
Cannabis sativa L. Miniature Inverted-Repeat Transposable-Element Landscapes in Wild-Type (JL) and Domesticated Genome (CBDRx)

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Discovery, Annotation, and Organization of Cannabis MITEs
2.2. Genome-Specific MITE Identification
2.3. Potential Uses of Genome-Specific MITE for Fingerprinting and Molecular Marker Development
3. Results
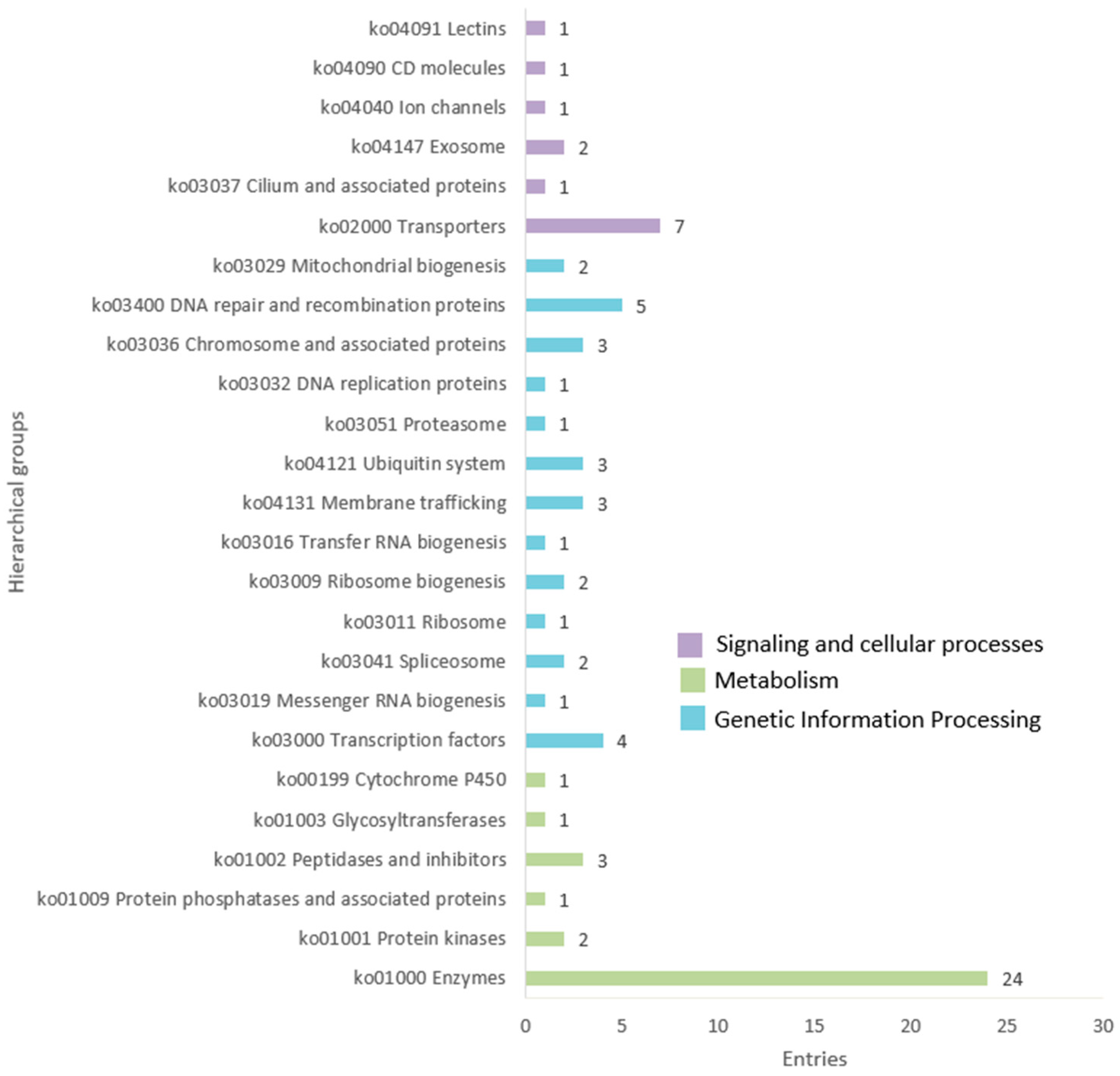
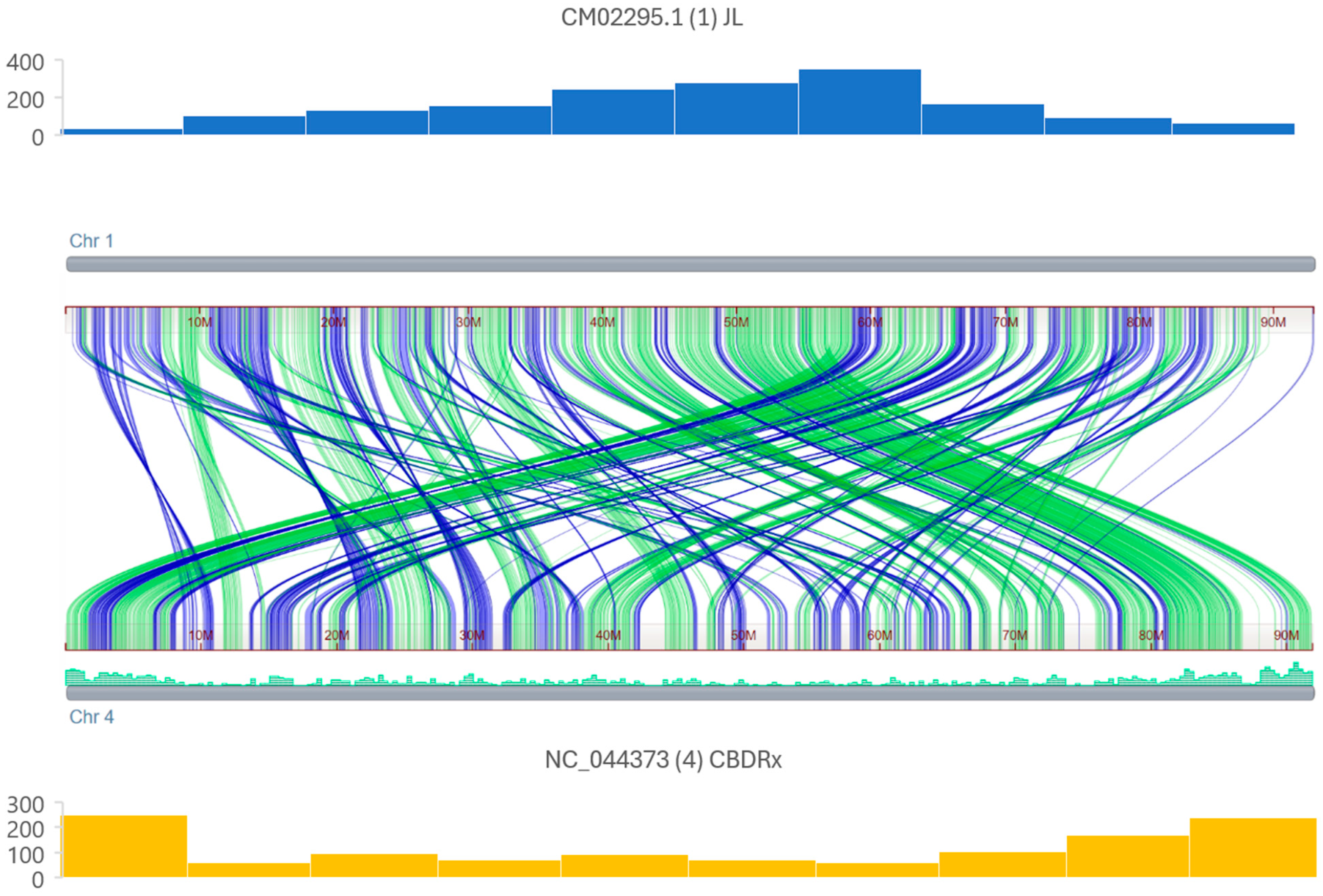
3.1. Discovery, Annotation, and Organization of Cannabis MITEs
3.2. Genome-Specific MITE Identification
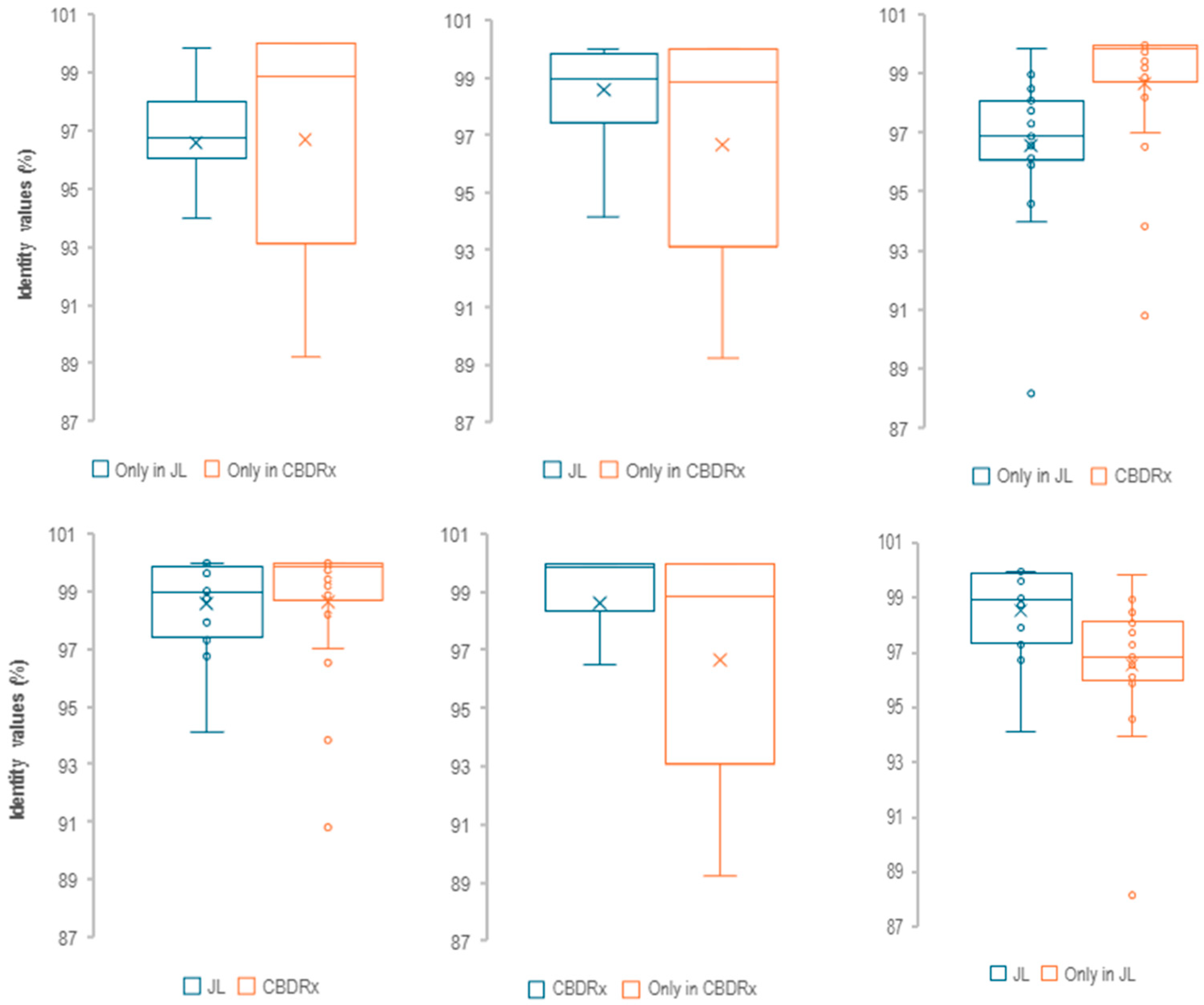
3.3. Finding Genome-Specific MITEs
3.4. Potential Uses of Genome-Specific MITEs for Fingerprinting and Molecular Marker Development
4. Discussion
4.1. Density of MITEs Within Cannabis Genomes
4.2. Potential of SNPs of Genome-Specific MITEs in Fingerprinting and Breeding
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Bakel, H.; Stout, J.M.; Cote, A.G.; Tallon, C.M.; Sharpe, A.G.; Hughes, T.R.; Page, J.E. The Draft Genome and Transcriptome of Cannabis sativa. Genome Biol. 2011, 12, R102. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Akiyama, Y.; Fukui, K.; Kamada, H.; Satoh, S. Characterization; Genome Sizes and Morphology of Sex Chromosomes in Hemp (Cannabis sativa L.). Cytologia 1998, 63, 459–464. [Google Scholar] [CrossRef]
- ElSohly, M.A.; Gul, W. Constituents of Cannabis sativa. In Handbook of Cannabis; Pertwee, R., Ed.; Oxford University Press: Oxford, UK, 2014; pp. 3–22. [Google Scholar]
- Clarke, R.C.; Merlin, M.D. Cannabis Domestication, Breeding History, Present-Day Genetic Diversity, and Future Prospects. CRC Crit. Rev. Plant Sci. 2016, 35, 293–327. [Google Scholar] [CrossRef]
- Rangwala, S.H.; Rudnev, D.V.; Ananiev, V.V.; Oh, D.-H.; Asztalos, A.; Benica, B.; Borodin, E.A.; Bouk, N.; Evgeniev, V.I.; Kodali, V.K.; et al. The NCBI Comparative Genome Viewer (CGV) Is an Interactive Visualization Tool for the Analysis of Whole-Genome Eukaryotic Alignments. PLoS Biol. 2024, 22, e3002405. [Google Scholar] [CrossRef]
- Cai, S.; Zhang, Z.; Huang, S.; Bai, X.; Huang, Z.; Zhang, Y.J.; Huang, L.; Tang, W.; Haughn, G.; You, S.; et al. CannabisGDB: A Comprehensive Genomic Database for Cannabis sativa L. Plant Biotechnol. J. 2021, 19, 857–859. [Google Scholar] [CrossRef]
- McKernan, K.; Helbert, Y.; Kane, L.T.; Ebling, H.; Zhang, L.; Liu, B.; Eaton, Z.; Sun, L.; Dimalanta, E.T.; Kingan, S.; et al. Cryptocurrencies and Zero Mode Wave Guides: An Unclouded Path to a More Contiguous Cannabis sativa L. Genome Assembly. OSF Prepr. 2018, 1–21. [Google Scholar] [CrossRef]
- Braich, S.; Baillie, R.C.; Spangenberg, G.C.; Cogan, N.O.I. A New and Improved Genome Sequence of Cannabis sativa. GigaByte 2020, 2020, gigabyte10. [Google Scholar] [CrossRef]
- Andersson, L.; Purugganan, M. Molecular Genetic Variation of Animals and Plants under Domestication. Proc. Natl. Acad. Sci. USA 2022, 119, e2122150119. [Google Scholar] [CrossRef]
- Feschotte, C.; Jiang, N.; Wessler, S.R. Plant Transposable Elements: Where Genetics Meets Genomics. Nat. Rev. Genet. 2002, 3, 329–341. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten Things You Should Know about Transposable Elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory Activities of Transposable Elements: From Conflicts to Benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Pulido, M.; Casacuberta, J.M. Transposable Element Evolution in Plant Genome Ecosystems. Curr. Opin. Plant Biol. 2023, 75, 102418. [Google Scholar] [CrossRef]
- Pandita, D.; Pandita, A. Plant Transposable Elements; Apple Academic Press: New York, NY, USA, 2023; ISBN 9781003315193. [Google Scholar]
- Bureau, T.E.; Wessler, S.R. Tourist: A Large Family of Small Inverted Repeat Elements Frequently Associated with Maize Genes. Plant Cell 1992, 4, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Fattash, I.; Rooke, R.; Wong, A.; Hui, C.; Luu, T.; Bhardwaj, P.; Yang, G. Miniature Inverted-Repeat Transposable Elements: Discovery, Distribution, and Activity. Genome 2013, 56, 475–486. [Google Scholar] [CrossRef]
- Jiang, N.; Feschotte, C.; Zhang, X.; Wessler, S.R. Using Rice to Understand the Origin and Amplification of Miniature Inverted Repeat Transposable Elements (MITEs). Curr. Opin. Plant Biol. 2004, 7, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Pegler, J.L.; Oultram, J.M.J.; Mann, C.W.G.; Carroll, B.J.; Grof, C.P.L.; Eamens, A.L. Miniature Inverted-Repeat Transposable Elements: Small DNA Transposons That Have Contributed to Plant MICRORNA Gene Evolution. Plants 2023, 12, 1101. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A Unified Classification System for Eukaryotic Transposable Elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Han, Y.; Wessler, S.R. MITE-Hunter: A Program for Discovering Miniature Inverted-Repeat Transposable Elements from Genomic Sequences. Nucleic Acids Res. 2010, 38, e199. [Google Scholar] [CrossRef]
- Bureau, T.E.; Wessler, S.R. Mobile Inverted-Repeat Elements of the Tourist Familyare Associated with the Genes of Many Cereal Grasses. Proc. Natl. Acad. Sci. USA 1994, 91, 1411–1415. [Google Scholar] [CrossRef]
- Lu, C.; Chen, J.; Zhang, Y.; Hu, Q.; Su, W.; Kuang, H. Miniature Inverted-Repeat Transposable Elements (MITEs) Have Been Accumulated through Amplification Bursts and Play Important Roles in Gene Expression and Species Diversity in Oryza sativa. Mol. Biol. Evol. 2012, 29, 1005–1017. [Google Scholar] [CrossRef]
- Benjak, A.; Boué, S.; Forneck, A.; Casacuberta, J.M. Recent Amplification and Impact of MITEs on the Genome of Grapevine (Vitis vinifera L.). Genome Biol. Evol. 2009, 1, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Crescente, J.M.; Zavallo, D.; Helguera, M.; Vanzetti, L.S. MITE Tracker: An Accurate Approach to Identify Miniature Inverted-Repeat Transposable Elements in Large Genomes. BMC Bioinform. 2018, 19, 348. [Google Scholar] [CrossRef]
- Xu, L.; Yuan, K.; Yuan, M.; Meng, X.; Chen, M.; Wu, J.; Li, J.; Qi, Y. Regulation of Rice Tillering by RNA-Directed DNA Methylation at Miniature Inverted-Repeat Transposable Elements. Mol. Plant 2020, 13, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Naito, K.; Zhang, F.; Tsukiyama, T.; Saito, H.; Hancock, C.N.; Richardson, A.O.; Okumoto, Y.; Tanisaka, T.; Wessler, S.R. Unexpected Consequences of a Sudden and Massive Transposon Amplification on Rice Gene Expression. Nature 2009, 461, 1130–1134. [Google Scholar] [CrossRef]
- Yin, S.; Wan, M.; Guo, C.; Wang, B.; Li, H.; Li, G.; Tian, Y.; Ge, X.; King, G.J.; Liu, K.; et al. Transposon Insertions within Alleles of BnaFLC.A10 and BnaFLC.A2 Are Associated with Seasonal Crop Type in Rapeseed. J. Exp. Bot. 2020, 71, 4729–4741. [Google Scholar] [CrossRef]
- Jeong, H.; Yun, Y.B.; Jeong, S.Y.; Cho, Y.; Kim, S. Characterization of Miniature Inverted Repeat Transposable Elements Inserted in the CitRWP Gene Controlling Nucellar Embryony and Development of Molecular Markers for Reliable Genotyping of CitRWP in Citrus Species. Sci. Hortic. 2023, 315, 112003. [Google Scholar] [CrossRef]
- Naito, K.; Cho, E.; Yang, G.; Campbell, M.A.; Yano, K.; Okumoto, Y.; Tanisaka, T.; Wessler, S.R. Dramatic Amplification of a Rice Transposable Element during Recent Domestication. Proc. Natl. Acad. Sci. USA 2006, 103, 17620–17625. [Google Scholar] [CrossRef]
- Venkatesh; Nandini, B. Miniature Inverted-Repeat Transposable Elements (MITEs), Derived Insertional Polymorphism as a Tool of Marker Systems for Molecular Plant Breeding. Mol. Biol. Rep. 2020, 47, 3155–3167. [Google Scholar] [CrossRef]
- von Zitzewitz, J.; Szűcs, P.; Dubcovsky, J.; Yan, L.; Francia, E.; Pecchioni, N.; Casas, A.; Chen, T.H.H.; Hayes, P.M.; Skinner, J.S. Molecular and Structural Characterization of Barley Vernalization Genes. Plant Mol. Biol. 2005, 59, 449–467. [Google Scholar] [CrossRef]
- Vaschetto, L.M. Miniature Inverted-Repeat Transposable Elements (MITEs) and Their Effects on the Regulation of Major Genes in Cereal Grass Genomes. Mol. Breed. 2016, 36, 30. [Google Scholar] [CrossRef]
- Dai, S.; Hou, J.; Qin, M.; Dai, Z.; Jin, X.; Zhao, S.; Dong, Y.; Wang, Y.; Wu, Z.; Lei, Z. Diversity and Association Analysis of Important Agricultural Trait Based on Miniature Inverted-Repeat Transposable Element Specific Marker in Brassica napus L. Oil Crop Sci. 2021, 6, 28–34. [Google Scholar] [CrossRef]
- Poretti, M.; Praz, C.R.; Meile, L.; Kälin, C.; Schaefer, L.K.; Schläfli, M.; Widrig, V.; Sanchez-Vallet, A.; Wicker, T.; Bourras, S. Domestication of High-Copy Transposons Underlays the Wheat Small RNA Response to an Obligate Pathogen. Mol. Biol. Evol. 2020, 37, 839–848. [Google Scholar] [CrossRef]
- Castanera, R.; Vendrell-Mir, P.; Bardil, A.; Carpentier, M.; Panaud, O.; Casacuberta, J.M. Amplification Dynamics of Miniature Inverted-repeat Transposable Elements and Their Impact on Rice Trait Variability. Plant J. 2021, 107, 118–135. [Google Scholar] [CrossRef]
- Ye, C.; Ji, G.; Liang, C. DetectMITE: A Novel Approach to Detect Miniature Inverted Repeat Transposable Elements in Genomes. Sci. Rep. 2016, 6, 19688. [Google Scholar] [CrossRef]
- Yang, G. MITE Digger, an Efficient and Accurate Algorithm for Genome Wide Discovery of Miniature Inverted Repeat Transposable Elements. BMC Bioinform. 2013, 14, 186. [Google Scholar] [CrossRef]
- Hu, J.; Zheng, Y.; Shang, X. MiteFinder: A Fast Approach to Identify Miniature Inverted-Repeat Transposable Elements on a Genome-Wide Scale. In Proceedings of the 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Kansas City, MO, USA, 13–16 November 2017; IEEE: Piscataway, NJ, USA, 2017; pp. 164–168. [Google Scholar]
- Satovic, E.; Cvitanic, E.T.; Plohl, M. Tools and Databases for Solving Problems in Detection and Identification of Repetitive DNA Sequences. Period Biol. 2020, 121–122, 7–14. [Google Scholar] [CrossRef]
- Gao, S.; Wang, B.; Xie, S.; Xu, X.; Zhang, J.; Pei, L.; Yu, Y.; Yang, W.; Zhang, Y. A High-Quality Reference Genome of Wild Cannabis sativa. Hortic. Res. 2020, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Grassa, C.J.; Weiblen, G.D.; Wenger, J.P.; Dabney, C.; Poplawski, S.G.; Timothy Motley, S.; Michael, T.P.; Schwartz, C.J. A New Cannabis Genome Assembly Associates Elevated Cannabidiol (CBD) with Hemp Introgressed into Marijuana. New Phytol. 2021, 230, 1665–1679. [Google Scholar] [CrossRef]
- Seabold, S.; Perktold, J. Statsmodels: Econometric and Statistical Modeling with Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y. KEGG Mapper for Inferring Cellular Functions from Protein Sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hu, Q.; Zhang, Y.; Lu, C.; Kuang, H. P-MITE: A Database for Plant Miniature Inverted-Repeat Transposable Elements. Nucleic Acids Res. 2014, 42, D1176–D1181. [Google Scholar] [CrossRef]
- Onetto, C.A.; Ward, C.M.; Borneman, A.R. The Genome Assembly of Vitis vinifera Cv. Shiraz. Aust. J. Grape Wine Res. 2023, 2023, 6686706. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking Transposable Element Annotation Methods for Creation of a Streamlined, Comprehensive Pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed]
- Rohilla, M.; Mazumder, A.; Saha, D.; Pal, T.; Begam, S.; Mondal, T.K. Genome-Wide Identification and Development of Miniature Inverted-Repeat Transposable Elements and Intron Length Polymorphic Markers in Tea Plant (Camellia sinensis). Sci. Rep. 2022, 12, 16233. [Google Scholar] [CrossRef]
- Keidar-Friedman, D.; Bariah, I.; Kashkush, K. Genome-Wide Analyses of Miniature Inverted-Repeat Transposable Elements Reveals New Insights into the Evolution of the Triticum-Aegilops Group. PLoS ONE 2018, 13, e0204972. [Google Scholar] [CrossRef]
- Boutanaev, A.M.; Osbourn, A.E. Multigenome Analysis Implicates Miniature Inverted-Repeat Transposable Elements (MITEs) in Metabolic Diversification in Eudicots. Proc. Natl. Acad. Sci. USA 2018, 115, E6650–E6658. [Google Scholar] [CrossRef]
- Li, R.; Yao, J.; Cai, S.; Fu, Y.; Lai, C.; Zhu, X.; Cui, L.; Li, Y. Genome-Wide Characterization and Evolution Analysis of Miniature Inverted-Repeat Transposable Elements in Barley (Hordeum vulgare). Front. Plant Sci. 2024, 15, E6650–E6658. [Google Scholar] [CrossRef]
- Liu, Y.; Tahir ul Qamar, M.; Feng, J.-W.; Ding, Y.; Wang, S.; Wu, G.; Ke, L.; Xu, Q.; Chen, L.-L. Comparative Analysis of Miniature Inverted–Repeat Transposable Elements (MITEs) and Long Terminal Repeat (LTR) Retrotransposons in Six Citrus Species. BMC Plant Biol. 2019, 19, 140. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly via Adaptive k -Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Istace, B.; Friedrich, A.; d’Agata, L.; Faye, S.; Payen, E.; Beluche, O.; Caradec, C.; Davidas, S.; Cruaud, C.; Liti, G.; et al. De Novo Assembly and Population Genomic Survey of Natural Yeast Isolates with the Oxford Nanopore MinION Sequencer. Gigascience 2017, 6, 1–13. [Google Scholar] [CrossRef]
- Chakraborty, M.; Baldwin-Brown, J.G.; Long, A.D.; Emerson, J.J. Contiguous and Accurate de Novo Assembly of Metazoan Genomes with Modest Long Read Coverage. Nucleic Acids Res. 2016, 44, e147. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Belaghzal, H.; Dekker, J.; Gibcus, J.H. Hi-C 2.0: An Optimized Hi-C Procedure for High-Resolution Genome-Wide Mapping of Chromosome Conformation. Methods 2017, 123, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and Accurate de Novo Genome Assembly from Long Uncorrected Reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Ghurye, J.; Pop, M.; Koren, S.; Bickhart, D.; Chin, C.-S. Scaffolding of Long Read Assemblies Using Long Range Contact Information. BMC Genom. 2017, 18, 527. [Google Scholar] [CrossRef]
- Tang, H.; Zhang, X.; Miao, C.; Zhang, J.; Ming, R.; Schnable, J.C.; Schnable, P.S.; Lyons, E.; Lu, J. ALLMAPS: Robust Scaffold Ordering Based on Multiple Maps. Genome Biol. 2015, 16, 3. [Google Scholar] [CrossRef]
- Hadagali, S.; Stelmach-Wityk, K.; Macko-Podgórni, A.; Cholin, S.; Grzebelus, D. Polymorphic Insertions of DcSto Miniature Inverted-Repeat Transposable Elements Reveal Genetic Diversity Structure within the Cultivated Carrot. J. Appl. Genet. 2024; Online ahead of print. [Google Scholar] [CrossRef]
- Chang, R.-Y.; O’Donoughue, L.S.; Bureau, T.E. Inter-MITE Polymorphisms (IMP): A High Throughput Transposon-Based Genome Mapping and Fingerprinting Approach. Theor. Appl. Genet. 2001, 102, 773–781. [Google Scholar] [CrossRef]
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebley, J. Identification of a Functional Transposon Insertion in the Maize Domestication Gene Tb1. Nat. Genet. 2011, 43, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y. Plant MITEs: Useful Tools for Plant Genetics and Genomics. Genom. Proteom. Bioinform. 2003, 1, 90–100. [Google Scholar] [CrossRef]
- VanBuren, R.; Man Wai, C.; Wang, X.; Pardo, J.; Yocca, A.E.; Wang, H.; Chaluvadi, S.R.; Han, G.; Bryant, D.; Edger, P.P.; et al. Exceptional Subgenome Stability and Functional Divergence in the Allotetraploid Ethiopian Cereal Teff. Nat. Commun. 2020, 11, 884. [Google Scholar] [CrossRef]
- Devos, K.M.; Qi, P.; Bahri, B.A.; Gimode, D.M.; Jenike, K.; Manthi, S.J.; Lule, D.; Lux, T.; Martinez-Bello, L.; Pendergast, T.H.; et al. Genome Analyses Reveal Population Structure and a Purple Stigma Color Gene Candidate in Finger Millet. Nat. Commun. 2023, 14, 3694. [Google Scholar] [CrossRef] [PubMed]
- Yim, W.C.; Swain, M.L.; Ma, D.; An, H.; Bird, K.A.; Curdie, D.D.; Wang, S.; Ham, H.D.; Luzuriaga-Neira, A.; Kirkwood, J.S.; et al. The Final Piece of the Triangle of U: Evolution of the Tetraploid Brassica Carinata Genome. Plant Cell 2022, 34, 4143–4172. [Google Scholar] [CrossRef]
- Wang, X.; Chen, S.; Ma, X.; Yssel, A.E.J.; Chaluvadi, S.R.; Johnson, M.S.; Gangashetty, P.; Hamidou, F.; Sanogo, M.D.; Zwaenepoel, A.; et al. Genome Sequence and Genetic Diversity Analysis of an Under-Domesticated Orphan Crop, White Fonio (Digitaria exilis). Gigascience 2021, 10, giab013. [Google Scholar] [CrossRef]
- Zavallo, D.; Crescente, J.M.; Gantuz, M.; Leone, M.; Vanzetti, L.S.; Masuelli, R.W.; Asurmendi, S. Genomic Re-Assessment of the Transposable Element Landscape of the Potato Genome. Plant Cell Rep. 2020, 39, 1161–1174. [Google Scholar] [CrossRef]
- Klai, K.; Zidi, M.; Chénais, B.; Denis, F.; Caruso, A.; Casse, N.; Mezghani Khemakhem, M. Miniature Inverted-Repeat Transposable Elements (MITEs) in the Two Lepidopteran Genomes of Helicoverpa armigera and Helicoverpa zea. Insects 2022, 13, 313. [Google Scholar] [CrossRef]
- Klai, K.; Chénais, B.; Zidi, M.; Djebbi, S.; Caruso, A.; Denis, F.; Confais, J.; Badawi, M.; Casse, N.; Mezghani Khemakhem, M. Screening of Helicoverpa armigera Mobilome Revealed Transposable Element Insertions in Insecticide Resistance Genes. Insects 2020, 11, 879. [Google Scholar] [CrossRef]
- Martelossi, J.; Forni, G.; Iannello, M.; Savojardo, C.; Martelli, P.L.; Casadio, R.; Mantovani, B.; Luchetti, A.; Rota-Stabelli, O. Wood Feeding and Social Living: Draft Genome of the Subterranean Termite Reticulitermes lucifugus (Blattodea; Termitoidae). Insect Mol. Biol. 2023, 32, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Depotter, J.R.L.; Ökmen, B.; Ebert, M.K.; Beckers, J.; Kruse, J.; Thines, M.; Doehlemann, G. High Nucleotide Substitution Rates Associated with Retrotransposon Proliferation Drive Dynamic Secretome Evolution in Smut Pathogens. Microbiol. Spectr. 2022, 10, e0034922. [Google Scholar] [CrossRef] [PubMed]
- Fouché, S.; Badet, T.; Oggenfuss, U.; Plissonneau, C.; Francisco, C.S.; Croll, D. Stress-Driven Transposable Element De-Repression Dynamics and Virulence Evolution in a Fungal Pathogen. Mol. Biol. Evol. 2020, 37, 221–239. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CBDRx | JL | ||||||
|---|---|---|---|---|---|---|---|
| Chr. Accession | Mb | MITEs | MITEs/Mb | Chr. Accession | Mb | MITEs | MITEs/Mb |
| NC_044371.1 | 101.21 | 1394 | 13 | CM022971.1 | 80.62 | 1934 | 23 |
| NC_044375.1 | 96.35 | 998 | 10 | CM022969.1 | 83.00 | 1327 | 15 |
| NC_044372.1 | 94.67 | 997 | 10 | CM022967.1 | 89.82 | 1370 | 15 |
| NC_044373.1 | 91.91 | 1201 | 13 | CM022965.1 | 93.00 | 1612 | 17 |
| NC_044374.1 | 88.18 | 1050 | 11 | CM022968.1 | 83.22 | 1320 | 15 |
| NC_044377.1 | 79.34 | 1099 | 13 | CM022970.1 | 82.47 | 1334 | 16 |
| NC_044378.1 | 71.24 | 844 | 11 | CM022973.1 | 69.09 | 1104 | 15 |
| NC_044379.1 | 64.62 | 929 | 14 | CM022974.1 | 54.53 | 1192 | 21 |
| NC_044376.1 | 61.56 | 861 | 13 | CM022972.1 | 70.97 | 1177 | 16 |
| NC_044370.1 | 104.99 | 1423 | 13 | CM022966.1 | 91.28 | 1791 | 19 |
| Totals | 854.49 | 10903 | 12.1 | 807.90 | 14444 | 17.2 1 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quiroga, M.; Crociara, C.; Schenfeld, E.; Fernández, F.D.; Crescente, J.; Vanzetti, L.; Helguera, M. Cannabis sativa L. Miniature Inverted-Repeat Transposable-Element Landscapes in Wild-Type (JL) and Domesticated Genome (CBDRx). Int. J. Plant Biol. 2025, 16, 40. https://doi.org/10.3390/ijpb16020040
Quiroga M, Crociara C, Schenfeld E, Fernández FD, Crescente J, Vanzetti L, Helguera M. Cannabis sativa L. Miniature Inverted-Repeat Transposable-Element Landscapes in Wild-Type (JL) and Domesticated Genome (CBDRx). International Journal of Plant Biology. 2025; 16(2):40. https://doi.org/10.3390/ijpb16020040
Chicago/Turabian StyleQuiroga, Mariana, Clara Crociara, Esteban Schenfeld, Franco Daniel Fernández, Juan Crescente, Leonardo Vanzetti, and Marcelo Helguera. 2025. "Cannabis sativa L. Miniature Inverted-Repeat Transposable-Element Landscapes in Wild-Type (JL) and Domesticated Genome (CBDRx)" International Journal of Plant Biology 16, no. 2: 40. https://doi.org/10.3390/ijpb16020040
APA StyleQuiroga, M., Crociara, C., Schenfeld, E., Fernández, F. D., Crescente, J., Vanzetti, L., & Helguera, M. (2025). Cannabis sativa L. Miniature Inverted-Repeat Transposable-Element Landscapes in Wild-Type (JL) and Domesticated Genome (CBDRx). International Journal of Plant Biology, 16(2), 40. https://doi.org/10.3390/ijpb16020040