
Screening Method for 22q11 Deletion Syndrome Involving the Use of TaqMan qPCR for TBX1 in Patients with Conotruncal Congenital Heart Disease

 , and
, and Highlights
- We propose an effective method for screening for 22q11.2 deletion syndrome using a TBX1 qPCR probe in patients with conotruncal congenital heart defects.
- We identified a 4.34% prevalence of 22q11.2 syndrome in a sample of Mexican patients with non-syndromic conotruncal congenital heart defects.
- These findings suggest a one-probe screening method for detecting 22q11.2 syndrome in patients with non-syndromic conotruncal congenital heart defects.
- They also highlight the underdiagnosis of 22q11.2 syndrome, which could be mitigated through the implementation of qPCR screening.
Abstract
:1. Introduction
2. Materials and Methods
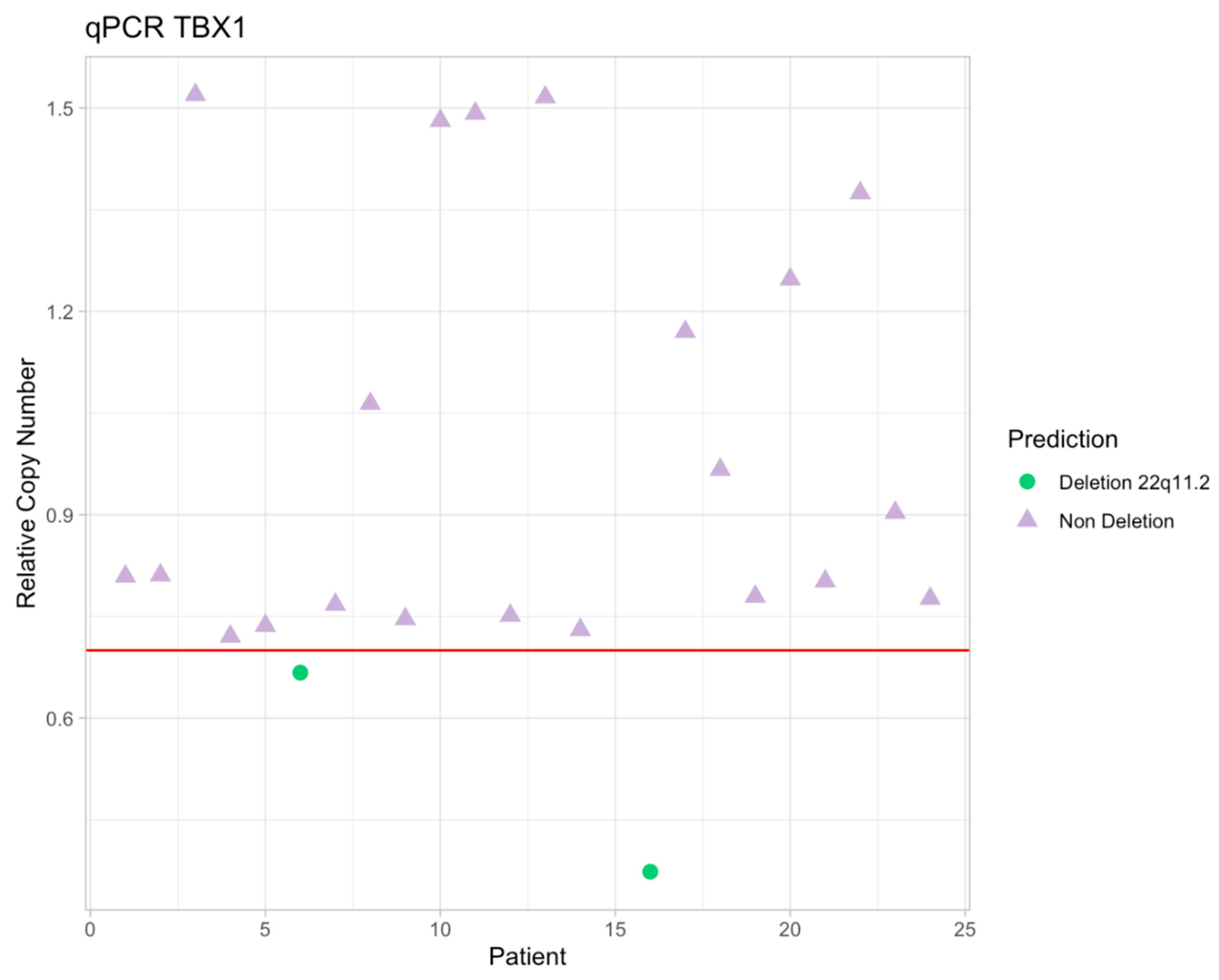
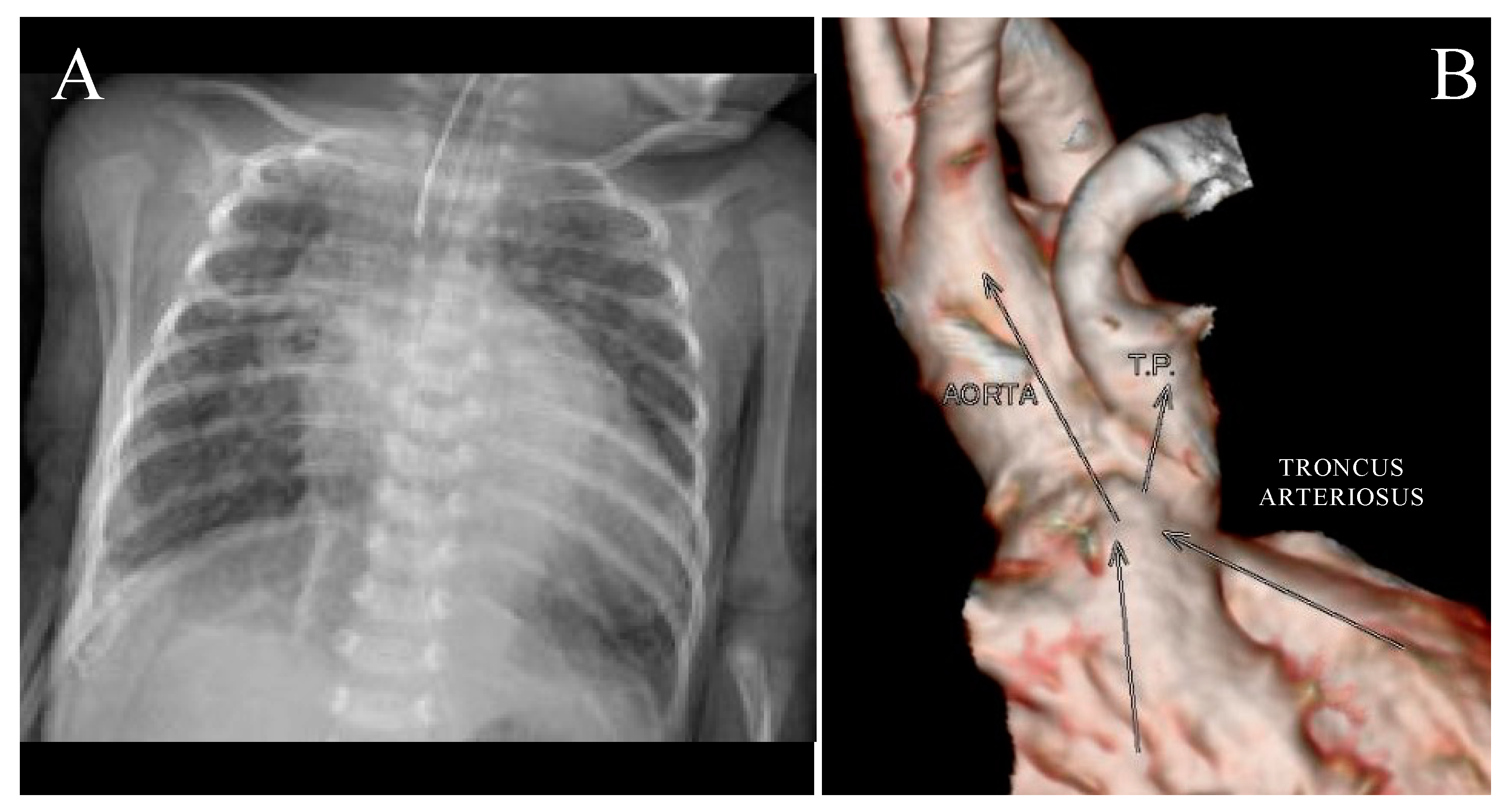
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blackburn, C.; Heude, E.; Tesarova, M.; Sefton, E.M.; Jullian, E.; Adachi, N.; Grimaldi, A.; Zikmund, T.; Kaiser, J.; Kardon, G.; et al. Unique Morphogenetic Signatures Define Mammalian Neck Muscles and Associated Connective Tissues. eLife 2018, 7, e40179. [Google Scholar] [CrossRef]
- Jiang, X.; Li, T.; Liu, S.; Fu, Q.; Li, F.; Chen, S.; Sun, K.; Xu, R.; Xu, Y. Variants in a Cis-Regulatory Element of TBX1 in Conotruncal Heart Defect Patients Impair GATA6-Mediated Transactivation. Orphanet J. Rare Dis. 2021, 16, 334. [Google Scholar] [CrossRef] [PubMed]
- Butensky, A.; de Rinaldis, C.P.; Patel, S.; Edman, S.; Bailey, A.; McGinn, D.E.; Zackai, E.; Crowley, T.B.; McDonald-McGinn, D.M.; Min, J.; et al. Cardiac Evaluation of Patients with 22q11.2 Duplication Syndrome. Am. J. Med. Genet. A 2021, 185, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Nissan, E.; Katz, U.; Levy-Shraga, Y.; Frizinsky, S.; Carmel, E.; Gothelf, D.; Somech, R. Clinical Features in a Large Cohort of Patients With 22q11.2 Deletion Syndrome. J. Pediatrics 2021, 238, 215–220.e5. [Google Scholar] [CrossRef]
- Khoshnood, B.; Lelong, N.; Houyel, L.; Thieulin, A.C.; Jouannic, J.M.; Magnier, S.; Delezoide, A.L.; Magny, J.F.; Rambaud, C.; Bonnet, D.; et al. Prevalence, Timing of Diagnosis and Mortality of Newborns with Congenital Heart Defects: A Population-Based Study. Heart 2012, 98, 1667–1673. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.; Zühlke, L.; Black, G.C.; Choy, M.K.; Li, N.; Keavney, B.D. Global Birth Prevalence of Congenital Heart Defects 1970–2017: Updated Systematic Review and Meta-Analysis of 260 Studies. Int. J. Epidemiol. 2019, 48, 455–463. [Google Scholar] [CrossRef]
- Johnson, T.R. Conotruncal Cardiac Defects: A Clinical Imaging Perspective. Pediatric Cardiol. 2010, 31, 430–437. [Google Scholar] [CrossRef]
- Unolt, M.; Versacci, P.; Anaclerio, S.; Lambiase, C.; Calcagni, G.; Trezzi, M.; Carotti, A.; Crowley, T.B.; Zackai, E.H.; Goldmuntz, E.; et al. Congenital Heart Diseases and Cardiovascular Abnormalities in 22q11.2 Deletion Syndrome: From Well-Established Knowledge to New Frontiers. Am. J. Med. Genet. A 2018, 176, 2087–2098. [Google Scholar] [CrossRef]
- Gao, S.; Li, X.; Amendt, B.A. Understanding the Role of Tbx1 as a Candidate Gene for 22q11.2 Deletion Syndrome. Curr. Allergy Asthma Rep. 2013, 13, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Huber, J.; Peres, V.C.; de Castro, A.L.; dos Santos, T.J.; da Fontoura Beltrão, L.; de Baumont, A.C.; Cossio, S.L.; Dalberto, T.P.; Riegel, M.; Cañedo, A.D.; et al. Molecular Screening for 22Q11.2 Deletion Syndrome in Patients With Congenital Heart Disease. Pediatric Cardiol. 2014, 35, 1356–1362. [Google Scholar] [CrossRef]
- Liao, H.C.; Liao, C.H.; Kao, S.M.; Chiang, C.C.; Chen, Y.J. Detecting 22q11.2 Deletion Syndrome in Newborns with Low T Cell Receptor Excision Circles from Severe Combined Immunodeficiency Screening. J. Pediatrics 2019, 204, 219–224.e1. [Google Scholar] [CrossRef] [PubMed]
- Tomita-Mitchell, A.; Mahnke, D.K.; Larson, J.M.; Ghanta, S.; Feng, Y.; Simpson, P.M.; Broeckel, U.; Duffy, K.; Tweddell, J.S.; Grossman, W.J.; et al. Multiplexed Quantitative Real-Time PCR to Detect 22q11.2 Deletion in Patients with Congenital Heart Disease. Physiol. Genom. 2010, 42A, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Review, C.; Communication, S.; Principles, G. World Medical Association Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. J. Am. Coll. Dent. 2014, 81, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.M.; John, P.; Fan, K.-H.; Bhatti, A.; Feingold, E.; Demirci, F.Y.; Kamboh, M.I. Association of VPREB1 Gene Copy Number Variation and Rheumatoid Arthritis Susceptibility. Dis. Markers 2020, 2020, 7189626. [Google Scholar] [CrossRef] [PubMed]
- Boom, R.; Sol, C.J.A.; Salimans, M.M.M.; Jansen, C.L.; Wertheim-Van Dillen, P.M.E.; Van Der Noordaa, J. Rapid and Simple Method for Purification of Nucleic Acids. J. Clin. Microbiol. 1990, 28, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Chung, W.K. Quantitative Analysis of Copy Number Variants Based on Real-Time LightCycler PCR. Curr. Protoc. Hum. Genet. 2014, 80, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pineda, T.; Zarante, I.; Paredes, A.C.; Rozo, J.P.; Reyes, M.C.; Moreno-Niño, O.M. CNVs in the 22q11.2 Chromosomal Region Should Be an Early Suspect in Infants with Congenital Cardiac Disease. Clin. Med. Insights: Cardiol. 2021, 15, 11795468211016870. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R; RStudio: Boston, MA, USA, 2021. [Google Scholar]
- Adachi, N.; Bilio, M.; Baldini, A.; Kelly, R.G. Cardiopharyngeal Mesoderm Origins of Musculoskeletal and Connective Tissues in the Mammalian Pharynx. Development 2020, 147, dev185256. [Google Scholar] [CrossRef]
- Fa, J.; Zhang, X.; Zhang, X.; Qi, M.; Zhang, X.; Fu, Q.; Xu, Z.; Gao, Y.; Wang, B. Long Noncoding RNA Lnc-TSSK2-8 Activates Canonical Wnt/β-Catenin Signaling Through Small Heat Shock Proteins HSPA6 and CRYAB. Front. Cell Dev. Biol. 2021, 9, 660576. [Google Scholar] [CrossRef]
- Choi, B.G.; Hwang, S.K.; Kwon, J.E.; Kim, Y.H. Array Comparative Genomic Hybridization as the First-Line Investigation for Neonates with Congenital Heart Disease: Experience in a Single Tertiary Center. Korean Circ. J. 2018, 48, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Maran, S.; Faten, S.A.; Lim, S.-H.E.; Lai, K.-S.; Ibrahim, W.P.W.; Ankathil, R.; Gan, S.H.; Tan, H.L.; Ielapi, N. Screening of 22q11.2DS Using Multiplex Ligation-Dependent Probe Amplification as an Alternative Diagnostic Method. BioMed Res. Int. 2020, 2020, 6945730. [Google Scholar] [CrossRef] [PubMed]
- Mastromoro, G.; Calcagni, G.; Vignaroli, W.; Anaclerio, S.; Pugnaloni, F.; Rinelli, G.; Secinaro, A.; Bordonaro, V.; Putotto, C.; Unolt, M.; et al. Crossed Pulmonary Arteries: An Underestimated Cardiovascular Variant with a Strong Association with Genetic Syndromes—A Report of 74 Cases with Systematic Review of the Literature. Am. J. Med. Genet. A 2022, 188, 2351–2359. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, A.; Lioncino, M.; Maratea, A.; Passariello, A.; Fusco, A.; Fratta, F.; Monda, E.; Caiazza, M.; Signore, G.; Esposito, A.; et al. Clinical Manifestations of 22q11.2 Deletion Syndrome. Heart Fail. Clin. 2022, 18, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.; Wolnik-Brzozowska, D.; Wisniewska, M.; Glazar, R.; Materna-Kiryluk, A.; Moszura, T.; Badura-Stronka, M.; Skolozdrzy, J.; Krawczynski, M.R.; Zeyland, J.; et al. Frequency of 22q11.2 Microdeletion in Children with Congenital Heart Defects in Western Poland. BMC Pediatrics 2010, 10, 88. [Google Scholar] [CrossRef] [Green Version]
- Sivertsen, Å.; Lie, R.T.; Wilcox, A.J.; Åbyholm, F.; Vindenes, H.; Haukanes, B.I.; Houge, G. Prevalence of Duplications and Deletions of the 22q11 DiGeorge Syndrome Region in a Population-Based Sample of Infants with Cleft Palate. Am. J. Med. Genet. A 2007, 143A, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Mlynarski, E.E.; Xie, M.; Taylor, D.; Sheridan, M.B.; Guo, T.; Racedo, S.E.; McDonald-McGinn, D.M.; Chow, E.W.C.; Vorstman, J.; Swillen, A.; et al. Rare Copy Number Variants and Congenital Heart Defects in the 22q11.2 Deletion Syndrome. Hum. Genet. 2016, 135, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Carotti, A.; Digilio, M.C.; Piacentini, G.; Saffirio, C.; di Donato, R.M.; Marino, B. Cardiac Defects and Results of Cardiac Surgery in 22q11.2 Deletion Syndrome. Dev. Disabil. Res. Rev. 2008, 14, 35–42. [Google Scholar] [CrossRef]
- Emanuel, B.S. Molecular Mechanisms and Diagnosis of Chromosome 22q11.2 Rearrangements. Dev. Disabil. Res. Rev. 2008, 14, 11–18. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Population | Screening Method | 22q11.2 Syndrome Frequency | Reference |
|---|---|---|---|
| Congenital Heart Disease | FISH | 14.94% | [25] |
| qPCR | 5% | [12] | |
| PCR-RFLP, MLPA, FISH | 1.27% | [10] | |
| aCGH | 4.94% | [21] | |
| MLPA | 4.76% | [22] | |
| MLPA | 21.9% | [17] | |
| Low T cell receptor excision circles | qPCR, MLPA | 10.7% | [11] |
| Hypocalcemia, cleft lip palate | MLPA | 1.8% | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos-Garcia, F.-J.; Castillo-Espinola, A.-M.; Medina-Escobedo, C.-E.; Zenteno, J.C.; Lara-Riegos, J.-C.; Rubio-Zapata, H.; Cruz-Robles, D.; Velazquez-Ibarra, A.-I. Screening Method for 22q11 Deletion Syndrome Involving the Use of TaqMan qPCR for TBX1 in Patients with Conotruncal Congenital Heart Disease. Cardiogenetics 2022, 12, 253-260. https://doi.org/10.3390/cardiogenetics12030024
Campos-Garcia F-J, Castillo-Espinola A-M, Medina-Escobedo C-E, Zenteno JC, Lara-Riegos J-C, Rubio-Zapata H, Cruz-Robles D, Velazquez-Ibarra A-I. Screening Method for 22q11 Deletion Syndrome Involving the Use of TaqMan qPCR for TBX1 in Patients with Conotruncal Congenital Heart Disease. Cardiogenetics. 2022; 12(3):253-260. https://doi.org/10.3390/cardiogenetics12030024
Chicago/Turabian StyleCampos-Garcia, Felix-Julian, Addy-Manuela Castillo-Espinola, Carolina-Elizabeth Medina-Escobedo, Juan C. Zenteno, Julio-Cesar Lara-Riegos, Hector Rubio-Zapata, David Cruz-Robles, and Ana-Isabel Velazquez-Ibarra. 2022. "Screening Method for 22q11 Deletion Syndrome Involving the Use of TaqMan qPCR for TBX1 in Patients with Conotruncal Congenital Heart Disease" Cardiogenetics 12, no. 3: 253-260. https://doi.org/10.3390/cardiogenetics12030024
APA StyleCampos-Garcia, F.-J., Castillo-Espinola, A.-M., Medina-Escobedo, C.-E., Zenteno, J. C., Lara-Riegos, J.-C., Rubio-Zapata, H., Cruz-Robles, D., & Velazquez-Ibarra, A.-I. (2022). Screening Method for 22q11 Deletion Syndrome Involving the Use of TaqMan qPCR for TBX1 in Patients with Conotruncal Congenital Heart Disease. Cardiogenetics, 12(3), 253-260. https://doi.org/10.3390/cardiogenetics12030024