Abstract
Anderson–Fabry Disease (AFD) is a rare, X-linked lysosomal storage disorder caused by a mutation in the α-Galactosidase A gene resulting in α-Galactosidase A enzyme (α-Gal A) deficiency. The metabolic defect leads to the progressive accumulation of glycosphingolipids and the structural and functional impairment of affected organs. Due to the inheritance pattern, male patients are hemizygous with more severe manifestations of the disease as compared to females who, in most cases, are heterozygous with delayed and variable clinical presentation caused by uneven X-chromosome inactivation. Fabry disease cases are often identified by targeted screening programs in high-risk groups, such as in patients with end-stage renal disease, premature stroke, or unexplained cardiomyopathy. Here, we describe a unique case of a homozygous female patient identified by a nationwide screening program in hypertrophic cardiomyopathy patients. Before the systematic screening, the patient had a diagnosis of hypertrophic obstructive cardiomyopathy and was treated accordingly, including with alcohol septal ablation to reduce the obstructive gradient. The confirmation of Fabry disease led to the discovery of the same variant in several members of her family. The identified variant was c.644A>G, p.Asn215Ser (p.N215S), which is known to cause predominant cardiac involvement with late onset of the disease. This variant is amenable to oral therapy with the small-molecule chaperone migalastat, which was started and then interrupted due to the recurrence of the patient’s migraine and then re-initiated again after two years. During this period, the patient received enzyme replacement therapy with agalsidase beta but developed progressively worsening venous access. Our case illustrates the importance of the systematic screening of patients with clinical evidence of hypertrophic cardiomyopathy in whom the routine diagnostic process fails to discover Fabry disease, in particular variants with late-onset cardiac manifestations. Many of the late-onset variants are amenable to orally active therapy with migalastat, which significantly improves the comfort of the treatment. Its long-term results are being analyzed by a large international “Follow-me” registry, which was designed to verify the validity of pivotal trials with migalastat in Fabry disease.
1. Introduction
Anderson–Fabry Disease (AFD) is a rare, X-linked lysosomal storage disorder. A mutation in the GLA gene results in α-Galactosidase A enzyme (α-Gal A) deficiency, followed by the accumulation of its substrates, glycosphingolipids, namely, globotriaosylceramide (Gb3). The overloaded lysosomes in different cells cause tissue malfunction and progressive organ failure. The clinical picture varies from the classical variant (with first signs during childhood and multiorgan involvement during young adulthood) to later-onset types, mainly characterized by single-organ manifestations, most frequently by cardiac involvement mimicking hypertrophic cardiomyopathy [1]. For two decades, the disease could be treated by enzyme replacement therapy. More recently, small-molecule chaperone treatment was introduced for patients carrying amenable mutations [1,2,3]. Since early diagnosis is of utmost importance for the achievement of optimal therapeutic response, many efforts have been made to improve timely diagnosis in affected individuals. One of the most rewarding strategies is based on high-risk population screening programs. Among those, screening in patients with unexplained cardiac hypertrophy or hypertrophic cardiomyopathy has led to the highest yields of Fabry disease detection [4,5]. This approach is relevant even in the case of diagnosing advanced stages of the disease since, on average, one newly diagnosed patient allows one to find another 3–4 family members affected by the disease who may benefit from prompt specific therapy initiation [6]. In males, the diagnosis is easily made by the detection of low α-Gal A activity. Female patients are in general heterozygous. Depending on X-chromosome inactivation skewness, residual activity may overlap with normal ranges, and diagnostic efforts should be combined with measurements of the specific biomarker (globotriaosylsphingosine—lyso-Gb3) or directly with GLA gene sequencing [7,8,9]. Cases of homozygosity in females are extremely rare.
2. Case Description
We would like to report the case of a patient who was identified through a nationwide screening program that enrolled consecutive unrelated patients suffering from hypertrophic cardiomyopathy (HCM). This screening was conducted in 16 out of 17 cardiac centers in the Czech Republic, covering specialized cardiology care from June 2017 to December 2018. All subjects were tested for α-Gal A activity and lyso-Gb3 levels using the dry blood spot method. In male patients with α-Gal A activity less than 1.2 µmol/h/L and in females with either low α-Gal A activity or lyso-Gb3 levels greater than 3.5 ng/mL, Fabry disease (FD) was suspected. Positive screening results were confirmed through genetic testing.
A 55-year-old woman tested positive for low enzyme activity and elevated lyso-Gb3 (Table 1), indicating a potential severe GLA variant. A subsequent genetic test revealed the presence of a single allele carrying a missense variant, potentially suggesting a hemizygous state, raising concerns about chromosomal changes such as Turner syndrome. However, the patient’s DNA analysis showed a rare case of homozygous status. There are only a few documented cases of female AFD patients who have two α-Gal A gene mutation alleles, and even fewer who are homozygous for one mutation. One such case was previously reported in Spain [7,8,9,10,11]. The pathogenic mutation identified was c.644A>G, p.Asn215Ser (N215S) in exon 5. This variant is a well-known and widespread mutation that usually results in predominant cardiac involvement [10].

Table 1.
Dried blood spot screening results.
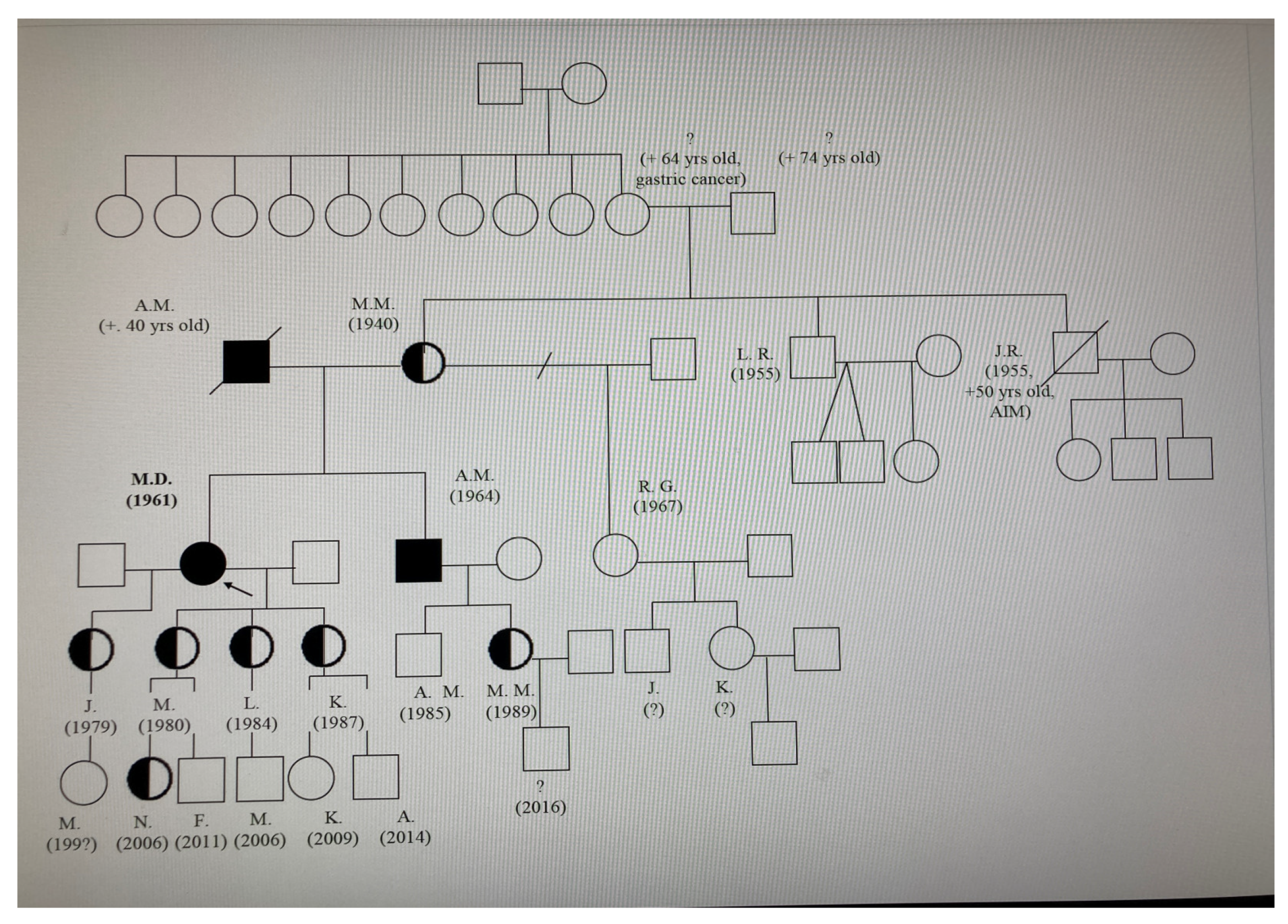
A diagram showing the family’s lineage was created (refer to Figure 1). Four members of the family agreed to undergo screening, and all were found to have the pathogenic variant through a DNA analysis.
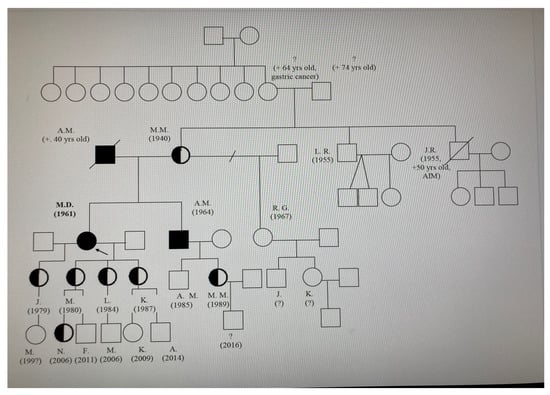
Figure 1.
Pedigree of the index patient (proband). Square: Represents a male individual in the family tree. Circle: Signifies a female individual. Line: Depicts the relationship between family members (e.g., marriage, parent-child connection). Black colour—full homozygote, half heterozygotes. Arrow—the proband.
In our index patient, the cardiovascular system was severely affected. The patient was followed up by her cardiologist from the age of 50 years. Echocardiography described progressive left ventricular hypertrophy, subsequently confirmed by a cardiac MRI scan at the age of 56. The patient also had a history of dynamic midventricular obstruction caused by dominant midventricular hypertrophy, treated by two sessions of successful alcohol septal ablation with subsequent substantial amelioration of her shortness of breath and chest pain (first alcohol septal ablation was performed at the age of 56 and repeated three years later).
Upon AFD diagnosis, the patient expressed symptoms of shortness of breath (NYHA II-III), occasional palpitations, and minimal ankle swelling that occurred mostly after prolonged standing. She did not experience any syncope, and her chest pain resolved after alcohol septal ablation. A physical examination showed no angiokeratomas, a slight apical heave outside the medioclavicular line, a regular heart rhythm, no audible murmurs, and no other signs of heart failure. Her blood pressure and heart rate consistently remained within normal limits.
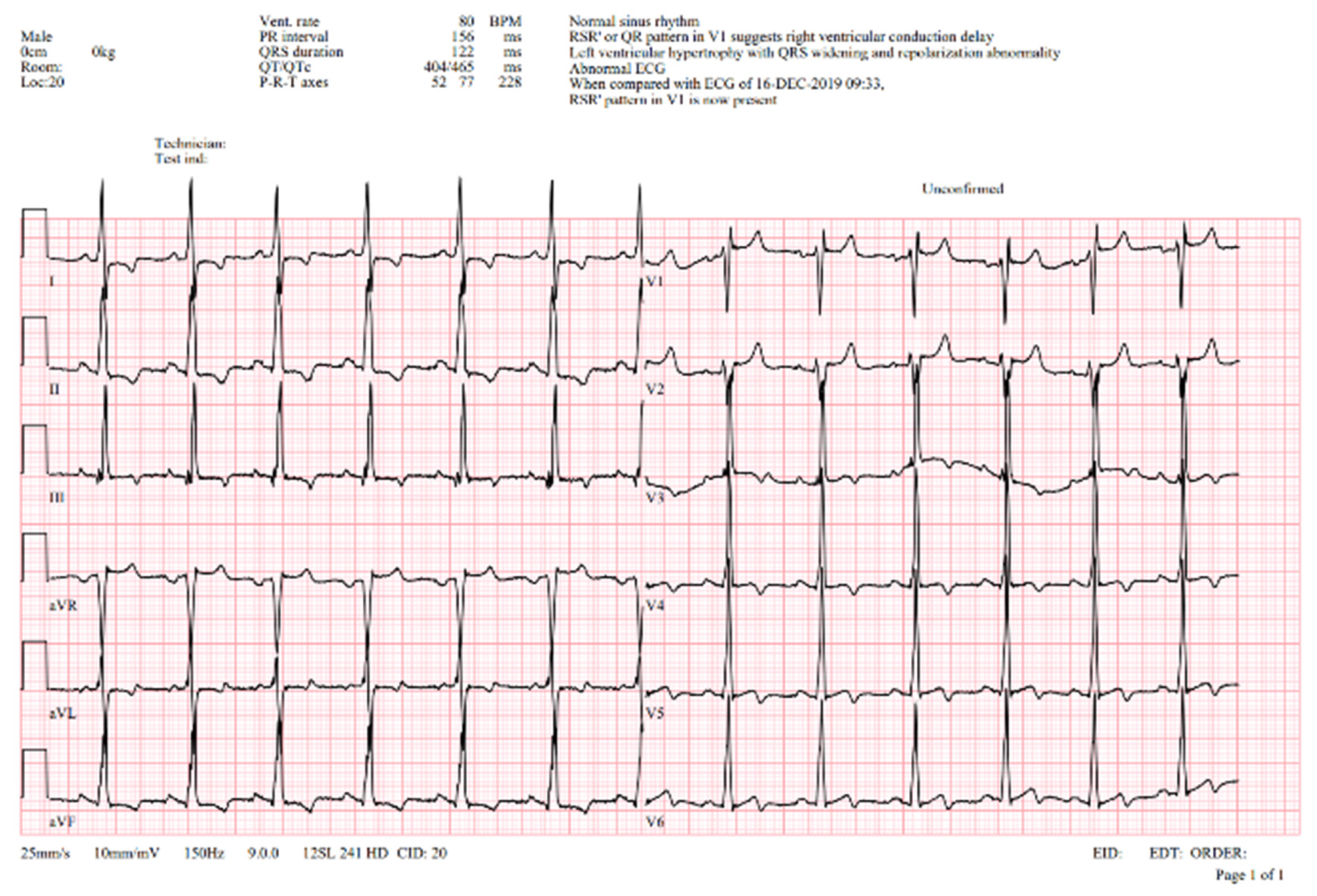
A 12-lead ECG showed signs of an incomplete right bundle branch block, a prolonged QT interval, voltage criteria for left ventricular hypertrophy, and widespread repolarization changes (Figure 2).
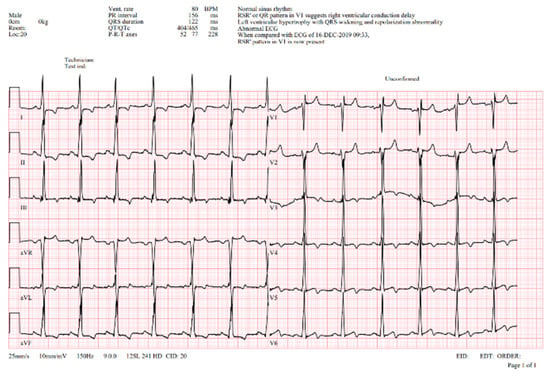
Figure 2.
ECG—abnormal ECG curve showing normal sinus rhythm, normal PQ interval (150 ms), left ventricular hypertrophy with QRS widening (122 ms) with right bundle branch pattern and repolarization abnormalities, prolonged QTc interval (Qtc 465 ms). V1-5Voltage criteria for hypertrophy with repolarization abnormalities.
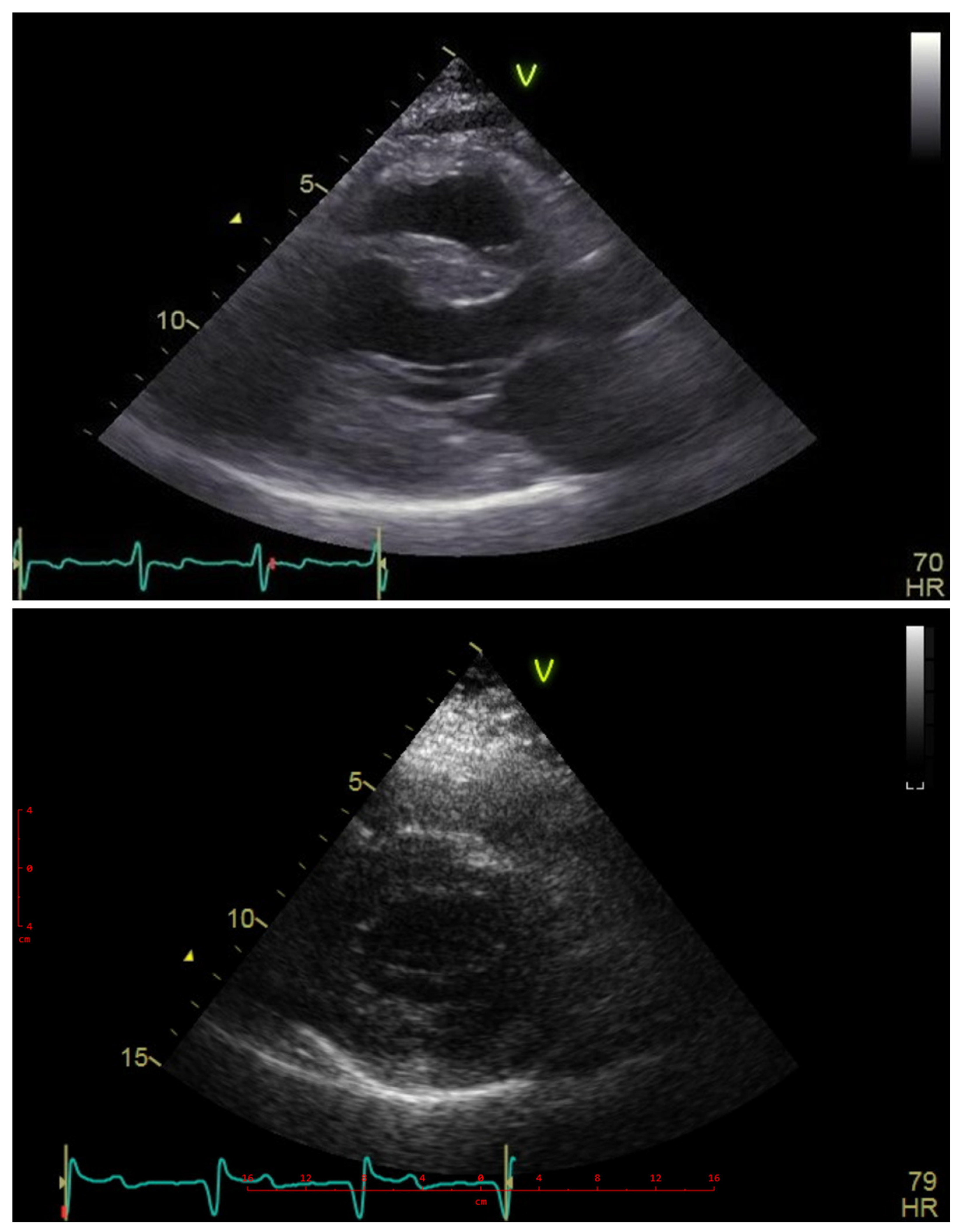
Her echocardiograms were of limited quality due to poor acoustic windows, as shown in Figure 3. The examination confirmed severe left ventricular hypertrophy, with the interventricular septum being thinner than the posterior wall. No obstructive gradient was detectable at rest or upon provocation maneuvers. The ejection fraction was normal, but Doppler indices suggested elevated filling pressures, and the left atrium was moderately dilated. The right ventricle was moderately hypertrophic and nondilated, with normal systolic function and filling pressures.
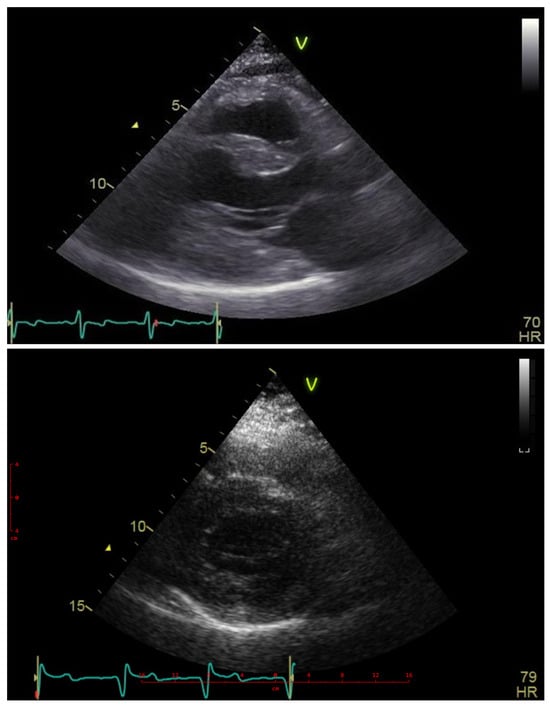
Figure 3.
Echocardiography—parasternal long- and short-axis views showing massive hypertrophy particularly within the posterior wall superseding the thickening of the interventricular septum. Blue line—ECG line, Heart Freq. 70 and 79 beats per minute.
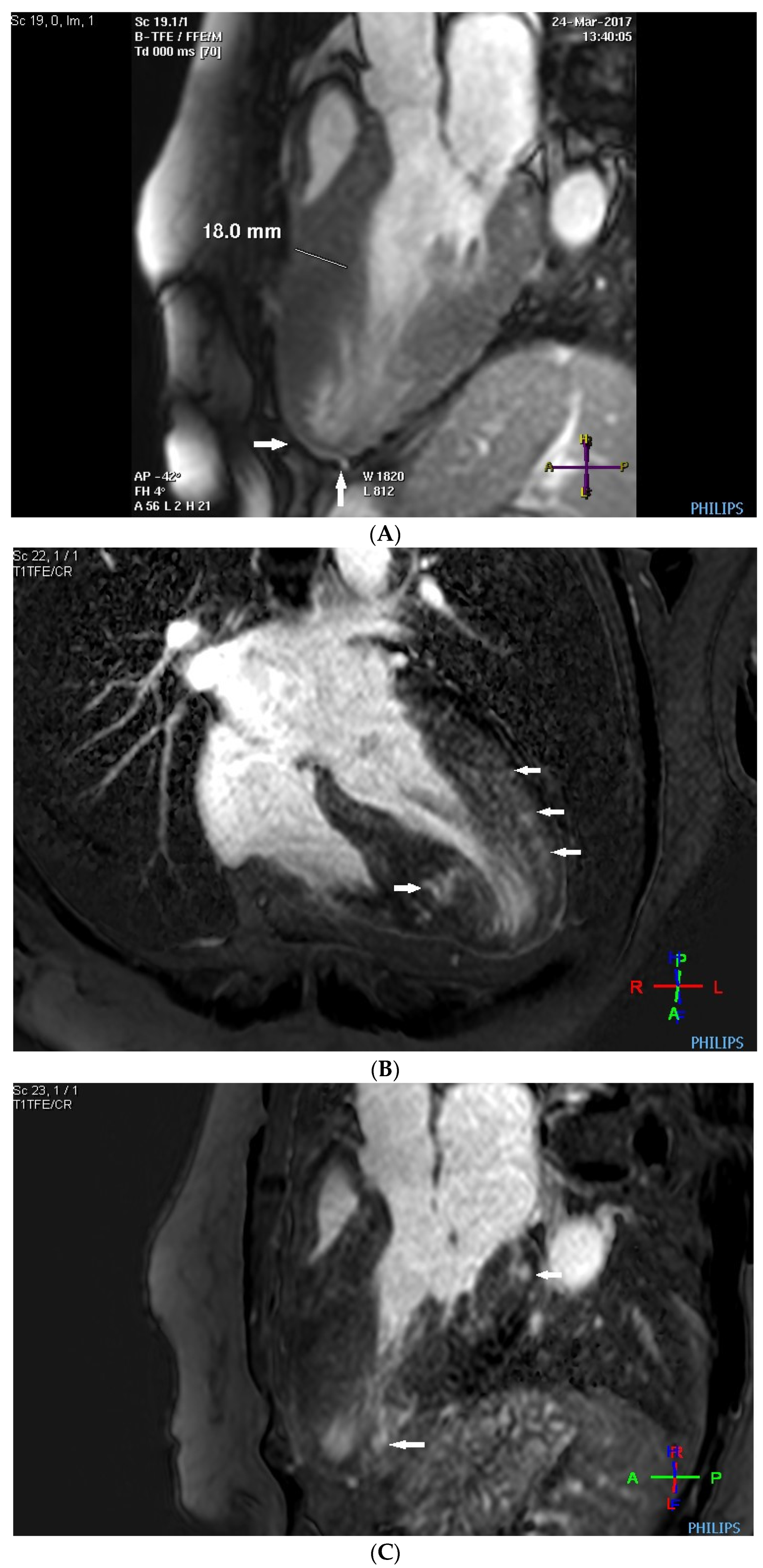
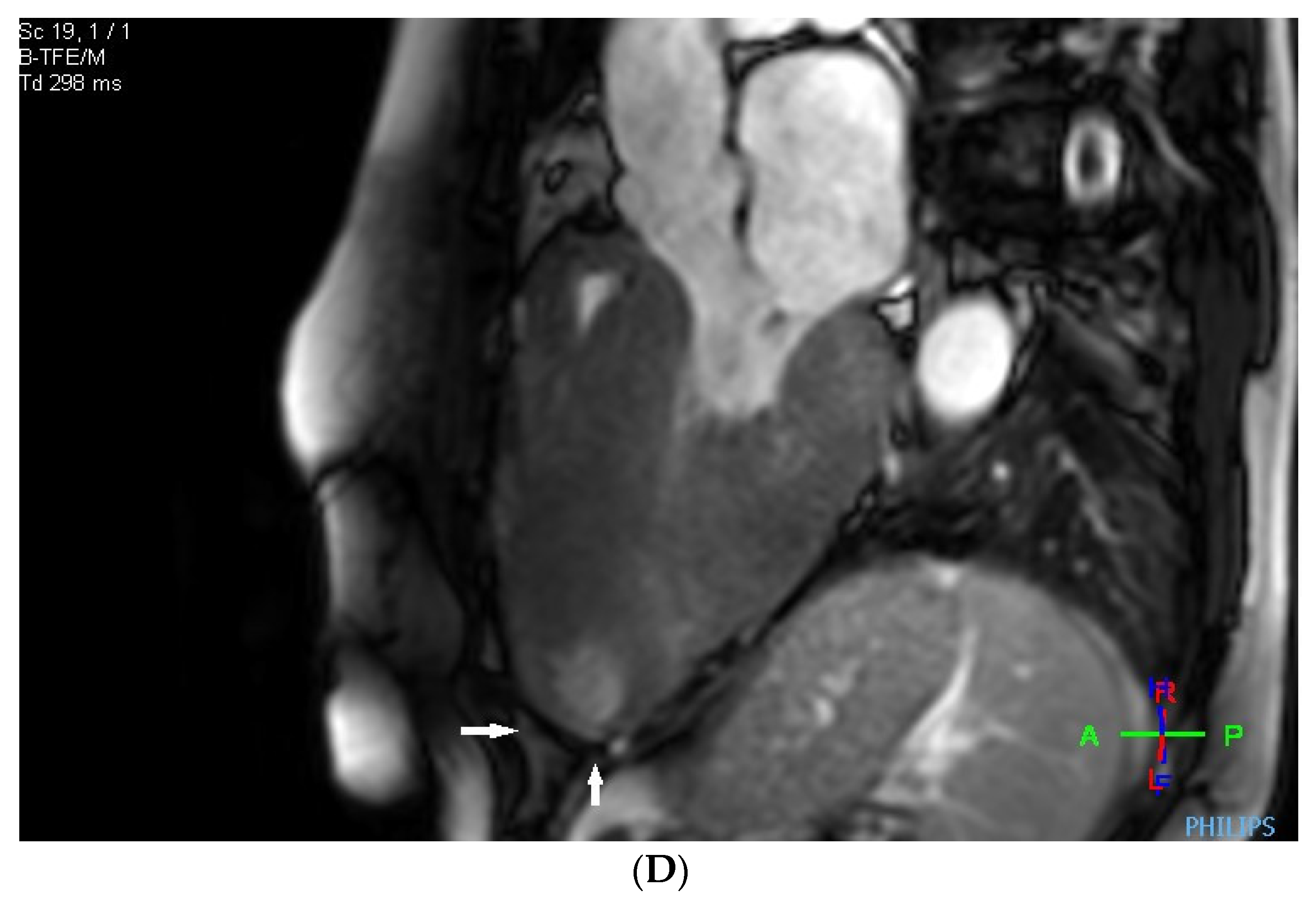
Before the alcohol septal ablation, cardiac magnetic resonance confirmed a good ejection fraction of the left ventricle. There was predominantly midventricular hypertrophy, including papillary muscles, with wall thinning in the apex due to the dynamic midventricular obstruction, as shown in Figure 4A,B. Late gadolinium enhancement revealed focal myocardial fibrosis in the inferoseptum and basal inferolateral wall, diffuse mid-wall fibrosis in the anterolateral wall, and a scar in the apical part of the inferior wall, as shown in Figure 4C,D.
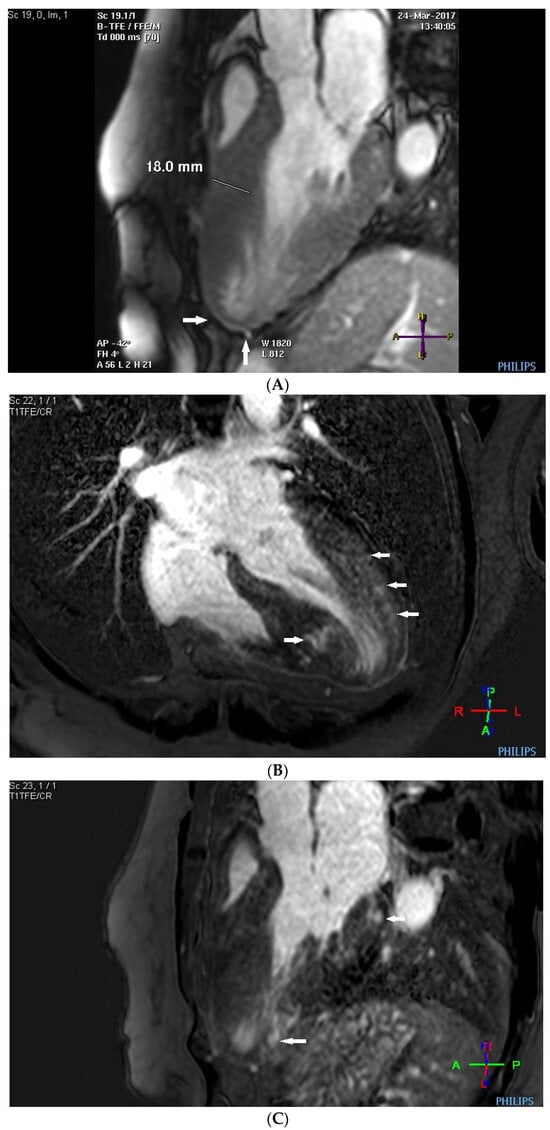
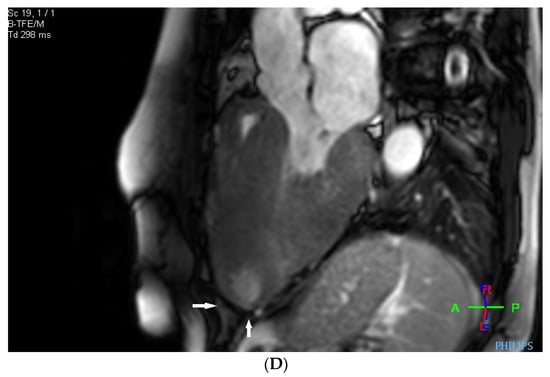
Figure 4.
(A–D) MRI scan. (A,B) Steady-state free precession cardiovascular magnetic resonance end-diastolic and end-systolic mages in long-axis views presenting dominant midventricular hypertrophy of left ventricle (including papillary muscles) and wall thinning in the apex (arrows) due to dynamic midventricular obstruction. (C,D) Late gadolinium enhancement in long-axis views demonstrating focal myocardial fibrosis in the inferoseptum and basal inferolateral wall, diffuse mid-wall fibrosis in the anterolateral wall, and scar in the apical part of inferior wall (arrows).
The patient was considered at high risk of sudden cardiac death due to the degree of hypertrophy and myocardial scarring, which resulted in cardioverter-defibrillator implantation.
The variant documented as N215S is primarily responsible for causing cardiac damage. However, even later-onset variants can exhibit a range of symptoms, including cerebrovascular issues like white matter lesions, tinnitus, vertigo, and premature stroke, as well as renal problems such as microalbuminuria and a lower GFR (Table 2).
Table 2.
BMI, MDRD, and renal and cardiac markers from 2018 to 2022.
The following may also be encountered: gastrointestinal problems like diarrhea, abdominal pain, and bloating; skin issues like angiokeratomas and hypohidrosis; and eye involvement like vessel tortuosity and cornea verticillata. In our patient, the estimated glomerular filtration rate was borderline normal with microalbuminuria between 12 and 24 mg/24 h, and conjunctival vessel tortuosity was observed (Figure 5). Moreover, she had a history of typical peripheral neuropathic pain (often described as acroparesthesias) and hypohidrosis.

Figure 5.
Eye involvement—conjunctival vessel tortuosity.
The N215S variant is characterized by a relatively high residual enzyme activity responding well to treatment with the small-molecule chaperone migalastat. In general, the treatment is well tolerated and proven to induce significant reductions in left ventricular mass [6]. After a year of undergoing migalastat treatment, the patient experienced a recurrence of severe migraine. As a result, she switched to intravenous infusion enzyme replacement therapy (agalsidase beta 1 mg/kg/every other week), which was well tolerated.
Due to limited venous access, a port was implanted but required removal after four months due to infection. The patient resumed therapy with migalastat, with no significant headaches or migraines.
After 4 years of therapy, there were no noticeable changes in her clinical condition. Nonetheless, the patient reported an improved quality of life, particularly the amelioration of exercise tolerance.
At the age of 62, the course of the disease was further complicated by sudden abdominal pain caused by intestinal ischemia causing paralytic ileus. An angioCT examination confirmed the occlusion of the celiac trunk, resulting in necrosis of the small intestine and most of the colon. Unfortunately, the outcome of the attempted surgery was fatal.
Three affected members of her family were thoroughly investigated, with some presenting typical signs and symptoms of FD. The most severe involvement was encountered in her 57-year-old brother, who has significant cardiac hypertrophy with heart failure, peripheral neuropathy, vessel tortuosities, and microalbuminuria and suffers from depression. Her 39-year-old niece has microalbuminuria and borderline left ventricular wall thickness, while her 30-year-old daughter remains asymptomatic. Therapy was started for the proband’s brother and niece.
3. Discussion
AFD is a disorder that affects lysosomes and is linked to the X chromosome. Female carriers of the disorder may exhibit a broad range of symptoms. Some heterozygous females may remain oligosymptomatic due to skewed X inactivation and mosaicism within tissues. In most females, the course of the disease is mitigated by at least some residual enzyme activity, more so in those predominantly expressing the wild-type allele over the mutated one. However, in the case of opposite skewness, the phenotype could be as severe as in males and resemble our special case [8].
However, in this case of rare homozygosity, the mitigation of the clinical manifestation by the remaining unaffected allele is impossible. In our patient, this was reflected by high lyso-Gb3 and low residual enzyme activity, showing values usually seen in male patients with late-onset variants. The course of the disease was also comparable to that in male patients with severe cardiac involvement leading to multiple complications.
Although several indices for the early recognition of AFD are known, including typical ECG features and imaging characteristics [12,13,14], late diagnosis is frequent and misclassification as HCM typical. This was clearly documented by our recent screening study among patients bearing the HCM diagnosis in tertiary centers [15]. Systematic screening among patients with HCM may reveal AFD earlier; however, as our study demonstrates, later-onset cardiac variants may be missed due to low lyso-Gb3 values and preserved residual enzyme activity. Unfortunately, in our patients, AFD was diagnosed late, already having irreversible cardiac changes, which prevented achieving a more favorable outcome despite chaperone and enzyme replacement therapy.
4. Conclusions
Although AFD is a rare inherited disease, screening programs in patients with HCM or unexplained left ventricular hypertrophy have shown that between 1% and 4% of cases are caused by AFD. With enzyme replacement or chaperone treatment available, it is crucial to diagnose the disease as early as possible to ensure better patient outcomes.
Author Contributions
Conceptualization and resources, G.D., D.Z. and A.L.; resources, A.L. data curation, G.D., J.J., M.V., P.R., A.T., M.P., D.Z. and A.L.; writing—original draft preparation, G.D. and A.L.; writing—review and editing, G.D., J.J., M.V., P.R., A.T., M.P. and D.Z.; supervision and funding acquisition, A.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by a grant from Sanofi/Genzyme GZ-2017-11701.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board /Ethics Committee/ of general University Hospital in Prague.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors obtained written informed consent from the patient’s family for publication. The authors would like to the patient’s family for their cooperation and agreement to publish their results.
Conflicts of Interest
G.D.: honoraria and consulting fees from Amicus Therapeutics, Sanofi, Chiesi and Takeda. SI the Bridge study PB-102-F30 NCT03018730. J.J.: nothing to declare. M.V.: nothing to declare. D.Z.: Speaking honoraria from Sanofi and Takeda. P.R.: Speaking honoraria from Sanofi and Takeda. A.T.: nothing to declare. M.P.: nothing to declare. A.L.: Fabry Registry Board member, honoraria and consulting fees from Amicus Therapeutics, Sanofi, Chiesi and Takeda. PI the Bridge study PB-102-F30 NCT03018730.
References
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Germain, D.P.; Fouilhoux, A.; Decramer, S.; Tardieu, M.; Pillet, P.; Fila, M.; Rivera, S.; Deschênes, G.; Lacombe, D. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin. Genet. 2019, 96, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, T.P.; Foresto, R.D.; Kirsztajn, G.M. Fabry disease: Genetics, pathology, and treatment. Rev. Assoc. Med. Bras. 2020, 66 (Suppl. S1), s10–s16. [Google Scholar] [CrossRef] [PubMed]
- Linhart, A.; Cecchi, F. Common presentation of rare diseases: Left ventricular hypertrophy and diastolic dysfunction. Int. J. Cardiol. 2018, 257, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Palecek, T.; Honzikova, J.; Poupetova, H.; Vlaskova, H.; Kuchynka, P.; Golan, L.; Magage, S.; Linhart, A. Prevalence of Fabry disease in male patients with unexplained left ventricular hypertrophy in primary cardiology practice: Prospective Fabry cardiomyopathy screening study (FACSS). J. Inherit. Metab. Dis. 2014, 37, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Elliott, P.M.; Falissard, B.; Fomin, V.V.; Hilz, M.J.; Jovanovic, A.; Kantola, I.; Linhart, A.; Mignani, R.; Namdar, M.; et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts. Mol. Genet. Metab. Rep. 2019, 19, 100454. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Mechtler, T.; Kasper, D.C.; Desnick, R.J. Correlation of Lyso-Gb3 levels in dried blood spots and sera from patients with classic and Later-Onset Fabry disease. Mol. Genet. Metab. 2017, 121, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Balendran, S.; Oliva, P.; Sansen, S.; Mechtler, T.P.; Streubel, B.; Cobos, P.N.; Lukacs, Z.; Kasper, D.C. Diagnostic strategy for females suspected of Fabry disease. Clin. Genet. 2020, 97, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Brand, E.; Burlina, A.; Cecchi, F.; Garman, S.C.; Kempf, J.; Laney, D.A.; Linhart, A.; Maródi, L.; Nicholls, K.; et al. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: A multicenter Fabry Registry study. Mol. Genet. Genom. Med. 2018, 6, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Marí; Coll, M.J.; Chabás, A. Molecular analysis in Fabry disease in Spain: Fifteen novel GLA mutations and identification of a homozygous female. Hum. Mutat. 2003, 22, 258. [Google Scholar] [CrossRef] [PubMed]
- Figliozzi, S. ECG-based score estimates the probability to detect Fabry Disease cardiac involvement. Int. J. Cardiol. 2021, 339, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Augusto, J.B. The myocardial phenotype of Fabry disease prehypertrophy and pre-detectable storage. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G. Standard ECG for differential diagnosis between Anderson-Fabry disease and hypertrophic cardiomyopathy. Heart 2022, 108, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Zemánek, D.; Januška, J.; Honěk, T.; Čurila, K.; Kubánek, M.; Šindelářová, Š.; Zahálková, L.; Klofáč, P.; Laštůvková, E.; Lichnerová, E.; et al. Nationwide screening of Fabry disease in patients with hypertrophic cardiomyopathy in Czech Republic. ESC Heart Fail. 2022, 9, 4160–4166. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).