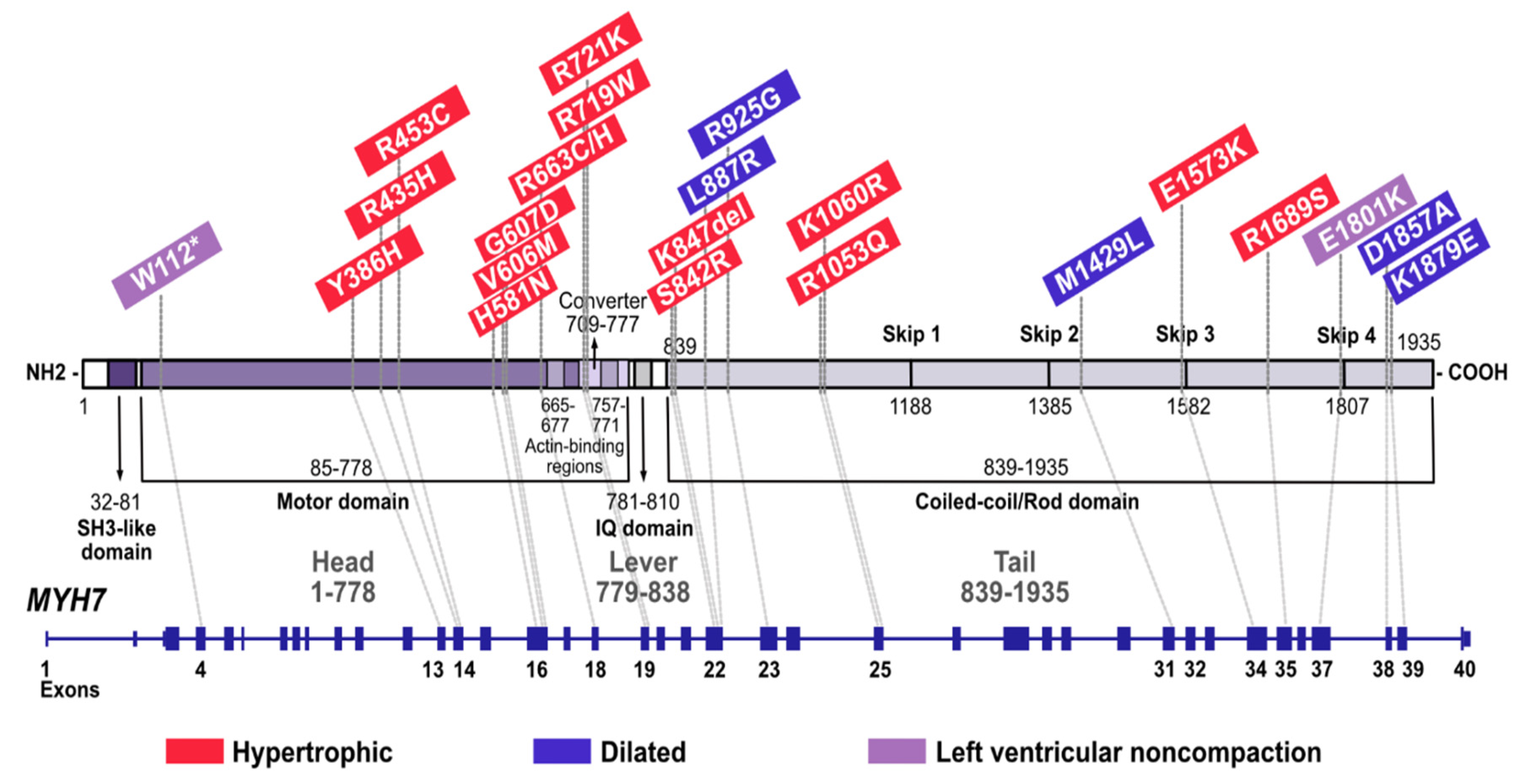
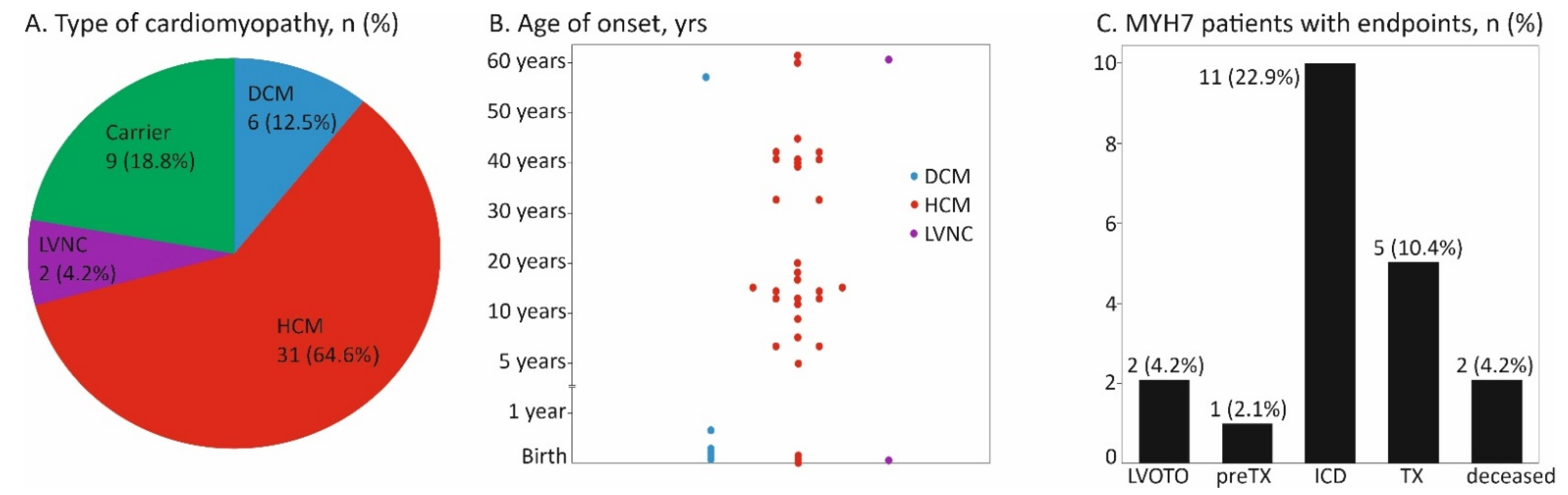
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brieler, J.; Breeden, M.A.; Tucker, J. Cardiomyopathy: An Overview. Am. Fam. Physician 2017, 96, 640–646. [Google Scholar] [PubMed]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart. J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart Association; et al. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [PubMed] [Green Version]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef]
- Wang, S.; Zou, Y.; Fu, C.; Xu, X.; Wang, J.; Song, L.; Wang, H.; Chen, J.; Wang, J.; Huan, T.; et al. Worse prognosis with gene mutations of beta-myosin heavy chain than myosin-binding protein C in Chinese patients with hypertrophic cardiomyopathy. Clin. Cardiol. 2008, 31, 114–118. [Google Scholar]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Tajsharghi, H.; Oldfors, A. Myosinopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 3–18. [Google Scholar] [CrossRef] [Green Version]
- von Knobelsdorff-Brenkenhoff, F.; Schulz-Menger, J. Role of cardiovascular magnetic resonance in the guidelines of the European Society of Cardiology. J. Cardiovasc. Magn. Reson. 2016, 18, 6. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar]
- Petersen, S.E.; Selvanayagam, J.B.; Wiesmann, F.; Robson, M.D.; Francis, J.M.; Anderson, R.H.; Watkins, H.; Neubauer, S. Left ventricular non-compaction: Insights from cardiovascular magnetic resonance imaging. J. Am. Coll. Cardiol. 2005, 46, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.A.; Riegel, B.; Bittner, V.; Nichols, J. Validity and reliability of the NYHA classes for measuring research outcomes in patients with cardiac disease. Heart Lung 2002, 31, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.D. The Ross classification for heart failure in children after 25 years: A review and an age-stratified revision. Pediatr. Cardiol. 2012, 33, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastiercritical-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar]
- Jääskeläinen, P.; Vangipurapu, J.; Raivo, J.; Kuulasmaa, T.; Heliö, T.; Aalto-Setälä, K.; Kaartinen, M.; Ilveskoski, E.; Vanninen, S.; Hämäläinen, L.; et al. Genetic basis and outcome in a nationwide study of Finnish patients with hypertrophic cardiomyopathy. ESC Heart Fail. 2019, 6, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, L.; Jalanko, A.; Varilo, T. Molecular genetics of the Finnish disease heritage. Hum. Mol. Genet. 1999, 8, 1913–1923. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of Hypertrophic Cardiomyopathy after 20 Years: Clinical Perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Lowey, S. Functional consequences of mutations in the myosin heavy chain at sites implicated in familial hypertrophic cardiomyopathy. Trends Cardiovasc. Med. 2002, 12, 348–354. [Google Scholar] [CrossRef]
- Gundapaneni, D.; Xu, J.; Root, D.D. High flexibility of the actomyosin cross-bridge resides in skeletal muscle myosin subfragment-2 as demonstrated by a new single-molecule assay. J. Struct. Biol. 2005, 149, 117–126. [Google Scholar] [CrossRef]
- Watkins, H. Hypertrophic cardiomyopathy: From molecular and genetic mechanisms to clinical management. Eur. Heart J. Suppl. 2001, 3, 43–50. [Google Scholar] [CrossRef]
- Opie, L.H.; Solaro, R.J. Myocardial contraction and relaxation. In Heart Physiology: From Cell to Circulation; Opie, L.H., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004; pp. 221–246. [Google Scholar]
- Buvoli, M.; Hamady, M.; Leinwand, L.A.; Knight, R. Bioinformatics assessment of beta-myosin mutations reveals myosin’s high sensitivity to mutations. Trends Cardiovasc. Med. 2008, 18, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.; Rutland, C.; Thomas, R.; Loughna, S. Cardiomyopathy: A systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 2010, 115, 49–60. [Google Scholar] [CrossRef] [PubMed]
- García-Giustiniani, D.; Arad, M.; Ortíz-Genga, M.; Barriales-Villa, R.; Fernández, X.; Rodríguez-García, I.; Mazzanti, A.; Veira, E.; Maneiro, E.; Rebolo, P.; et al. Phenotype and prognostic correlations of the converter region mutations affecting the β myosin heavy chain. Heart 2015, 101, 1047–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, J.; Winkler, G.; Schulte, I.; Scholz, T.; McKenna, W.; Brenner, B.; Kraft, T. Mutation of the myosin converter domain alters cross-bridge elasticity. Proc. Natl. Acad. Sci. USA 2002, 99, 3557–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norrish, G.; Field, E.; Mcleod, K.; Ilina, M.; Stuart, G.; Bhole, V.; Uzun, O.; Brown, E.; Daubeney, P.E.F.; Lota, A.; et al. Clinical presentation and survival of childhood hypertrophic cardiomyopathy: A retrospective study in the United Kingdom. Eur. Heart J. 2019, 40, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Alexander, P.M.A.; Nugent, A.W.; Daubeney, P.E.F.; Lee, K.J.; Sleeper, L.A.; Schuster, T.; Turner, C.; Davis, A.M.; Semsarian, C.; Colan, S.D.; et al. Long-Term Outcomes of Hypertrophic Cardiomyopathy Diagnosed During Childhood: Results from a National Population-Based Study. Circulation 2018, 138, 29–36. [Google Scholar] [CrossRef]
- Heliö, K.; Kangas-Kontio, T.; Weckström, S.; Vanninen, S.U.M.; Aalto-Setälä, K.; Alastalo, T.P.; Myllykangas, S.; Heliö, T.M.; Koskenvuo, J.W. DSP p.(Thr2104Glnfs*12) variant presents variably with early onset severe arrhythmias and left ventricular cardiomyopathy. BMC Med. Genet. 2020, 21, 19. [Google Scholar] [CrossRef] [Green Version]
- Mestroni, L.; Maisch, B.; McKenna, W.J.; Schwartz, K.; Charron, P.; Rocco, C.; Tesson, F.; Richter, A.; Wilke, A.; Komajda, M. Guidelines for the study of familial dilated cardiomyopathies. Eur. Heart J. 1999, 20, 93–102. [Google Scholar] [CrossRef]
- Cox, G.F.; Sleeper, L.A.; Lowe, A.M.; Towbin, J.A.; Colan, S.D.; Orav, E.J.; Lurie, P.R.; Messere, J.E.; Wilkinson, J.D.; Lipshultz, S.E. Factors associated with establishing a causal diagnosis for children with cardiomyopathy. Pediatrics 2006, 118, 1519–1531. [Google Scholar] [CrossRef] [Green Version]
- Colan, S.D.; Lipshultz, S.E.; Lowe, A.M.; Sleeper, L.A.; Messere, J.; Cox, G.F.; Lurie, P.R.; Orav, E.J.; Towbin, J.A. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: Findings from the Pediatric Cardiomyopathy Registry. Circulation 2007, 115, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. ACCF/AHA Guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: Executive summary. Circulation 2011, 124, 2761–2796. [Google Scholar] [CrossRef]
- Norrish, G.; Jager, J.; Field, E.; Quinn, E.; Fell, H.; Lord, E.; Cicerchia, M.N.; Ochoa, J.P.; Cervi, E.; Elliott, P.M.; et al. Yield of Clinical Screening for Hypertrophic Cardiomyopathy in Child First-Degree Relatives. Circulation 2019, 140, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e533–e557. [Google Scholar] [PubMed]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESCEndorsed by Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [PubMed] [Green Version]
- Norrish, G.; Qu, C.; Field, E.; Cervi, E.; Khraiche, D.; Klaassen, S.; Ojala, T.H.; Sinagra, G.; Yamazawa, H.; Marrone, C.; et al. External validation of the HCM Risk-Kids model for predicting sudden cardiac death in childhood hypertrophic cardiomyopathy. Eur. J. Prev. Cardiol. 2021, 42, zwab181. [Google Scholar] [CrossRef] [PubMed]
- Mathew, J.; Zahavich, L.; Lafreniere-Roula, M.; Wilson, J.; George, K.; Benson, L.; Bowdin, S.; Mital, S. Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin. Genet. 2018, 93, 310–319. [Google Scholar] [CrossRef] [Green Version]
- Al-Wakeel-Marquard, N.; Degener, F.; Herbst, C.; Kühnisch, J.; Dartsch, J.; Schmitt, B.; Kuehne, T.; Messroghli, D.; Berger, F.; Klaassen, S. RIKADA Study Reveals Risk Factors in Pediatric Primary Cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e012531. [Google Scholar] [CrossRef] [Green Version]

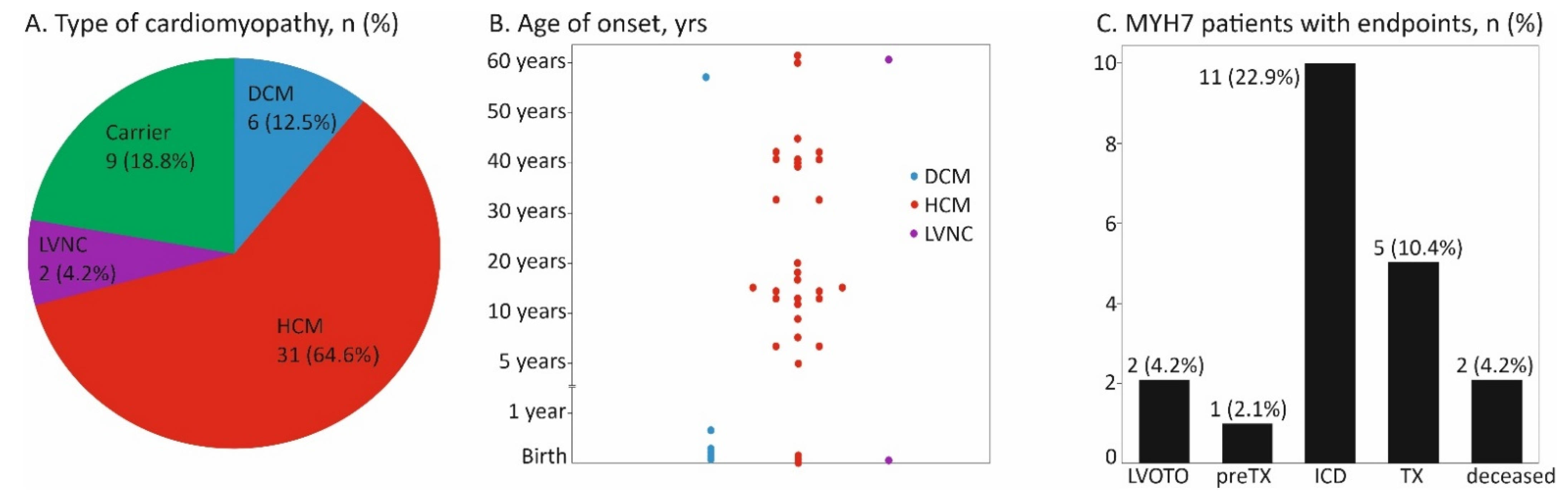

{kind=link}
{kind=link}
| Gene | Sex | CMP | Age at the Time of Dg (Yrs) | Classification | Endpoint |
|---|---|---|---|---|---|
| 0–1 years | |||||
| c. 2773 A > C, p. (R925G) | female | DCM | 0.03 | pathogenic | |
| c. 2773 A > C, p. (R925G) | female | DCM | 0.08 | pathogenic | deceased |
| c. 5570 A > C, p. (D1857A) | male | DCM | 0.13 | VUS | |
| c. 5635 A > G, p. (K1879E) | male | DCM | 0.33 | pathogenic | TX |
| c. 2660 T > G, p. (L887R) | female | DCM | 0.67 | pathogenic | |
| c. 1156 T > C, p. (Y386H) | male | HCM | 0,01 | likely pathogenic | lvoto |
| c. 3158 G > A, p. (R1053Q) | male | HCM | 0.25 | pathogenic | |
| c. 1357 C > T, p. (R453C) | female | HCM | 0.33 | pathogenic | ICD |
| c. 5401 > A, p. (E1801 K) | male | LVNC | 0,05 | pathogenic | preTX |
| c. 3179 A > G, p. (K1060R) | male | carrier | 0.33 | VUS | |
| c. 3179 A > G, p. (K1060R) | female | carrier | 0.33 | VUS | |
| c. 4717 G > A, p. (E1573K) | male | carrier | 0.75 | VUS | |
| 1–12 years | |||||
| c. 2526 T > A, p. (S842R) | female | HCM | 5 | pathogenic | TX |
| c. 1816 G > A, p. (V606M) | female | HCM | 7 | pathogenic | |
| c. 1357 C > T, p. (R453C) | female | HCM | 7 | pathogenic | ICD |
| c. 3158 G > A, p. (R1053Q) | male | HCM | 7.5 | pathogenic | |
| c. 1357 C > T p. (R453C) | male | HCM | 9 | pathogenic | ICD |
| c. 1987 C > T, p. (R663C) | female | HCM | 12 | pathogenic | |
| c. 2155 C > T, p. (R719W) | female | HCM | 12 | pathogenic | ICD |
| 1816 G > A, p. (V606M) | male | HCM | 12 | pathogenic | |
| c. 1987 C > T, p. (R663C) | female | carrier | 2 | pathogenic | |
| c. 1987 C > T, p. (R663C) | male | carrier | 4 | pathogenic | |
| c. 3158 G > A, p. (R1053Q) | male | carrier | 4.5 | pathogenic | |
| c. 1988 G > A, p. (R663H) | male | carrier | 6 | pathogenic | |
| c. 3158 G > A, p. (R1053Q) | male | carrier | 12 | pathogenic | |
| c. 2539_2541delAAG, p. (K847del) | female | carrier | 12 | likely pathogenic | |
| >12 years | |||||
| c. 4285 A > T, p. (M1429L) | female | DCM | 57 | VUS | ICD |
| c. 2155 C > T, p. (R719W) | female | HCM | 15 | pathogenic | ICD |
| 1816 G > A, p. (V606M) | female | HCM | 14.5 | pathogenic | LVOTO + ICD |
| c. 1358 G > A, p. (R435H) | male | HCM | 14.5 | pathogenic | ICD |
| c. 1741 C > A, p. (H581N) | female | HCM | 15 | VUS | ICD |
| c. 2155 C > T, p. (R719W) | female | HCM | 15 | pathogenic | |
| c. 1820 G > A, p. (G607D) | male | HCM | 16 | VUS | ICD |
| c. 2155 C > T, p. (R719W) | female | HCM | 17 | pathogenic | TX |
| c. 3158 G > A, p. (R1053Q) | male | HCM | 18 | pathogenic | |
| c. 2162 G > A, p. (R721K) | male | HCM | 20 | VUS | TX |
| c. 1816 G > A, p. (V606M) | male | HCM | 33 | pathogenic | deceased |
| c. 3158 G > A, p. (R1053Q) | male | HCM | 37 | pathogenic | |
| c. 3179 A > G, p. (K1060R) | male | HCM | 38 | VUS | |
| c. 1987 C > T, p. (R663C) | male | HCM | 40 | pathogenic | |
| c. 1816 G- > A, p. (V606M) | male | HCM | 41 | pathogenic | LVOTO |
| c. 1988 G- > A, p. (R663H) | male | HCM | 41 | pathogenic | ICD |
| c. 3158 G > A, p. (R1053Q) | male | HCM | 43 | pathogenic | |
| c. 3158 G > A, p. (R1053Q) | male | HCM | 43 | pathogenic | |
| c. 3158 G > A, p. (R1053Q) | female | HCM | 45 | pathogenic | ICD |
| c. 3158 G > A, p. (R1053Q) | female | HCM | 60 | pathogenic | |
| c. 1816 G > A, p. (V606M) | male | HCM | 62 | pathogenic | |
| c. 335 G > A, p. (W112 *) | female | LVNC | 61 | VUS |
| Whole Cohort | 0–1 Years | 1–12 Years | >12 Years | p-Value | |
|---|---|---|---|---|---|
| Age of onset, years | 18.2 (±19.1) | 0.27 (±0.24) | 8.2 (±3.5) | 35.6 (±16.3) | |
| Female sex, n (%) | 21 (43.8) | 5 (23.8) | 7 (33.3) | 9 (42.9) | 0.961 |
| FHx CMP, n (%) | 27 (56.3) | 8 (29.6) | 12 (44.4) | 7 (25.9) | 0.015 |
| FHx SCD, n (%) | 5 (10.9) | 2 (40.0) | 1 (20.0) | 2 (40.0) | 0.707 |
| Unexplained syncope, n (%) | 4 (9.8) | 0 (0) | 0 (0) | 4 (100) | 0.044 |
| Neurological deficits, n (%) | 5 (10.4) | 1 (20.0) | 3 (60.0) | 1 (20.0) | 0.166 |
| B Blockers (at the time of diagnosing), n (%) | 26 (54.2) | 8 (30.8) | 6 (23.1) | 12 (46.2) | 0.647 |
| Echocardiography | |||||
| LVMWT, mm | 13.0 (±8.4) | 5.7 (±3.9) | 10.1 (±4.7) | 18.8 (±8.4) | |
| LVEDD, mm | 39.3 (±10.3) | 30.2 (±8.2) | 35.0 (±8.1) | 46.8 (±6.8) | |
| Ejection fraction, % | 64.6 (±12.2) | 50.0 (±20.5) | 72.6 (±4.7) | 66.7 (±12.2) | |
| CMR, n (%) | 26 (55.3) | 9 (34.6) | 4 (15.4) | 13 (50.0) | |
| LGE, % [median (range)] | 4.9 (0–24) | 0 (0–24) | 7.5 (0–15) | 10.0 (0–24) | |
| GLS, % [median (range)] | −22.8 [−10–(−33)] | −24.4 [−18–(−30)] | −23.2 [−15–(−27)] | −19.1 [−10–(−33)] | |
| Cardiomyopathies, n (%) | |||||
| HCM | 31 (64.6) | 3 (9.7) | 9 (29.0) | 19 (61.3) | <0.001 |
| DCM | 6 (12.5) | 5 (83.3) | 0 (0) | 1 (16.7) | <0.001 |
| LVNC | 2 (4.2) | 1 (50) | 0 (0) | 1 (50) | 0.613 |
| Carrier | 9 (20.0) | 3 (33.3) | 6 (66.7) | 0 (0) | 0.005 |
| Endpoint, n (%) | 21 (43.8) | 5 (23.8) | 5 (23.8) | 11 (52.4) | 0.517 |
| Cardiac transplant, n (%) | 5 (10.4) | 1 (20.0) | 1 (20.0) | 3 (60.0) | 0.734 |
| Pre-transplant, n (%) | 1 (2.1) | 1 (100) | 0 (0) | 0 (0) | 0.216 |
| Death, n (%) | 2 (4.2) | 1 (50) | 0 (0) | 1 (50) | 0.551 |
| LVOT obstruction, n (%) | 4 (8.3) | 2 (50) | 0 (0) | 2 (50) | 0.287 |
| ICD implantation, n (%) | 11 (22.9) | 1 (9.1) | 4 (36.4) | 6 (54.5) | 0.378 |
| Time from diagnose to the endpoint, years | 8.0 (±8.6) | 3.1 (±5.4) | 6.1 (±3.3) | 11.0 (±10.4) | 0.204 |
| Time from birth to endpoint, years | 25.9 (±21.0) | 3.3 (±5.5) | 14.9 (±3.5) | 41.1 (±17.1) | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vepsäläinen, T.; Heliö, T.; Vasilescu, C.; Martelius, L.; Weckström, S.; Koskenvuo, J.; Hiippala, A.; Ojala, T. MYH7 Genotype–Phenotype Correlation in a Cohort of Finnish Patients. Cardiogenetics 2022, 12, 122-132. https://doi.org/10.3390/cardiogenetics12010013
Vepsäläinen T, Heliö T, Vasilescu C, Martelius L, Weckström S, Koskenvuo J, Hiippala A, Ojala T. MYH7 Genotype–Phenotype Correlation in a Cohort of Finnish Patients. Cardiogenetics. 2022; 12(1):122-132. https://doi.org/10.3390/cardiogenetics12010013
Chicago/Turabian StyleVepsäläinen, Teemu, Tiina Heliö, Catalina Vasilescu, Laura Martelius, Sini Weckström, Juha Koskenvuo, Anita Hiippala, and Tiina Ojala. 2022. "MYH7 Genotype–Phenotype Correlation in a Cohort of Finnish Patients" Cardiogenetics 12, no. 1: 122-132. https://doi.org/10.3390/cardiogenetics12010013
APA StyleVepsäläinen, T., Heliö, T., Vasilescu, C., Martelius, L., Weckström, S., Koskenvuo, J., Hiippala, A., & Ojala, T. (2022). MYH7 Genotype–Phenotype Correlation in a Cohort of Finnish Patients. Cardiogenetics, 12(1), 122-132. https://doi.org/10.3390/cardiogenetics12010013