
Comprehensive Microbiological and Metagenomic Analysis of the Guillain–Barré Syndrome Outbreak in Lima, 2019
 , , , , , , , ,
, , , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Samples
2.1.1. Rectal Swabs
2.1.2. Blood and Oropharyngeal Samples
2.2. Genomic and Metagenomic Sequencing
2.2.1. Genomic Sequencing
2.2.2. Metagenomic Sequencing
2.3. Bioinformatic Analysis
2.3.1. Bacterial Genome Analysis
2.3.2. Metagenomic Analysis
3. Results
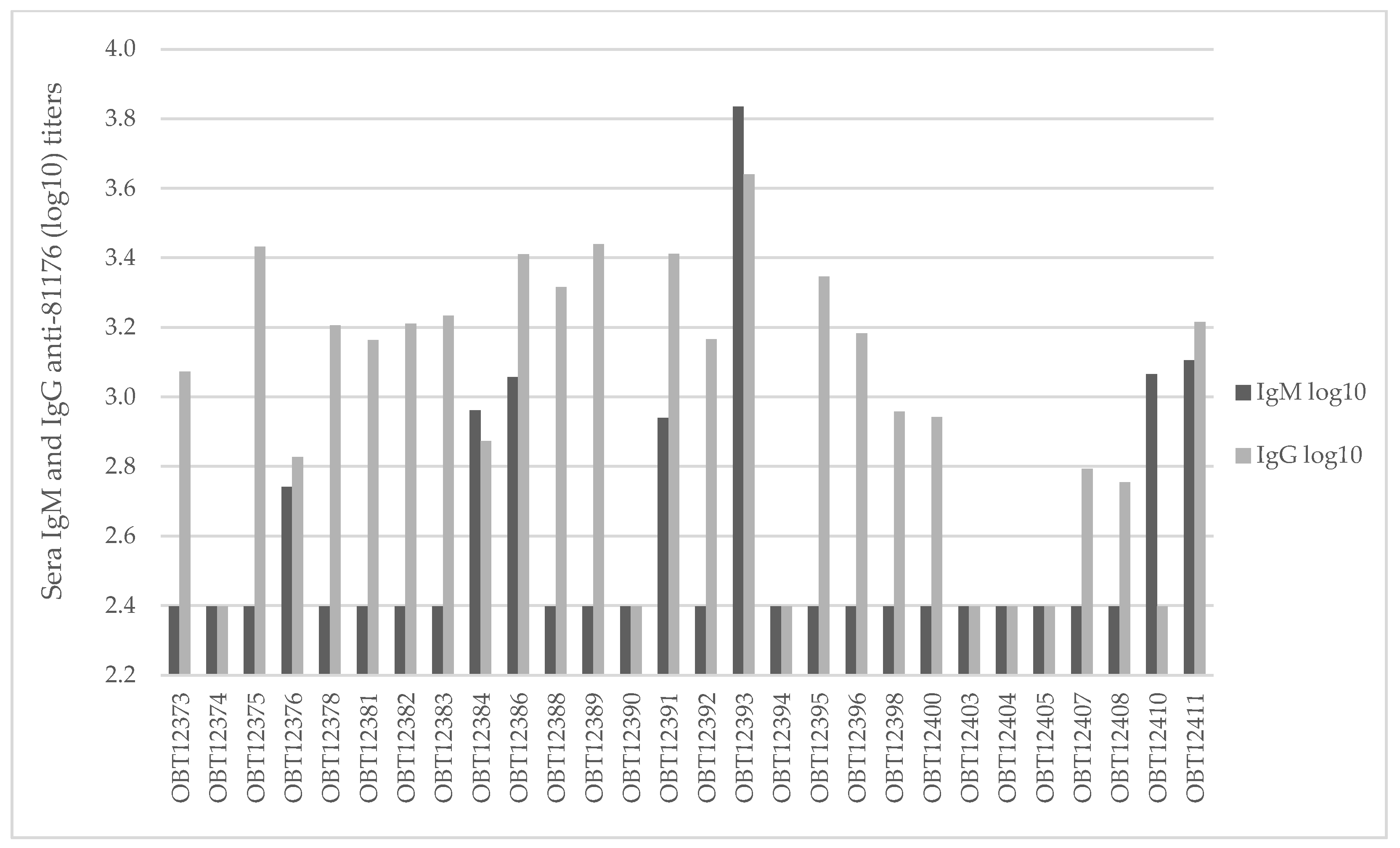
3.1. Bacterial and Viral Isolation and Characterization
3.2. Campylobacter jejuni Isolation and Characterization
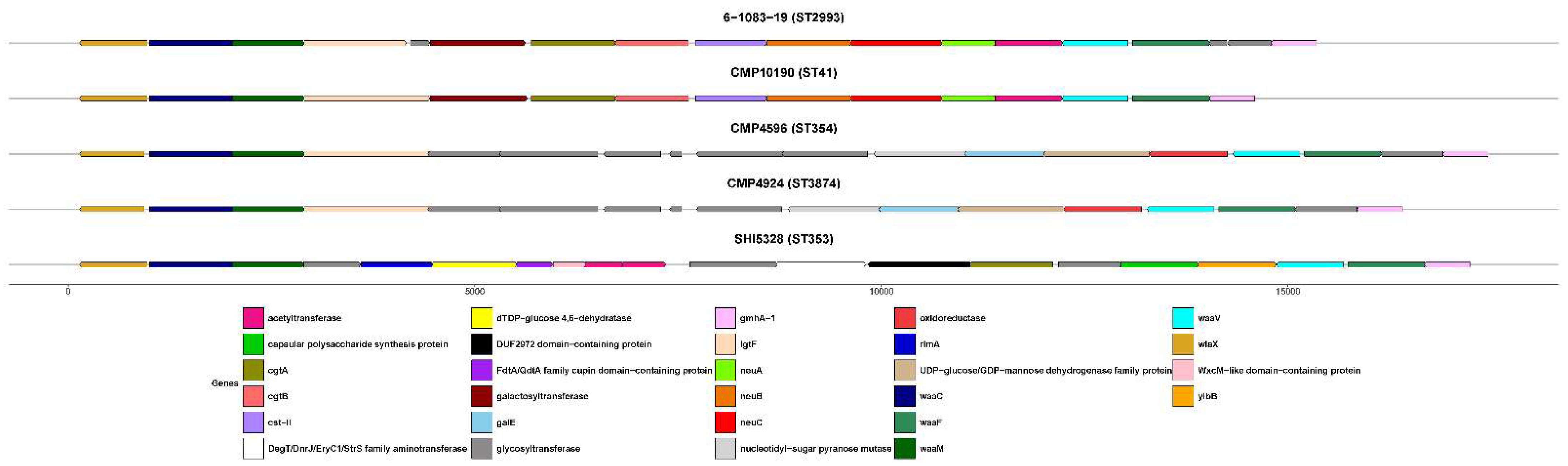
Genomic Characterization
3.3. Metagenomic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDP | Acute inflammatory demyelinating polyradiculoneuropathy. |
| AMAN | Acute motor axonal neuropathy. |
| AR | Antibiotic resistance |
| ATCC | American-Type Culture Collection. |
| CBA | Columbia blood agar. |
| CCDA | Charcoal Cefoperazone Deoxycholate Agar. |
| cgMLST | Core genome multi-locus sequence typing. |
| CHIKV | Chikungunya virus. |
| CLSI | Clinical and Laboratory Standards Institute. |
| CSF | Cerebral spinal fluid. |
| DENV | Dengue virus. |
| DNA | Deoxyribonucleic acid. |
| EBV | Epstein–Barr Virus. |
| ELISA | Enzyme-linked immunosorbent assay. |
| GBS | Guillain–Barré syndrome |
| HEp-2 | Human epithelial type-2 cell line. |
| HMAF | Hyperimmune mouse ascitic fluid |
| HIV | Human Immunodeficiency Virus |
| HNCH | Hospital Nacional Cayetano Heredia |
| IF | Immunofluorescence assay |
| LPC | Lactose positive colonies |
| LOS | Lipo-oligosaccharides |
| MDCK | Madin-Darby Canine Kidney cell line |
| MIC | Minimum inhibitory concentration |
| MLST | Multi-locus sequence typing. |
| MFS | Miller Fisher syndrome. |
| NAMRU S | U.S. Naval Medical Research Unit SOUTH. |
| OD | Optical density. |
| PCR | Polymerase chain reaction. |
| QC | Quality control. |
| RNA | Ribonucleic acid. |
| SS | Salmonella–Shigella. |
| ST | Sequence type. |
| TCBS | Thiosulfate–Citrate–Bile Salts–Sucrose. |
| TGB | Thioglycolate broth. |
| TNA | Total nucleic acid. |
| VF | Virulence factor. |
| Vero-76 | African Green Monkey cell line. |
| ZIKV | Zika virus. |
References
- Goodfellow, J.A.; Willison, H.J. Chapter Twelve—Gangliosides and Autoimmune Peripheral Nerve Diseases. In Progress in Molecular Biology and Translational Science; Schnaar, R.L., Lopez, P.H.H., Eds.; Gangliosides in Health and Disease; Academic Press: Cambridge, MA, USA, 2018; Volume 156, pp. 355–382. [Google Scholar]
- Shahrizaila, N.; Lehmann, H.C.; Kuwabara, S. Guillain-Barré Syndrome. The Lancet 2021, 397, 1214–1228. [Google Scholar] [CrossRef] [PubMed]
- Willison, H.J.; Jacobs, B.C.; Doorn, P.A. van Guillain-Barré Syndrome. The Lancet 2016, 388, 717–727. [Google Scholar] [CrossRef]
- van den Berg, B.; Walgaard, C.; Drenthen, J.; Fokke, C.; Jacobs, B.C.; van Doorn, P.A. Guillain–Barré Syndrome: Pathogenesis, Diagnosis, Treatment and Prognosis. Nat. Rev. Neurol. 2014, 10, 469–482. [Google Scholar] [CrossRef]
- Leonhard, S.E.; van der Eijk, A.A.; Andersen, H.; Antonini, G.; Arends, S.; Attarian, S.; Barroso, F.A.; Bateman, K.J.; Batstra, M.R.; Benedetti, L.; et al. An International Perspective on Preceding Infections in Guillain-Barré Syndrome. Neurology 2022, 99, e1299–e1313. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, Y.; Rojas, M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Monsalve, D.M.; Gershwin, M.E.; Anaya, J.-M. Guillain–Barré Syndrome, Transverse Myelitis and Infectious Diseases. Cell. Mol. Immunol. 2018, 15, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Willison, H.J. The Immunobiology of Guillain-Barré Syndromes. J. Peripher. Nerv. Syst. 2005, 10, 94–112. [Google Scholar] [CrossRef]
- Kuijf, M.L.; Godschalk, P.C.R.; Gilbert, M.; Endtz, H.P.; Tio-Gillen, A.P.; Ang, C.W.; van Doorn, P.A.; Jacobs, B.C. Origin of Ganglioside Complex Antibodies in Guillain–Barré Syndrome. J. Neuroimmunol. 2007, 188, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Shahrizaila, N.; Yuki, N. Guillain-Barré Syndrome Animal Model: The First Proof of Molecular Mimicry in Human Autoimmune Disorder. BioMed Res. Int. 2011, 2011, 829129. [Google Scholar] [CrossRef]
- Willison, H.J.; Goodyear, C.S. Glycolipid Antigens and Autoantibodies in Autoimmune Neuropathies. Trends Immunol. 2013, 34, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.W.; Willison, H.J.; Nachamkin, I.; Li, C.Y.; Veitch, J.; Ung, H.; Wang, G.R.; Liu, R.C.; Cornblath, D.R.; Asbury, A.K.; et al. Anti-GD1a Antibody Is Associated with Axonal but Not Demyelinating Forms of Guillain-Barré Syndrome. Ann. Neurol. 1999, 45, 168–173. [Google Scholar] [CrossRef]
- Jacobs, B.C.; van Doorn, P.A.; Tio-Gillen, A.P.; Visser, L.H.; van der Meché, F.G.A.; Schmitz, P.I.M.; Herbrink, P.; Hooijkaas, H. Campylobacter Jejuni Infections and Anti-GM1 Antibodies in Guillain-Barré Syndrome. Ann. Neurol. 1996, 40, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, Q.; He, L.; Meng, F.; Gu, Y.; Zheng, M.; Gong, Y.; Wang, P.; Ruan, F.; Zhou, L.; et al. Association Study Between an Outbreak of Guillain-Barre Syndrome in Jilin, China, and Preceding Campylobacter Jejuni Infection. Foodborne Pathog. Dis. 2010, 7, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Parkhill, J.; Wren, B.W.; Mungall, K.; Ketley, J.M.; Churcher, C.; Basham, D.; Chillingworth, T.; Davies, R.M.; Feltwell, T.; Holroyd, S.; et al. The Genome Sequence of the Food-Borne Pathogen Campylobacter Jejuni Reveals Hypervariable Sequences. Nature 2000, 403, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Guerry, P.; Szymanski, C.M.; Prendergast, M.M.; Hickey, T.E.; Ewing, C.P.; Pattarini, D.L.; Moran, A.P. Phase Variation of Campylobacter Jejuni 81-176 Lipooligosaccharide Affects Ganglioside Mimicry and Invasiveness In Vitro. Infect. Immun. 2002, 70, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Karlyshev, A.V.; Champion, O.L.; Churcher, C.; Brisson, J.-R.; Jarrell, H.C.; Gilbert, M.; Brochu, D.; St Michael, F.; Li, J.; Wakarchuk, W.W.; et al. Analysis of Campylobacter Jejuni Capsular Loci Reveals Multiple Mechanisms for the Generation of Structural Diversity and the Ability to Form Complex Heptoses. Mol. Microbiol. 2005, 55, 90–103. [Google Scholar] [CrossRef]
- Papri, N.; Islam, Z.; Leonhard, S.E.; Mohammad, Q.D.; Endtz, H.P.; Jacobs, B.C. Guillain–Barré Syndrome in Low-Income and Middle-Income Countries: Challenges and Prospects. Nat. Rev. Neurol. 2021, 17, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Capasso, A.; Ompad, D.C.; Vieira, D.L.; Wilder-Smith, A.; Tozan, Y. Incidence of Guillain-Barré Syndrome (GBS) in Latin America and the Caribbean before and during the 2015–2016 Zika Virus Epidemic: A Systematic Review and Meta-Analysis. PLoS Negl. Trop. Dis. 2019, 13, e0007622. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Triggers of Guillain–Barré Syndrome: Campylobacter Jejuni Predominates. Int. J. Mol. Sci. 2022, 23, 14222. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jiménez-Arango, J.A.; Zea-Vera, A.F.; González-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuñiga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain–Barré Syndrome Associated with Zika Virus Infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Munayco, C.V.; Gavilan, R.G.; Ramirez, G.; Loayza, M.; Miraval, M.L.; Whitehouse, E.; Gharpure, R.; Soares, J.; Soplopuco, H.V.; Sejvar, J. Large Outbreak of Guillain-Barré Syndrome, Peru, 2019. Emerg. Infect. Dis. 2019, 26, 2778–2780. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.P.; Leonhard, S.E.; Halstead, S.K.; Cuba, M.A.; Castañeda, C.C.; Dioses, J.A.; Tipismana, M.A.; Abanto, J.T.; Llanos, A.; Gourlay, D.; et al. Guillain-Barré Syndrome Outbreak in Peru 2019 Associated With Campylobacter Jejuni Infection. Neurol. Neuroimmunol. Neuroinflammation 2021, 8, e952. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S.A.; Winch, P.J.; Leontsini, E.; Torres Gayoso, C.; López Romero, S.; Gilman, R.H.; Oberhelman, R.A. Domestic Poultry-Raising Practices in a Peruvian Shantytown: Implications for Control of Campylobacter Jejuni-Associated Diarrhea. Acta Trop. 2003, 86, 41–54. [Google Scholar] [CrossRef]
- Lee, G.; Pan, W.; Yori, P.P.; Olortegui, M.P.; Tilley, D.; Gregory, M.; Oberhelman, R.; Burga, R.; Chavez, C.B.; Kosek, M. Symptomatic and Asymptomatic Campylobacter Infections Associated with Reduced Growth in Peruvian Children. PLoS Negl. Trop. Dis. 2013, 7, e2036. [Google Scholar] [CrossRef]
- Guion, C.E.; Ochoa, T.J.; Walker, C.M.; Barletta, F.; Cleary, T.G. Detection of Diarrheagenic Escherichia Coli by Use of Melting-Curve Analysis and Real-Time Multiplex PCR. J. Clin. Microbiol. 2008, 46, 1752–1757. [Google Scholar] [CrossRef]
- Dekeyser, P.; Gossuin-Detrain, M.; Butzler, J.P.; Sternon, J. Acute Enteritis Due to Related Vibrio: First Positive Stool Cultures. J. Infect. Dis. 1972, 125, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Poly, F.; Serichantalergs, O.; Kuroiwa, J.; Pootong, P.; Mason, C.; Guerry, P.; Parker, C.T. Updated Campylobacter Jejuni Capsule PCR Multiplex Typing System and Its Application to Clinical Isolates from South and Southeast Asia. PLoS ONE 2015, 10, e0144349. [Google Scholar] [CrossRef] [PubMed]
- M100 Ed34 | Performance Standards for Antimicrobial Susceptibility Testing, 34th Edition. Available online: https://clsi.org/standards/products/microbiology/documents/m100/ (accessed on 2 August 2024).
- Forshey, B.M.; Guevara, C.; Laguna-Torres, V.A.; Cespedes, M.; Vargas, J.; Gianella, A.; Vallejo, E.; Madrid, C.; Aguayo, N.; Gotuzzo, E.; et al. Arboviral Etiologies of Acute Febrile Illnesses in Western South America, 2000–2007. PLoS Negl. Trop. Dis. 2010, 4, e787. [Google Scholar] [CrossRef] [PubMed]
- Forshey, B.M.; Laguna-Torres, V.A.; Vilcarromero, S.; Bazan, I.; Rocha, C.; Morrison, A.C.; Stoddard, S.T.; Alegre, Y.; Gomez, J.; Scott, T.W.; et al. Epidemiology of Influenza-like Illness in the Amazon Basin of Peru, 2008–2009. Influenza Other Respir. Viruses 2010, 4, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Watts, D.M.; Lavera, V.; Callahan, J.; Rossi, C.; Oberste, M.S.; Roehrig, J.T.; Cropp, C.B.; Karabatsos, N.; Smith, J.F.; Gubler, D.J.; et al. Venezuelan Equine Encephalitis and Oropouche Virus Infections among Peruvian Army Troops in the Amazon Region of Peru. Am. J. Trop. Med. Hyg. 1997, 56, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Innis, B.L.; Nisalak, A.; Nimmannitya, S.; Kusalerdchariya, S.; Chongswasdi, V.; Suntayakorn, S.; Puttisri, P.; Hoke, C.H. An Enzyme-Linked Immunosorbent Assay to Characterize Dengue Infections Where Dengue and Japanese Encephalitis Co-Circulate. Am. J. Trop. Med. Hyg. 1989, 40, 418–427. [Google Scholar] [CrossRef]
- Ansari, M.Z.; Shope, R.E.; Malik, S. Evaluation of Vero Cell Lysate Antigen for the ELISA of Flaviviruses. J. Clin. Lab. Anal. 1993, 7, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.D.; Reynolds, N.D.; Pike, B.L.; Espinoza, N.M.; Kuroiwa, J.; Jani, V.; Ríos, P.A.; Nunez, R.G.; Yori, P.P.; Bernal, M.; et al. Distribution of Capsular Types of Campylobacter Jejuni Isolates from Symptomatic and Asymptomatic Children in Peru. Am. J. Trop. Med. Hyg. 2019, 101, 541. [Google Scholar] [CrossRef] [PubMed]
- Quino, W.; Caro-Castro, J.; Mestanza, O.; Hurtado, V.; Zamudio, M.L.; Cruz-Gonzales, G.; Gavilan, R.G. Emergence and Molecular Epidemiology of Campylobacter Jejuni ST-2993 Associated with a Large Outbreak of Guillain-Barré Syndrome in Peru. Microbiol. Spectr. 2022, 10, e01187-22. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. Fastqc: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 2 August 2024).
- Bushnell, B. BBDuk: Adapter/Quality Trimming and Filtering; University of California: La Jolla, CA, USA, 2014. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN Analysis of Metagenomic Data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLOS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive Visualization of de Novo Genome Assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Chain, P.S.G.; Grafham, D.V.; Fulton, R.S.; FitzGerald, M.G.; Hostetler, J.; Muzny, D.; Ali, J.; Birren, B.; Bruce, D.C.; Buhay, C.; et al. Genome Project Standards in a New Era of Sequencing. Science 2009, 326, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Li, P.-E.; Lo, C.-C.; Anderson, J.J.; Davenport, K.W.; Bishop-Lilly, K.A.; Xu, Y.; Ahmed, S.; Feng, S.; Mokashi, V.P.; Chain, P.S.G. Enabling the Democratization of the Genomics Revolution with a Fully Integrated Web-Based Bioinformatics Platform. Nucleic Acids Res. 2017, 45, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, J.; Gibson, M.K.; Franzosa, E.A.; Segata, N.; Dantas, G.; Huttenhower, C. High-Specificity Targeted Functional Profiling in Microbial Communities with ShortBRED. PLOS Comput. Biol. 2015, 11, e1004557. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genomics 2011, 12, 402. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence With Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Shakya, M.; Ahmed, S.A.; Davenport, K.W.; Flynn, M.C.; Lo, C.-C.; Chain, P.S.G. Standardized Phylogenetic and Molecular Evolutionary Analysis Applied to Species across the Microbial Tree of Life. Sci. Rep. 2020, 10, 1723. [Google Scholar] [CrossRef]
- Stamatakis, A.; Ludwig, T.; Meier, H. RAxML-III: A Fast Program for Maximum Likelihood-Based Inference of Large Phylogenetic Trees. Bioinformatics 2005, 21, 456–463. [Google Scholar] [CrossRef]
- Jolley, K.; Bray, J.; Maiden, M. Open-Access Bacterial Population Genomics: BIGSdb Software, the PubMLST.Org Website and Their Applications. Wellcome Open Res. 2018, 3. [Google Scholar] [CrossRef]
- Campylobacter Jejuni/Coli. Available online: https://pubmlst.org/organisms/campylobacter-jejunicoli (accessed on 2 August 2024).
- Rees, J.H.; Soudain, S.E.; Gregson, N.A.; Hughes, R.A.C. Campylobacter Jejuni Infection and Guillain–Barré Syndrome. N. Engl. J. Med. 1995, 333, 1374–1379. [Google Scholar] [CrossRef]
- Mori, M.; Kuwabara, S.; Miyake, M.; Noda, M.; Kuroki, H.; Kanno, H.; Ogawara, K.; Hattori, T. Haemophilus Influenzae Infection and Guillain–Barré Syndrome. Brain 2000, 123, 2171–2178. [Google Scholar] [CrossRef] [PubMed]
- Sreelakshmi, V.; Pattanaik, A.; Marate, S.; Mani, R.S.; Pai, A.R.; Mukhopadhyay, C. Guillain-Barré Syndrome (GBS) with Antecedent Chikungunya Infection: A Case Report and Literature Review. Neurol. Res. Pract. 2024, 6, 21. [Google Scholar] [CrossRef]
- Lebrun, G.; Chadda, K.; Reboux, A.-H.; Martinet, O.; Gaüzère, B.-A. Guillain-Barré Syndrome after Chikungunya Infection. Emerg. Infect. Dis. J. 2009, 15, 495. [Google Scholar] [CrossRef] [PubMed]
- Sivadon-Tardy, V.; Orlikowski, D.; Porcher, R.; Sharshar, T.; Durand, M.-C.; Enouf, V.; Rozenberg, F.; Caudie, C.; Annane, D.; van der Werf, S.; et al. Guillain-Barré Syndrome and Influenza Virus Infection. Clin. Infect. Dis. 2009, 48, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Grose, C.; Feorino, P. Epstein-Barr Virus and Guillain-Barré Syndrome. The Lancet 1972, 300, 1285–1287. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wu, G.; Lim, E.S.; Droit, L.; Krishnamurthy, S.; Barouch, D.H.; Virgin, H.W.; Wang, D. VirusSeeker, a Computational Pipeline for Virus Discovery and Virome Composition Analysis. Virology 2017, 503, 21–30. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; University of California: La Jolla, CA, USA, 2014. [Google Scholar]
- Parker, C.T.; Huynh, S.; Heikema, A.P.; Cooper, K.K.; Miller, W.G. Complete Genome Sequences of Campylobacter Jejuni Strains RM3196 (233.94) and RM3197 (308.95) Isolated from Patients with Guillain-Barré Syndrome. Genome Announc. 2015, 3, 10–1128. [Google Scholar] [CrossRef]
- Zhang, M.; He, L.; Li, Q.; Sun, H.; Gu, Y.; You, Y.; Meng, F.; Zhang, J. Genomic Characterization of the Guillain-Barre Syndrome-Associated Campylobacter Jejuni ICDCCJ07001 Isolate. PLoS ONE 2010, 5, e15060. [Google Scholar] [CrossRef]
- Gilbert, M.; Karwaski, M.-F.; Bernatchez, S.; Young, N.M.; Taboada, E.; Michniewicz, J.; Cunningham, A.-M.; Wakarchuk, W.W. The Genetic Bases for the Variation in the Lipo-Oligosaccharide of the Mucosal Pathogen, Campylobacter Jejuni: Biosynthesis of Sialylated Ganglioside Mimics in the Core Oligosaccharide *. J. Biol. Chem. 2002, 277, 327–337. [Google Scholar] [CrossRef]
- Yuki, N. Guillain–Barré Syndrome and Anti-Ganglioside Antibodies: A Clinician-Scientist’s Journey. Proc. Jpn. Acad. Ser. B 2012, 88, 299–326. [Google Scholar] [CrossRef]
- Platts-Mills, J.A.; Liu, J.; Rogawski, E.T.; Kabir, F.; Lertsethtakarn, P.; Siguas, M.; Khan, S.S.; Praharaj, I.; Murei, A.; Nshama, R.; et al. Use of Quantitative Molecular Diagnostic Methods to Assess the Aetiology, Burden, and Clinical Characteristics of Diarrhoea in Children in Low-Resource Settings: A Reanalysis of the MAL-ED Cohort Study. Lancet Glob. Health 2018, 6, e1309–e1318. [Google Scholar] [CrossRef] [PubMed]
- Amour, C.; Gratz, J.; Mduma, E.; Svensen, E.; Rogawski, E.T.; McGrath, M.; Seidman, J.C.; McCormick, B.J.J.; Shrestha, S.; Samie, A.; et al. Epidemiology and Impact of Campylobacter Infection in Children in 8 Low-Resource Settings: Results From the MAL-ED Study. Clin. Infect. Dis. 2016, 63, 1171–1179. [Google Scholar] [CrossRef]
- Heikema, A.P.; Islam, Z.; Horst-Kreft, D.; Huizinga, R.; Jacobs, B.C.; Wagenaar, J.A.; Poly, F.; Guerry, P.; van Belkum, A.; Parker, C.T.; et al. Campylobacter Jejuni Capsular Genotypes Are Related to Guillain–Barré Syndrome. Clin. Microbiol. Infect. 2015, 21, 852.e1–852.e9. [Google Scholar] [CrossRef] [PubMed]
- Islam, Z.; van Belkum, A.; Wagenaar, J.A.; Cody, A.J.; Boer, A.G.d.; Tabor, H.; Jacobs, B.C.; Talukder, K.A.; Endtz, H.P. Comparative Genotyping of Campylobacter Jejuni Strains from Patients with Guillain-Barré Syndrome in Bangladesh. PLoS ONE 2009, 4, e7257. [Google Scholar] [CrossRef] [PubMed]
- Islam, Z.; van Belkum, A.; Wagenaar, J.A.; Cody, A.J.; de Boer, A.G.; Sarker, S.K.; Jacobs, B.C.; Talukder, K.A.; Endtz, H.P. Comparative Population Structure Analysis of Campylobacter Jejuni from Human and Poultry Origin in Bangladesh. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 2173–2181. [Google Scholar] [CrossRef]
- Parker, C.T.; Horn, S.T.; Gilbert, M.; Miller, W.G.; Woodward, D.L.; Mandrell, R.E. Comparison of Campylobacter Jejuni Lipooligosaccharide Biosynthesis Loci from a Variety of Sources. J. Clin. Microbiol. 2005, 43, 2771–2781. [Google Scholar] [CrossRef]
- Hameed, A.; Woodacre, A.; Machado, L.R.; Marsden, G.L. An Updated Classification System and Review of the Lipooligosaccharide Biosynthesis Gene Locus in Campylobacter Jejuni. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Maguiña Vargas, C. Lecciones Aprendidas Del Brote de Guillain-Barré Durante El 2019. Acta Médica Peru. 2019, 36, 183–184. [Google Scholar] [CrossRef]
- Jo, Y.-S.; Choi, J.-Y.; Chung, H.; Kim, Y.; Na, S.-J. Recurrent Guillain-Barré Syndrome Following Urinary Tract Infection by Escherichia Coli. J. Korean Med. Sci. 2018, 33, e29. [Google Scholar] [CrossRef]
- Kono, Y.; Nishitarumizu, K.; Higashi, T.; Funakoshi, K.; Odaka, M. Rapidly Progressive Guillain-Barré Syndrome Following Escherichia Coli Infection. Intern. Med. 2007, 46, 589–591. [Google Scholar] [CrossRef]
- Koga, M.; Yuki, N.; Hirata, K.; Morimatsu, M.; Mori, M.; Kuwabara, S. Anti-GM1 Antibody IgG Subclass. Neurology 2003, 60, 1514–1518. [Google Scholar] [CrossRef]
- Su, Y.-C.; Resman, F.; Hörhold, F.; Riesbeck, K. Comparative Genomic Analysis Reveals Distinct Genotypic Features of the Emerging Pathogen Haemophilus Influenzae Type f. BMC Genomics 2014, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Koike, S.; Hirata, K.; Yuki, N. Ambiguous Value of Haemophilus Influenzae Isolation in Guillain-Barré and Fisher Syndromes. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1736–1738. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, A.L.; Thakur, K.T. Acute Inflammatory Demyelinating Polyradiculoneuropathy Following Malaria. J. Clin. Neurosci. 2014, 21, 704–706. [Google Scholar] [CrossRef]
- Gangula, R.S.; Stanley, W.; Vandanapu, A.; Prabhu, M. Guillain-Barre Syndrome with Falciparum Malaria and Scrub Typhus Mixed Infection-An Unusual Combination. J. Clin. Diagn. Res. 2017, 11, OD10. [Google Scholar] [CrossRef] [PubMed]
- Kanjalkar, M.; Karnad, D.R.; Narayana, R.V.; Shah, P.U. Guillain-Barre Syndrome Following Malaria. J. Infect. 1999, 38, 48–50. [Google Scholar] [CrossRef]
- Praveen, K.a.S.; Subrahmanyam, D.K.S. A Rare Cause of Guillain-Barre Syndrome. Int. J. Nutr. Pharmacol. Neurol. Dis. 2011, 1, 204. [Google Scholar] [CrossRef]
- Wijesundere, A. Guillain-Barré Syndrome in Plasmodium Falciparum Malaria. Postgrad. Med. J. 1992, 68, 376–377. [Google Scholar] [CrossRef] [PubMed]
- Padmini, R.; Maheshwari, M.C.P. Vivax Malaria Complicated by Peripheral Neuropathy with Electrophysiological Studies. J. Assoc. Physicians India 1980, 28, 152–156. [Google Scholar]
- Cao-Lormeau, V.-M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré Syndrome Outbreak Associated with Zika Virus Infection in French Polynesia: A Case-Control Study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Lima, M.E.d.S.; Bachur, T.P.R.; Aragão, G.F. Guillain-Barre Syndrome and Its Correlation with Dengue, Zika and Chikungunya Viruses Infection Based on a Literature Review of Reported Cases in Brazil. Acta Trop. 2019, 197, 105064. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Correa, J.; Siqueira, I.C.d.; Mota, S.; Rosário, M.S.d.; Jesus, P.A.P.d.; Alcantara, L.C.J.; Ernst, J.D.; Rodriguez, A. Anti-Ganglioside Antibodies in Patients with Zika Virus Infection-Associated Guillain-Barré Syndrome in Brazil. PLoS Negl. Trop. Dis. 2019, 13, e0007695. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, C.; Vasconcelos, P.F.C.; Cabezas, C.; Vasconcelos, P.F.C. Creciente Amenaza de Enfermedades Emergentes y Reemergentes: Arbovirus y Enfermedades Transmitidas Por Vectores En Las Américas. Rev. Peru. Med. Exp. Salud Publica 2024, 41, 4–6. [Google Scholar] [CrossRef]
- Morrison, A.C.; Paz-Soldan, V.A.; Vazquez-Prokopec, G.M.; Lambrechts, L.; Elson, W.H.; Barrera, P.; Astete, H.; Briesemeister, V.; Leguia, M.; Jenkins, S.A.; et al. Quantifying Heterogeneities in Arbovirus Transmission: Description of the Rationale and Methodology for a Prospective Longitudinal Study of Dengue and Zika Virus Transmission in Iquitos, Peru (2014–2019). PLoS ONE 2023, 18, e0273798. [Google Scholar] [CrossRef] [PubMed]
- Briefing Note: Increase in Cases Guillain-Barré Syndrome Peru—PAHO/WHO | Pan American Health Organization. Available online: https://www.paho.org/en/documents/briefing-note-increase-cases-guillain-barre-syndrome-peru (accessed on 3 July 2024).
- Díaz-Soto, S.; Chavez, K.; Chaca, A.; Alanya, J.; Tirado-Hurtado, I. Outbreak of Guillain-Barre Syndrome in Peru. eNeurologicalSci 2019, 14, 89–90. [Google Scholar] [CrossRef]
- Pachas, P.; Donaires, F.; Gavilán, R.G.; Quino, W.; Vidal, M.; Cabezas, C.; García, M.; Huaringa, M.; Peceros, F.; Valdivia, F.; et al. Agentes infecciosos en muestras biológicas de pacientes con síndrome de Guillain-Barré en Perú, 2018-2019. Rev. Peru. Med. Exp. Salud Pública 2020, 37, 681–688. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome | Genome Length (bp) | # CDS | GC% | # tRNA | # VF | # AR | % Reads Mapped | Average Coverage Depth (X) |
|---|---|---|---|---|---|---|---|---|
| OBT12377 * | 1,627,623 | 1629 | 30.6 | 44 | 125 | 5 | 99.7 | 1333 |
| OBT12386 * | 1,627,622 | 1625 | 30.6 | 44 | 125 | 5 | 99.7 | 932 |
| OBT12390 * | 1,627,621 | 1626 | 30.6 | 44 | 125 | 5 | 99.6 | 1156 |
| OBT12393 * | 1,627,618 | 1626 | 30.6 | 44 | 125 | 5 | 100.0 | 595 |
| 6-1083-19 | 1,627,620 | 1625 | 30.6 | 44 | 125 | 5 | 97.5 | 134 |
| 6-1197-19 | 1,627,619 | 1629 | 30.6 | 44 | 125 | 5 | 99.4 | 168 |
| 6-897-19 | 1,628,038 | 1627 | 30.3 | 44 | 122 | 5 | 98.4 | 162 |
| 6-1198-19 | 1,638,300 | 1634 | 30.4 | 46 | 123 | 5 | 96.4 | 144 |
| CMP10190 | 1,667,545 | 1681 | 30.6 | 49 | 124 | 4 | 98.4 | 89 |
| CMP10201 | 1,667,160 | 1681 | 30.6 | 45 | 124 | 4 | 99.1 | 115 |
| CMP4596 | 1,653,172 | 1706 | 30.4 | 42 | 133 | 6 | 96.5 | 169 |
| CMP4850 | 1,664,740 | 1683 | 30.5 | 44 | 123 | 4 | 97.4 | 147 |
| CMP4924 | 1,703,186 | 1769 | 30.2 | 42 | 134 | 5 | 92.4 | 164 |
| CMP9838 | 1,667,235 | 1681 | 30.6 | 45 | 119 | 5 | 99.3 | 181 |
| CMP9934 | 1,666,416 | 1681 | 30.6 | 45 | 119 | 5 | 99.2 | 111 |
| CMP10096 | 1,661,894 | 1683 | 30.6 | 41 | 117 | 5 | 91.5 | 111 |
| SHI3134 | 1,666,121 | 1676 | 30.5 | 44 | 125 | 3 | 98.9 | 179 |
| SHI4648 | 1,666,109 | 1679 | 30.5 | 44 | 125 | 3 | 95.4 | 157 |
| SHI5328 | 1,701,559 | 1736 | 30.3 | 44 | 131 | 4 | 99.8 | 176 |
| SHI9672 | 1,666,509 | 1676 | 30.5 | 48 | 125 | 4 | 99.2 | 96 |
| SHI3157 | 1,666,090 | 1680 | 30.4 | 42 | 123 | 4 | 97.3 | 200 |
| SHI5837 | 1,642,933 | 1646 | 29.9 | 45 | 131 | 5 | 96.5 | 129 |
| Sample | MLST-Designated Sequence Type | Diseases Caused by MLST-Designated Sequence Type | Closest Profile (cgMLST) | Loci Matched to Closest Profile |
|---|---|---|---|---|
| OBT12377 | 2993 | GBS, gastroenteritis | cgST-29793 | 90.2 |
| OBT12386 | 2993 | GBS, gastroenteritis | cgST-29793 | 90.2 |
| OBT12390 | 2993 | GBS, gastroenteritis | cgST-29793 | 90.2 |
| OBT12393 | 2993 | GBS, gastroenteritis | cgST-29793 | 90.2 |
| 6-897-19 | 2993 | GBS, gastroenteritis | cgST-29793 | 90.2 |
| 6-1083-19 | 2993 | GBS, gastroenteritis | cgST-29793 | 90.2 |
| 6-1197-19 | 2993 | GBS, gastroenteritis | cgST-29793 | 90.2 |
| 6-1198-19 | 2993 | GBS, gastroenteritis | cgST-29793 | 90.1 |
| CMP4596 | 354 | Gastroenteritis, systemic disease | cgST-30119 | 80.7 |
| CMP4850 | 2993 | GBS, gastroenteritis | cgST-29793 | 90.3 |
| CMP4924 | 3874 | Gastroenteritis | cgST-17417 | 73.9 |
| CMP9838 | 41 | Gastroenteritis | cgST-4301 | 37.8 |
| CMP9934 | 41 | Gastroenteritis | cgST-4301 | 37.8 |
| CMP10096 | 41 | Gastroenteritis | cgST-4301 | 37.8 |
| CMP10190 | 41 | Gastroenteritis | cgST-4301 | 37.8 |
| CMP10201 | 41 | Gastroenteritis | cgST-4301 | 37.8 |
| SHI3134 | 2993 | GBS, gastroenteritis | cgST-29793 | 89.9 |
| SHI3157 | 2993 | GBS, gastroenteritis | cgST-29793 | 89.8 |
| SHI4648 | 2993 | GBS, gastroenteritis | cgST-29793 | 90.3 |
| SHI5328 | 353 | Gastroenteritis, systemic disease | cgST-30854 | 73.9 |
| SHI5837 | 353 | Gastroenteritis, systemic disease | cgST-30854 | 72.6 |
| SHI9672 | 2993 | GBS, gastroenteritis | cgST-29793 | 90.4 |
| Sample | Length of LOS (bp) | # of CDS | Closest Reference (NCBI Accession #) | Percent Identity |
|---|---|---|---|---|
| 6-897-19 1 | 15,370 | 18 | OBT12390 (CP059160.1) | 100.0 |
| 6-1083-19 1 | 15,370 | 18 | 100.0 | |
| 6-1197-19 1 | 15,370 | 18 | 100.0 | |
| 6-1198-19 * | 15,370 | 18 | 99.0 | |
| SHI3134 1 | 15,370 | 18 | 100.0 | |
| SHI3157 1 | 15,370 | 18 | 100.0 | |
| SHI4648 1 | 15,370 | 18 | 100.0 | |
| SHI9672 1 | 15,370 | 18 | 100.0 | |
| CMP4850 1 | 15,370 | 18 | 99.0 | |
| CMP9838 2 | 14,610 | 15 | HF5-4A-4 (CP007188.1) | 97.7 |
| CMP9934 | 14,610 | 15 | 97.7 | |
| CMP10096 | 14,610 | 15 | 97.7 | |
| CMP10190 2 | 14,610 | 15 | 97.7 | |
| CMP10201 2 | 14,610 | 15 | 97.7 | |
| CMP4596 | 17,482 | 18 | CLB104 (CP034393.1) | 99.8 |
| CMP4924 | 16,427 | 17 | AR-0412 (CP044173.1) | 97.3 |
| SHI5328 | 17,255 | 20 | AR-0414 (CP044169.1) | 100.0 |
| SHI5837 | 17,257 | 20 | 100.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, J.D.; Ramos, M.; Cruz, C.; Long, K.A.; Voegtly, L.J.; Meza, R.; Espinoza, N.; Ttito, A.R.; Cáceres, H.U.; Cuentas, A.L.; et al. Comprehensive Microbiological and Metagenomic Analysis of the Guillain–Barré Syndrome Outbreak in Lima, 2019. Microbiol. Res. 2024, 15, 1826-1844. https://doi.org/10.3390/microbiolres15030122
Rojas JD, Ramos M, Cruz C, Long KA, Voegtly LJ, Meza R, Espinoza N, Ttito AR, Cáceres HU, Cuentas AL, et al. Comprehensive Microbiological and Metagenomic Analysis of the Guillain–Barré Syndrome Outbreak in Lima, 2019. Microbiology Research. 2024; 15(3):1826-1844. https://doi.org/10.3390/microbiolres15030122
Chicago/Turabian StyleRojas, Jesús D., Mariana Ramos, Cristopher Cruz, Kyle A. Long, Logan J. Voegtly, Rina Meza, Nereyda Espinoza, Ana Ramos Ttito, Hugo Umeres Cáceres, Alejandro Llanos Cuentas, and et al. 2024. "Comprehensive Microbiological and Metagenomic Analysis of the Guillain–Barré Syndrome Outbreak in Lima, 2019" Microbiology Research 15, no. 3: 1826-1844. https://doi.org/10.3390/microbiolres15030122
APA StyleRojas, J. D., Ramos, M., Cruz, C., Long, K. A., Voegtly, L. J., Meza, R., Espinoza, N., Ttito, A. R., Cáceres, H. U., Cuentas, A. L., Meza, Y., Troncos, G., Poly, F. M., Paskey, A. C., Lueder, M. R., Rice, G. K., Cer, R. Z., Bishop-Lilly, K. A., Silva, M., & Grogl, M. (2024). Comprehensive Microbiological and Metagenomic Analysis of the Guillain–Barré Syndrome Outbreak in Lima, 2019. Microbiology Research, 15(3), 1826-1844. https://doi.org/10.3390/microbiolres15030122