
Gene Therapy for Inherited Arrhythmia Syndromes

Abstract
1. Introduction
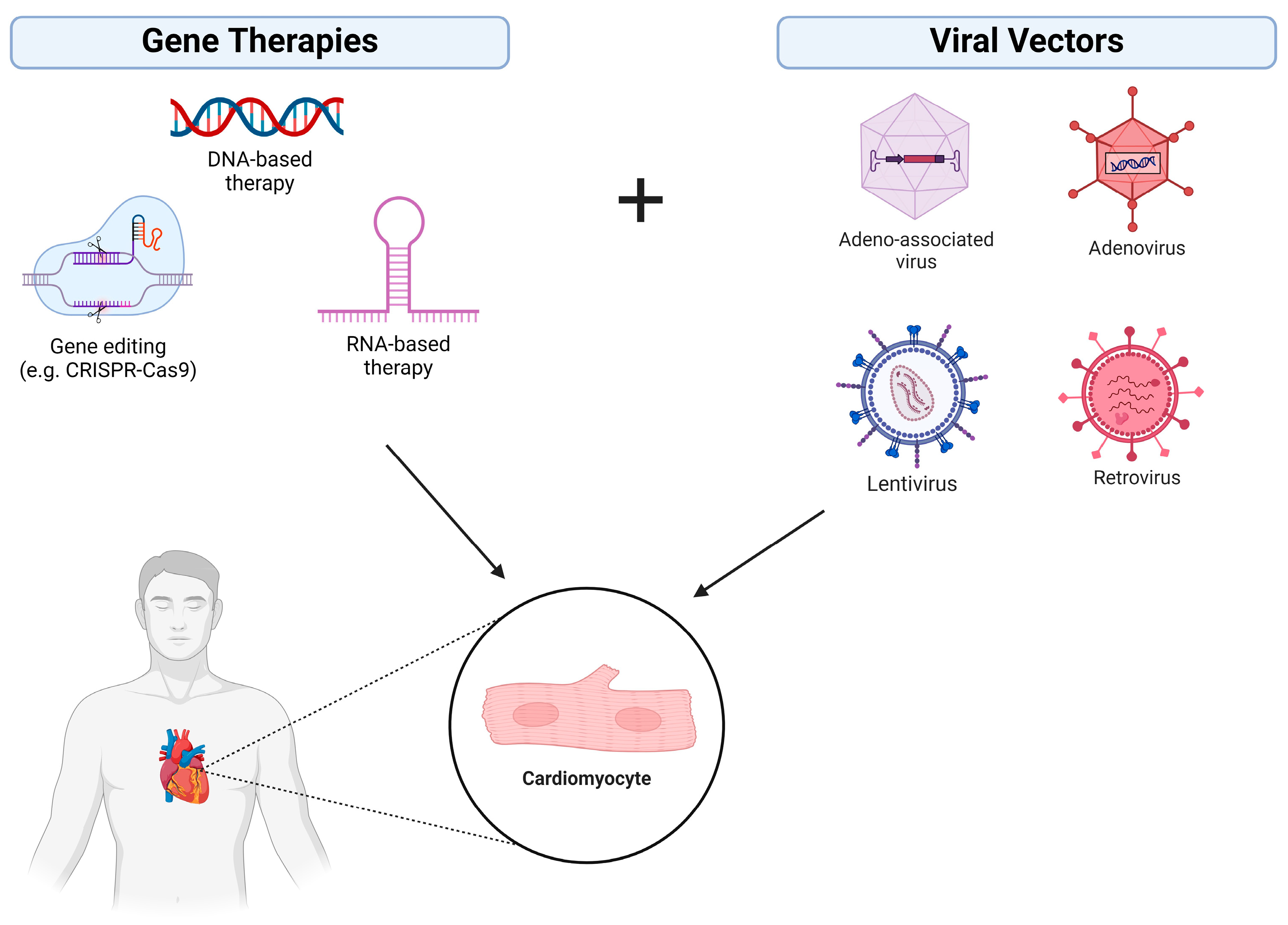
1.1. Gene Augmentation/Replacement
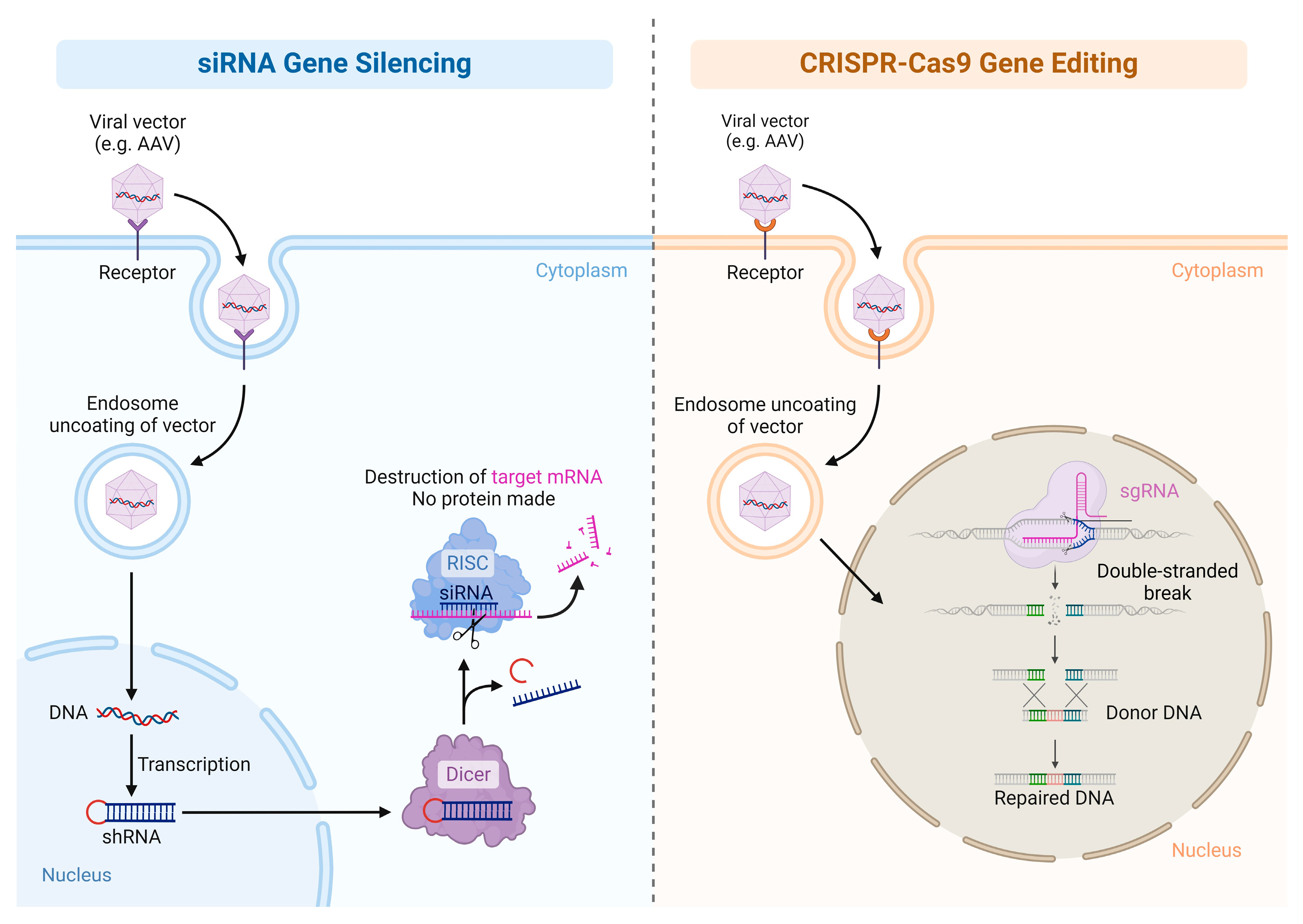
1.2. Gene Inhibition Therapy
1.3. Gene-Editing Therapy
2. Gene Therapy Vectors—The Evolution, Where We Are, and Where We Are Going
2.1. Viral Vectors
2.2. Non-Viral Vectors
3. Basic Science and Animal Studies Pertaining to Gene Therapies and Inherited Arrhythmia Syndromes
3.1. Long QT Syndrome
3.1.1. Jervell and Lange-Niellson Syndrome, Anderson Tawil Syndrome, and Timothy Syndrome
3.1.2. Management and Gene Therapy for LQTS
3.2. Catecholaminergic Polymorphic Ventricular Tachycardia
3.3. Brugada Syndrome
3.4. Arrhythmogenic Right Ventricular Cardiomyopathy
4. Discussion
4.1. Integration of Gene Therapy with Clinical Medicine
4.2. Limitations of Gene Therapy
4.3. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwartz, P.J.; Ackerman, M.J.; Antzelevitch, C.; Bezzina, C.R.; Borggrefe, M.; Cuneo, B.F.; Wilde, A.A.M. Inherited Cardiac Arrhythmias. Nat. Rev. Dis. Primer 2020, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Belete, T.M. The Current Status of Gene Therapy for the Treatment of Cancer. Biol. Targets Ther. 2021, 15, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Kaji, E.H.; Leiden, J.M. Gene and Stem Cell Therapies. JAMA 2001, 285, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Argirò, A.; Ding, J.; Adler, E. Gene Therapy for Heart Failure and Cardiomyopathies. Rev. Esp. Cardiol. Engl. Ed. 2023, 76, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Nóbrega, C.; Mendonça, L.; Matos, C.A. Gene Therapy Strategies: Gene Augmentation. In A Handbook of Gene and Cell Therapy; Nóbrega, C., Mendonça, L., Matos, C.A., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 117–126. ISBN 978-3-030-41333-0. [Google Scholar]
- Gerhardt, M.J.; Priglinger, C.S.; Rudolph, G.; Hufendiek, K.; Framme, C.; Jägle, H.; Salchow, D.J.; Anschütz, A.; Michalakis, S.; Priglinger, S.G. Gene Therapy with Voretigene Neparvovec Improves Vision and Partially Restores Electrophysiological Function in Pre-School Children with Leber Congenital Amaurosis. Biomedicines 2022, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Ozelo, M.C.; Mahlangu, J.; Pasi, K.J.; Giermasz, A.; Leavitt, A.D.; Laffan, M.; Symington, E.; Quon, D.V.; Wang, J.-D.; Peerlinck, K.; et al. Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. N. Engl. J. Med. 2022, 386, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Pipe, S.W.; Leebeek, F.W.; Recht, M.; Key, N.S.; Castaman, G.; Miesbach, W.; Lattimore, S.; Peerlinck, K.; Van der Valk, P.; Coppens, M.; et al. Gene Therapy with Etranacogene Dezaparvovec for Hemophilia B. N. Engl. J. Med. 2023, 388, 706–718. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.-J. Therapeutic siRNA: State of the Art. Signal Transduct. Target. Ther. 2020, 5, 1–25. [Google Scholar] [CrossRef]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro Story and the Clinical Translation of Nanomedicines Containing Nucleic Acid-Based Drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria | New England Journal of Medicine. Available online: https://www.nejm.org/doi/full/10.1056/NEJMoa1913147 (accessed on 12 May 2024).
- Hoess, R.H.; Abremski, K. The Cre-Lox Recombination System. In Nucleic Acids and Molecular Biology 4; Eckstein, F., Lilley, D.M.J., Eds.; Springer: Berlin/Heidelberg, Germany, 1990; pp. 99–109. ISBN 978-3-642-84150-7. [Google Scholar]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of Genome Editing Technology in the Targeted Therapy of Human Diseases: Mechanisms, Advances and Prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef]
- Gupta, R.M.; Musunuru, K. Expanding the Genetic Editing Tool Kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef]
- Guilinger, J.P.; Pattanayak, V.; Reyon, D.; Tsai, S.Q.; Sander, J.D.; Joung, J.K.; Liu, D.R. Broad Specificity Profiling of TALENs Results in Engineered Nucleases With Improved DNA Cleavage Specificity. Nat. Methods 2014, 11, 429–435. [Google Scholar] [CrossRef]
- Barrangou, R. The Roles of CRISPR–Cas Systems in Adaptive Immunity and Beyond. Curr. Opin. Immunol. 2015, 32, 36–41. [Google Scholar] [CrossRef]
- Frangoul, H.; Locatelli, F.; Sharma, A.; Bhatia, M.; Mapara, M.; Molinari, L.; Wall, D.; Liem, R.I.; Telfer, P.; Shah, A.J.; et al. Exagamglogene Autotemcel for Severe Sickle Cell Disease. N. Engl. J. Med. 2024, 390, 1649–1662. [Google Scholar] [CrossRef]
- FDA Commissioner. FDA Approves First Gene Therapies to Treat Patients with Sickle Cell Disease. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease (accessed on 12 May 2024).
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Chalberg, T.W.; Schaffer, D.V. Viral Vectors for Gene Therapy: Translational and Clinical Outlook. Annu. Rev. Biomed. Eng. 2015, 17, 63–89. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Prondzynski, M.; Carrier, L.; Pu, W.T. Gene Therapy for Inherited Arrhythmias. Cardiovasc. Res. 2020, 116, 1635–1650. [Google Scholar] [CrossRef]
- Romano, G. Development of Safer Gene Delivery Systems to Minimize the Risk of Insertional Mutagenesis-Related Malignancies: A Critical Issue for the Field of Gene Therapy. ISRN Oncol. 2012, 2012, 616310. [Google Scholar] [CrossRef]
- Zylberberg, C.; Gaskill, K.; Pasley, S.; Matosevic, S. Engineering Liposomal Nanoparticles for Targeted Gene Therapy. Gene Ther. 2017, 24, 441–452. [Google Scholar] [CrossRef]
- Nsairat, H.; Alshaer, W.; Odeh, F.; Esawi, E.; Khater, D.; Bawab, A.A.; El-Tanani, M.; Awidi, A.; Mubarak, M.S. Recent Advances in Using Liposomes for Delivery of Nucleic Acid-Based Therapeutics. OpenNano 2023, 11, 100132. [Google Scholar] [CrossRef]
- Evers, M.J.W.; Du, W.; Yang, Q.; Kooijmans, S.A.A.; Vink, A.; van Steenbergen, M.; Vader, P.; de Jager, S.C.A.; Fuchs, S.A.; Mastrobattista, E.; et al. Delivery of Modified mRNA to Damaged Myocardium by Systemic Administration of Lipid Nanoparticles. J. Control. Release 2022, 343, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Taylor, A.; Shi, H.; Zhou, F.; Li, P.; Joshi, J.; Zhu, W.; Wang, S. Peptide-Guided Nanoparticle Drug Delivery for Cardiomyocytes. Biology 2024, 13, 47. [Google Scholar] [CrossRef]
- Labonia, M.C.I.; Estape Senti, M.; Van Der Kraak, P.H.; Brans, M.A.D.; Deshantri, A.K.; Dokter, I.; Schiffelers, R.M.; Sluijter, J.P.G.; Vader, P. Effective Cardiac mRNA Delivery Using Lipid Nanoparticles. Eur. Heart J. 2023, 44, ehad655.3301. [Google Scholar] [CrossRef]
- Khawajakhail, R.; Khan, R.U.; Gondal, M.U.R.; Toru, H.K.; Malik, M.; Iqbal, A.; Malik, J.; Faraz, M.; Awais, M. Advancements in Gene Therapy Approaches for Atrial Fibrillation: Targeted Delivery, Mechanistic Insights and Future Prospects. Curr. Probl. Cardiol. 2024, 49, 102431. [Google Scholar] [CrossRef]
- Williams, P.D.; Kingston, P.A. Plasmid-Mediated Gene Therapy for Cardiovascular Disease. Cardiovasc. Res. 2011, 91, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Dotzler, S.M.; Kim, C.S.J.; Gendron, W.A.C.; Zhou, W.; Ye, D.; Bos, J.M.; Tester, D.J.; Barry, M.A.; Ackerman, M.J. Suppression-Replacement KCNQ1 Gene Therapy for Type 1 Long QT Syndrome. Circulation 2021, 143, 1411–1425. [Google Scholar] [CrossRef]
- Bains, S.; Zhou, W.; Dotzler, S.M.; Martinez, K.; Kim, C.J.; Tester, D.J.; Ye, D.; Ackerman, M.J. Suppression and Replacement Gene Therapy for KCNH2-Mediated Arrhythmias. Circ. Genomic Precis. Med. 2022, 15, e003719. [Google Scholar] [CrossRef]
- Denegri, M.; Bongianino, R.; Lodola, F.; Boncompagni, S.; De Giusti, V.C.; Avelino-Cruz, J.E.; Liu, N.; Persampieri, S.; Curcio, A.; Esposito, F.; et al. Single Delivery of an Adeno-Associated Viral Construct to Transfer the CASQ2 Gene to Knock-In Mice Affected by Catecholaminergic Polymorphic Ventricular Tachycardia Is Able to Cure the Disease From Birth to Advanced Age. Circulation 2014, 129, 2673–2681. [Google Scholar] [CrossRef]
- Kurtzwald-Josefson, E.; Yadin, D.; Harun-Khun, S.; Waldman, M.; Aravot, D.; Shainberg, A.; Eldar, M.; Hochhauser, E.; Arad, M. Viral Delivered Gene Therapy to Treat Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT2) in Mouse Models. Heart Rhythm 2017, 14, 1053–1060. [Google Scholar] [CrossRef]
- Bongianino, R.; Denegri, M.; Mazzanti, A.; Lodola, F.; Vollero, A.; Boncompagni, S.; Fasciano, S.; Rizzo, G.; Mangione, D.; Barbaro, S.; et al. Allele-Specific Silencing of Mutant mRNA Rescues Ultrastructural and Arrhythmic Phenotype in Mice Carriers of the R4496C Mutation in the Ryanodine Receptor Gene (RYR2). Circ. Res. 2017, 121, 525–536. [Google Scholar] [CrossRef]
- Pan, X.; Philippen, L.; Lahiri, S.K.; Lee, C.; Park, S.H.; Word, T.A.; Li, N.; Jarrett, K.E.; Gupta, R.; Reynolds, J.O.; et al. In Vivo Ryr2 Editing Corrects Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2018, 123, 953–963. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Caballero, A.; Wang, S.; Ai, Y.; Hylind, R.J.; Lu, F.; Heims-Waldron, D.A.; Chambers, K.D.; Zhang, D.; Abrams, D.J.; et al. Gene Therapy for Catecholaminergic Polymorphic Ventricular Tachycardia by Inhibition of Ca2+/Calmodulin-Dependent Kinase II. Circulation 2019, 140, 405–419. [Google Scholar] [CrossRef]
- Yu, G.; Chakrabarti, S.; Tischenko, M.; Chen, A.-L.; Wang, Z.; Cho, H.; French, B.A.; Naga Prasad, S.V.; Chen, Q.; Wang, Q.K. Gene Therapy Targeting Protein Trafficking Regulator MOG1 in Mouse Models of Brugada Syndrome, Arrhythmias, and Mild Cardiomyopathy. Sci. Transl. Med. 2022, 14, eabf3136. [Google Scholar] [CrossRef]
- Bradford, W.H.; Zhang, J.; Gutierrez-Lara, E.J.; Liang, Y.; Do, A.; Wang, T.-M.; Nguyen, L.; Mataraarachchi, N.; Wang, J.; Gu, Y.; et al. Plakophilin 2 Gene Therapy Prevents and Rescues Arrhythmogenic Right Ventricular Cardiomyopathy in a Mouse Model Harboring Patient Genetics. Nat. Cardiovasc. Res. 2023, 2, 1246–1261. [Google Scholar] [CrossRef]
- van Opbergen, C.J.; Narayanan, B.; Sacramento, C.B.; Stiles, K.M.; Mishra, V.; Frenk, E.; Ricks, D.; Chen, G.; Zhang, M.; Yarabe, P.; et al. AAV-Mediated Delivery of Plakophilin-2a Arrests Progression of Arrhythmogenic Right Ventricular Cardiomyopathy in Murine Hearts: Preclinical Evidence Supporting Gene Therapy in Humans. Circ. Genom. Precis. Med. 2024, 17, e004305. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Crotti, L.; Insolia, R. Long-QT Syndrome. Circ. Arrhythm. Electrophysiol. 2012, 5, 868–877. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Crotti, L. QTc Behavior During Exercise and Genetic Testing for the Long-QT Syndrome. Circulation 2011, 124, 2181–2184. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Amin, A.S.; Postema, P.G. Diagnosis, Management and Therapeutic Strategies for Congenital Long QT Syndrome. Heart 2022, 108, 332–338. [Google Scholar] [CrossRef]
- Tranebjærg, L.; Samson, R.A.; Green, G.E. Jervell and Lange-Nielsen Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Schwartz, P.J.; Spazzolini, C.; Crotti, L.; Bathen, J.; Amlie, J.P.; Timothy, K.; Shkolnikova, M.; Berul, C.I.; Bitner-Glindzicz, M.; Toivonen, L.; et al. The Jervell and Lange-Nielsen Syndrome: Natural History, Molecular Basis, and Clinical Outcome. Circulation 2006, 113, 783–790. [Google Scholar] [CrossRef]
- Veerapandiyan, A.; Statland, J.M.; Tawil, R. Andersen-Tawil Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Jiang, C.; Zhang, Y. Current Updates on Arrhythmia within Timothy Syndrome: Genetics, Mechanisms and Therapeutics. Expert Rev. Mol. Med. 2023, 25, e17. [Google Scholar] [CrossRef]
- Moss, A.J.; Zareba, W.; Hall, W.J.; Schwartz, P.J.; Crampton, R.S.; Benhorin, J.; Vincent, G.M.; Locati, E.H.; Priori, S.G.; Napolitano, C.; et al. Effectiveness and Limitations of β-Blocker Therapy in Congenital Long-QT Syndrome. Circulation 2000, 101, 616–623. [Google Scholar] [CrossRef]
- Went, T.R.; Sultan, W.; Sapkota, A.; Khurshid, H.; Qureshi, I.A.; Jahan, N.; Tara, A.; Win, M.; Wiltshire, D.A.; Kannan, A.; et al. A Systematic Review on the Role of Βeta-Blockers in Reducing Cardiac Arrhythmias in Long QT Syndrome Subtypes 1-3. Cureus 2021, 13, e17632. [Google Scholar] [CrossRef]
- Saadeh, K.; Shivkumar, K.; Jeevaratnam, K. Targeting the β-Adrenergic Receptor in the Clinical Management of Congenital Long QT Syndrome. Ann. N. Y. Acad. Sci. 2020, 1474, 27–46. [Google Scholar] [CrossRef]
- Ghzally, Y.; Mahajan, K. Implantable Defibrillator. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Leenhardt, A.; Denjoy, I.; Guicheney, P. Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Arrhythm. Electrophysiol. 2012, 5, 1044–1052. [Google Scholar] [CrossRef]
- Györke, S. Molecular Basis of Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Rhythm 2009, 6, 123–129. [Google Scholar] [CrossRef]
- Sumitomo, N.; Harada, K.; Nagashima, M.; Yasuda, T.; Nakamura, Y.; Aragaki, Y.; Saito, A.; Kurosaki, K.; Jouo, K.; Koujiro, M.; et al. Catecholaminergic Polymorphic Ventricular Tachycardia: Electrocardiographic Characteristics and Optimal Therapeutic Strategies to Prevent Sudden Death. Heart 2003, 89, 66–70. [Google Scholar] [CrossRef]
- Peltenburg, P.J.; Kallas, D.; Bos, J.M.; Lieve, K.V.V.; Franciosi, S.; Roston, T.M.; Denjoy, I.; Sorensen, K.B.; Ohno, S.; Roses-Noguer, F.; et al. An International Multicenter Cohort Study on β-Blockers for the Treatment of Symptomatic Children with Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2022, 145, 333–344. [Google Scholar] [CrossRef]
- Pérez, P.R.; Hylind, R.J.; Roston, T.M.; Bezzerides, V.J.; Abrams, D.J. Gene Therapy for Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Lung Circ. 2023, 32, 790–797. [Google Scholar] [CrossRef]
- El Sayed, M.; Goyal, A.; Callahan, A.L. Brugada Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Antzelevitch, C. Genetic Basis of Brugada Syndrome. Heart Rhythm Off. J. Heart Rhythm Soc. 2007, 4, 756–757. [Google Scholar] [CrossRef]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right Ventricular Cardiomyopathy and Sudden Death in Young People. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef]
- Marcus, F.I.; Edson, S.; Towbin, J.A. Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy. J. Am. Coll. Cardiol. 2013, 61, 1945–1948. [Google Scholar] [CrossRef]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the Desmosomal Protein Plakophilin-2 Are Common in Arrhythmogenic Right Ventricular Cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- Kirchner, F.; Schuetz, A.; Boldt, L.-H.; Martens, K.; Dittmar, G.; Haverkamp, W.; Thierfelder, L.; Heinemann, U.; Gerull, B. Molecular Insights into Arrhythmogenic Right Ventricular Cardiomyopathy Caused by Plakophilin-2 Missense Mutations. Circ. Cardiovasc. Genet. 2012, 5, 400–411. [Google Scholar] [CrossRef]
- Rasmussen, T.B.; Nissen, P.H.; Palmfeldt, J.; Gehmlich, K.; Dalager, S.; Jensen, U.B.; Kim, W.Y.; Heickendorff, L.; Mølgaard, H.; Jensen, H.K.; et al. Truncating Plakophilin-2 Mutations in Arrhythmogenic Cardiomyopathy Are Associated with Protein Haploinsufficiency in Both Myocardium and Epidermis. Circ. Cardiovasc. Genet. 2014, 7, 230–240. [Google Scholar] [CrossRef]
- Corrado, D.; Wichter, T.; Link, M.S.; Hauer, R.; Marchlinski, F.; Anastasakis, A.; Bauce, B.; Basso, C.; Brunckhorst, C.; Tsatsopoulou, A.; et al. Treatment of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: An International Task Force Consensus Statement. Eur. Heart J. 2015, 36, 3227–3237. [Google Scholar] [CrossRef]
- Sasaki, N.; Kok, C.Y.; Westhaus, A.; Alexander, I.E.; Lisowski, L.; Kizana, E. In Search of Adeno-Associated Virus Vectors with Enhanced Cardiac Tropism for Gene Therapy. Heart Lung Circ. 2023, 32, 816–824. [Google Scholar] [CrossRef]
- Breton, C.; Furmanak, T.; Avitto, A.N.; Smith, M.K.; Latshaw, C.; Yan, H.; Greig, J.A.; Wilson, J.M. Increasing the Specificity of AAV-Based Gene Editing through Self-Targeting and Short-Promoter Strategies. Mol. Ther. 2021, 29, 1047–1056. [Google Scholar] [CrossRef]
- Grosch, M.; Schraft, L.; Chan, A.; Küchenhoff, L.; Rapti, K.; Ferreira, A.-M.; Kornienko, J.; Li, S.; Radke, M.H.; Krämer, C.; et al. Striated Muscle-Specific Base Editing Enables Correction of Mutations Causing Dilated Cardiomyopathy. Nat. Commun. 2023, 14, 3714. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Li, D.; Wang, N.; Gruber, J.; Lo, A.W.; Conti, R.M. The Estimated Annual Financial Impact of Gene Therapy in the United States. Gene Ther. 2023, 30, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of Clinical Trial Success Rates and Related Parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.D.; Panzer, A.D.; Kim, D.D.; Margaretos, N.M.; Neumann, P.J. Variation in US Private Health Plans’ Coverage of Orphan Drugs. Am. J. Manag. Care 2019, 25, 508–512. [Google Scholar] [PubMed]
- Abramochkin, D.; Li, B.; Zhang, H.; Kravchuk, E.; Nesterova, T.; Glukhov, G.; Shestak, A.; Zaklyazminskaya, E.; Sokolova, O.S. Novel Gain-of-Function Mutation in the Kv11.1 Channel Found in the Patient with Brugada Syndrome and Mild QTc Shortening. Biochem. Mosc. 2024, 89, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.M.; Macías, Á.; Moreno-Manuel, A.I.; Gutiérrez, L.K.; Vera-Pedrosa, M.L.; Martínez-Carrascoso, I.; Pérez, P.S.; Robles, J.M.; Bermúdez-Jiménez, F.J.; Díaz-Agustín, A.; et al. Extracellular Cysteine Disulfide Bond Break at Cys122 Disrupts PIP2-Dependent Kir2.1 Channel Function and Leads to Arrhythmias in Andersen-Tawil Syndrome. bioRxiv 2023. [Google Scholar] [CrossRef]
- Pirruccello, J.P.; Di Achille, P.; Nauffal, V.; Nekoui, M.; Friedman, S.F.; Klarqvist, M.D.R.; Chaffin, M.D.; Weng, L.-C.; Cunningham, J.W.; Khurshid, S.; et al. Genetic Analysis of Right Heart Structure and Function in 40,000 People. Nat. Genet. 2022, 54, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, E.; Bouzarelou, D.; Tsaousis, G.; Papathanasiou, A.; Vogiatzi, G.; Vlachopoulos, C.; Miliou, A.; Papachristou, P.; Prappa, E.; Servos, G.; et al. Application of next Generation Sequencing in Cardiology: Current and Future Precision Medicine Implications. Front. Cardiovasc. Med. 2023, 10, 1202381. [Google Scholar] [CrossRef]
- Deng, H.; Huang, W.; Zhang, Z. Nanotechnology Based CRISPR/Cas9 System Delivery for Genome Editing: Progress and Prospect. Nano Res. 2019, 12, 2437–2450. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Q.; Zhang, X.; Huang, H.; Tang, S.; Chai, Y.; Xu, Z.; Li, M.; Chen, X.; Liu, J.; et al. Recent Advances in Exosome-Mediated Nucleic Acid Delivery for Cancer Therapy. J. Nanobiotechnol. 2022, 20, 279. [Google Scholar] [CrossRef]
- Nijak, A.; Saenen, J.; Labro, A.J.; Schepers, D.; Loeys, B.L.; Alaerts, M. iPSC-Cardiomyocyte Models of Brugada Syndrome—Achievements, Challenges and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 2825. [Google Scholar] [CrossRef] [PubMed]
- Okata, S.; Yuasa, S.; Suzuki, T.; Ito, S.; Makita, N.; Yoshida, T.; Li, M.; Kurokawa, J.; Seki, T.; Egashira, T.; et al. Embryonic Type Na+ Channel β-Subunit, SCN3B Masks the Disease Phenotype of Brugada Syndrome. Sci. Rep. 2016, 6, 34198. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.C.; Cui, M.; Chemello, F.; Li, H.; Chen, K.; Tan, W.; Atmanli, A.; McAnally, J.R.; Zhang, Y.; Xu, L.; et al. Base Editing Correction of Hypertrophic Cardiomyopathy in Human Cardiomyocytes and Humanized Mice. Nat. Med. 2023, 29, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, M.; Masood, M.F.; Javed, I.; Adil, H.; Faraz, M.A.; Bhat, R.R.; Fatima, M.; Abdelkhalek, A.M.; Buccilli, B.; Raza, S.; et al. Unlocking the Mysteries, Bridging the Gap, and Unveiling the Multifaceted Potential of Stem Cell Therapy for Cardiac Tissue Regeneration: A Narrative Review of Current Literature, Ethical Challenges, and Future Perspectives. Cureus 2023, 15, e41533. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, Y.; Singh, R.; Wang, Z.Z. Arrhythmogenic Risks of Stem Cell Replacement Therapy for Cardiovascular Diseases. J. Cell. Physiol. 2020, 235, 6257–6267. [Google Scholar] [CrossRef] [PubMed]
- Schwartze, J.T.; Havenga, M.; Bakker, W.A.M.; Bradshaw, A.C.; Nicklin, S.A. Adenoviral Vectors for Cardiovascular Gene Therapy Applications: A Clinical and Industry Perspective. J. Mol. Med. Berl. Ger. 2022, 100, 875–901. [Google Scholar] [CrossRef]
- Center for Biologics Evaluation and Research. Cellular & Gene Therapy Guidances; FDA: Rockville, MD, USA, 2023.
- Spartalis, M.; Spartalis, E.; Siasos, G. Inherited Arrhythmias and Gene Therapy: Are There Any Ethical Considerations to Take into Account? World J. Cardiol. 2023, 15, 623–626. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Gene Therapy Strategy | Description | Technologies | Clinical Applications |
|---|---|---|---|
| Gene augmentation/replacement | Replacement of a defective gene with a functional version | Viral vectors (e.g., adenovirus, AAV, lentivirus), non-viral vectors (e.g., LNPs) | Spinal muscular atrophy 1, hemophilia A, hemophilia B, leber congenital amourosis |
| Gene inhibition | Downregulation of disease associated gene expression | siRNA, miRNA. shRNA | hATTR polyneuropathy, hepatic porphyria |
| Gene editing | Precise genome modification in living cells | ZFNs, TALENs, CRISPR-Cas9 | Sickle cell disease, acute lymphoblastic leukemia |
| Disease | Study | Vector | Gene Therapy Type | Disease Model |
|---|---|---|---|---|
| LQT1 | Dotzler et al. (2021) [32] | Lentivirus | SupRep—short hairpin RNA + cDNA | iPSC-CM |
| LQT2 | Bains et al. (2022) [33] | AAV9 | SupRep—short hairpin RNA + cDNA | iPSC-CM |
| CPVT | Denegri et al. (2014) [34] | AAV9 | cDNA | CASQ2 knock-out mice |
| Kurtzwald-Josefson et al. (2017) [35] | AAV9 | cDNA | CASQ2 knock-out mice | |
| Bongianino et al. (2017) [36] | AAV9 | siRNA | RyR2R4496C/+ mice | |
| Pan et al. (2018) [37] | AAV9 | CRISPR-Cas9 | RyR2R176Q/+ mice | |
| Bezzerides et al. (2019) [38] | AAV9 | Inhibitory peptide | RyR2R176Q/+ mice | |
| BrS | Yu et al. (2022) [39] | AAV9 | Gene replacement | hiCM |
| ARVC | Bradford et al. (2023) [40] | AAV9 | Gene replacement | PKP2 RNA splice acceptor mutation (PKP2 IVS10-1G>C) |
| van Opbergen et al. (2023) [41] | AAV9 | Gene replacement | PKP2 knock-out mice |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leong, C.J.; Sharma, S.; Seth, J.; Dave, A.; Abdul Ghafoor, A.A.; Laksman, Z. Gene Therapy for Inherited Arrhythmia Syndromes. Cardiogenetics 2024, 14, 132-148. https://doi.org/10.3390/cardiogenetics14030011
Leong CJ, Sharma S, Seth J, Dave A, Abdul Ghafoor AA, Laksman Z. Gene Therapy for Inherited Arrhythmia Syndromes. Cardiogenetics. 2024; 14(3):132-148. https://doi.org/10.3390/cardiogenetics14030011
Chicago/Turabian StyleLeong, Cameron J., Sohat Sharma, Jayant Seth, Archan Dave, Abdul Aziz Abdul Ghafoor, and Zachary Laksman. 2024. "Gene Therapy for Inherited Arrhythmia Syndromes" Cardiogenetics 14, no. 3: 132-148. https://doi.org/10.3390/cardiogenetics14030011
APA StyleLeong, C. J., Sharma, S., Seth, J., Dave, A., Abdul Ghafoor, A. A., & Laksman, Z. (2024). Gene Therapy for Inherited Arrhythmia Syndromes. Cardiogenetics, 14(3), 132-148. https://doi.org/10.3390/cardiogenetics14030011