
Usher Syndrome


Abstract
:1. Introduction
2. Materials and Methods
3. Genetics of USH
3.1. USH1
3.1.1. MYO7A—Myosin VIIa, USH1B
3.1.2. PCDH15—Protocadherin-15, USH1F
3.1.3. USH1C—Harmonin
3.1.4. USH1G (SANS)—USH Type-1G Protein, USH1G
3.1.5. CDH23—Cadherin-23, USH1D
3.1.6. CIB2, USH1J?
3.1.7. ESPN—Espin
3.1.8. USH1E, USH1H, and USH1K
3.2. USH2
3.2.1. USH2A—Usherin
3.2.2. ADGRV1 (GPR98 or VLGR1)—Adhesion G-Protein Coupled Receptor V1, USH2C
3.2.3. WHRN—Whirlin, USH2D
3.2.4. PDZD7, A Modifier Gene?
3.3. USH3
3.3.1. CLRN1—Clarin-1, USH3A
3.3.2. HARS1—Histidine—tRNA Ligase, Cytoplasmic, USH3B?
3.3.3. ABHD12—Lysophosphatidylserine Lipase ABHD12, USH3?
3.4. Atypical Usher or Unclear Role
3.4.1. CEP250—Centrosome-Associated Protein CEP250, Atypical Usher
3.4.2. CEP78—Centrosomal Protein of 78 kDa, Atypical Usher
3.4.3. ARSG, Arylsulfatase G, Atypical Usher
4. Epidemiology
5. Diagnosis and Prognosis
5.1. Hearing Loss
5.2. Balance
5.3. Vision Loss
5.4. Prognosis
6. Usher Type 4?
7. Rehabilitation
7.1. Hearing
7.2. Vision
7.3. Balance
7.4. Physical and Psychological Comorbidity
8. Treatment and Future Perspectives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Colman, A.M. A Dictionary of Psychology; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Koenekoop, R.; Arriaga, M.; Trzupek, K.M.; Lentz, J. Usher Syndrome Type I. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1999. [Google Scholar]
- Koenekoop, R.K.; Arriaga, M.A.; Trzupek, K.M.; Lentz, J. Usher Syndrome Type II. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1999. [Google Scholar]
- Reiners, J.; Nagel-Wolfrum, K.; Jurgens, K.; Marker, T.; Wolfrum, U. Molecular basis of human Usher syndrome: Deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 2006, 83, 97–119. [Google Scholar] [CrossRef] [PubMed]
- Usher, C.H. On the inheritance of Retinitis pigmentosa with notes of cases. R. Lond. Opthalmol. Hosp. Rep. 1914, 19, 130–236. [Google Scholar]
- Booth, K.T.; Kahrizi, K.; Babanejad, M.; Daghagh, H.; Bademci, G.; Arzhangi, S.; Zareabdollahi, D.; Duman, D.; El-Amraoui, A.; Tekin, M.; et al. Variants in CIB2 cause DFNB48 and not USH1J. Clin. Genet. 2018, 93, 812–821. [Google Scholar] [CrossRef]
- Cesca, F.; Bettella, E.; Polli, R.; Leonardi, E.; Aspromonte, M.C.; Sicilian, B.; Stanzial, F.; Benedicenti, F.; Sensi, A.; Ciorba, A.; et al. Frequency of Usher gene mutations in non-syndromic hearing loss: Higher variability of the Usher phenotype. J. Hum. Genet. 2020, 65, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.Y.; Scarborough, J.D.; Zheng, Y.; Yu, H.; Choi, D.; Gillespie, P.G. Digenic inheritance of deafness caused by 8J allele of myosin-VIIA and mutations in other Usher I genes. Hum. Mol. Genet. 2012, 21, 2588–2598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.Y.; Yan, D.; Ouyang, X.M.; Du, L.L.; Yu, H.; Chang, B.; Johnson, K.R.; Liu, X.Z. Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Hum. Mol. Genet. 2005, 14, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Castiglione, A.; Busi, M.; Martini, A. Syndromic hearing loss: An update. Hear. Balance Commun. 2013, 11, 146–159. [Google Scholar] [CrossRef]
- Busi, M.; Castiglione, A.; Masieri, M.T.; Ravani, A.; Guaran, V.; Astolfi, L.; Trevisi, P.; Ferlini, A.; Martini, A. Novel mutations in the SLC26A4 gene. Int. J. Pediatric Otorhinolaryngol. 2012, 76, 1249–1254. [Google Scholar] [CrossRef]
- Castiglione, A.; Melchionda, S.; Carella, M.; Trevisi, P.; Bovo, R.; Manara, R.; Martini, A. EYA1-related disorders: Two clinical cases and a literature review. Int. J. Pediatric Otorhinolaryngol. 2014, 78, 1201–1210. [Google Scholar] [CrossRef]
- Gerber, S.; Bonneau, D.; Gilbert, B.; Munnich, A.; Dufier, J.L.; Rozet, J.M.; Kaplan, J. USH1A: Chronicle of a slow death. Am. J. Hum. Genet. 2006, 78, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Hmani, M.; Ghorbel, A.; Boulila-Elgaied, A.; Ben Zina, Z.; Kammoun, W.; Drira, M.; Chaabouni, M.; Petit, C.; Ayadi, H. A novel locus for Usher syndrome type II, USH2B, maps to chromosome 3 at p23-24.2. Eur. J. Hum. Genet. 1999, 7, 363–367. [Google Scholar] [CrossRef]
- Hmani-Aifa, M.; Ben Arab, S.; Kharrat, K.; Orten, D.J.; Boulila-Elgaied, A.; Drira, M.; Hachicha, S.; Kimberling, W.J.; Ayadi, H. Distinctive audiometric features between USH2A and USH2B subtypes of Usher syndrome. J. Med. Genet. 2002, 39, 281–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hmani-Aifa, M.; Benzina, Z.; Zulfiqar, F.; Dhouib, H.; Shahzadi, A.; Ghorbel, A.; Rebai, A.; Soderkvist, P.; Riazuddin, S.; Kimberling, W.J.; et al. Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family. Eur. J. Hum. Genet. 2009, 17, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Qu, C.; Liang, F.; Long, Q.; Zhao, M.; Shang, H.; Fan, L.; Wang, L.; Foster, J., 2nd; Yan, D.; Liu, X. Genetic screening revealed usher syndrome in a paediatric Chinese patient. Hear. Balance Commun. 2017, 15, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Toms, M.; Pagarkar, W.; Moosajee, M. Usher syndrome: Clinical features, molecular genetics and advancing therapeutics. Ther. Adv. Ophthalmol. 2020, 12, 2515841420952194. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.; Gasparini, P.; Girotto, G. Molecular testing for the study of non-syndromic hearing loss. Hear. Balance Commun. 2020, 18, 270–277. [Google Scholar] [CrossRef]
- Fuster-Garcia, C.; Garcia-Bohorquez, B.; Rodriguez-Munoz, A.; Aller, E.; Jaijo, T.; Millan, J.M.; Garcia-Garcia, G. Usher Syndrome: Genetics of a Human Ciliopathy. Int. J. Mol. Sci. 2021, 22, 6723. [Google Scholar] [CrossRef]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Usher Syndrome. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2018; pp. 167–170. [Google Scholar]
- Tanimoto, M.; Ota, Y.; Inoue, M.; Oda, Y. Origin of inner ear hair cells: Morphological and functional differentiation from ciliary cells into hair cells in zebrafish inner ear. J. Neurosci. 2011, 31, 3784–3794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherson, D.R. Sensory Hair Cells: An Introduction to Structure and Physiology. Integr. Comp. Biol. 2018, 58, 282–300. [Google Scholar] [CrossRef] [PubMed]
- El-Amraoui, A.; Petit, C. Usher I syndrome: Unravelling the mechanisms that underlie the cohesion of the growing hair bundle in inner ear sensory cells. J. Cell Sci. 2005, 118, 4593–4603. [Google Scholar] [CrossRef] [Green Version]
- El-Amraoui, A.; Petit, C. The retinal phenotype of Usher syndrome: Pathophysiological insights from animal models. C. R. Biol. 2014, 337, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Ashmore, J.F. Hair cells. Sci. Prog. 1988, 72, 139–153. [Google Scholar] [PubMed]
- Chaïb, H.; Kaplan, J.; Gerber, S.; Vincent, C.; Ayadi, H.; Slim, R.; Munnich, A.; Weissenbach, J.; Petit, C. A newly identified locus for Usher syndrome type I, USH1E, maps to chromosome 21q21. Hum. Mol. Genet. 1997, 6, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.; Zallocchi, M. Usher protein functions in hair cells and photoreceptors. Int. J. Biochem. Cell Biol. 2014, 46, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Tian, G.; Zhou, Y.; Hajkova, D.; Miyagi, M.; Dinculescu, A.; Hauswirth, W.W.; Palczewski, K.; Geng, R.; Alagramam, K.N.; Isosomppi, J.; et al. Clarin-1, encoded by the Usher Syndrome III causative gene, forms a membranous microdomain: Possible role of clarin-1 in organizing the actin cytoskeleton. J. Biol. Chem. 2009, 284, 18980–18993. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zou, J.; Shen, Z.; Song, E.; Yang, J. Whirlin interacts with espin and modulates its actin-regulatory function: An insight into the mechanism of Usher syndrome type II. Hum. Mol. Genet. 2012, 21, 692–710. [Google Scholar] [CrossRef] [Green Version]
- Michel, V.; Pepermans, E.; Boutet de Monvel, J.; England, P.; Nouaille, S.; Aghaie, A.; Delhommel, F.; Wolff, N.; Perfettini, I.; Hardelin, J.P.; et al. Interaction of protocadherin-15 with the scaffold protein whirlin supports its anchoring of hair-bundle lateral links in cochlear hair cells. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Ahmed, Z.M.; Riazuddin, S.; Khan, S.N.; Friedman, P.L.; Riazuddin, S.; Friedman, T.B. USH1H, a novel locus for type I Usher syndrome, maps to chromosome 15q22-23. Clin. Genet. 2009, 75, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Dad, S.; Østergaard, E.; Thykjaer, T.; Albrectsen, A.; Ravn, K.; Rosenberg, T.; Møller, L.B. Identification of a novel locus for a USH3 like syndrome combined with congenital cataract. Clin. Genet. 2010, 78, 388–397. [Google Scholar] [CrossRef]
- Jaworek, T.J.; Bhatti, R.; Latief, N.; Khan, S.N.; Riazuddin, S.; Ahmed, Z.M. USH1K, a novel locus for type I Usher syndrome, maps to chromosome 10p11.21-q21.1. J. Hum. Genet. 2012, 57, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Tahani, N.; Maffei, P.; Dollfus, H.; Paisey, R.; Valverde, D.; Milan, G.; Han, J.C.; Favaretto, F.; Madathil, S.C.; Dawson, C.; et al. Consensus clinical management guidelines for Alstrom syndrome. Orphanet J. Rare Dis. 2020, 15, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ozanturk, A.; Marshall, J.D.; Collin, G.B.; Duzenli, S.; Marshall, R.P.; Candan, S.; Tos, T.; Esen, I.; Taskesen, M.; Cayir, A.; et al. The phenotypic and molecular genetic spectrum of Alstrom syndrome in 44 Turkish kindreds and a literature review of Alstrom syndrome in Turkey. J. Hum. Genet. 2015, 60, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kimberling, W.J. Clinical and Genetic Heterogeneity of Usher Syndrome. Audiol. Med. 2003, 1, 67–70. [Google Scholar] [CrossRef]
- Moller, C.G.; Kimberling, W.J.; Davenport, S.L.; Priluck, I.; White, V.; Biscone-Halterman, K.; Odkvist, L.M.; Brookhouser, P.E.; Lund, G.; Grissom, T.J. Usher syndrome: An otoneurologic study. Laryngoscope 1989, 99, 73–79. [Google Scholar] [PubMed]
- Schaefer, G.B.; Bodensteiner, J.B.; Thompson, J.N., Jr.; Kimberling, W.J.; Craft, J.M. Volumetric neuroimaging in Usher syndrome: Evidence of global involvement. Am. J. Med. Genet. 1998, 79, 1–4. [Google Scholar] [CrossRef]
- Ciorba, A.; Schrott-Fisher, A.; Berto, A.; Glueckert, R.; Janecke, A.; Martini, A. Histopathological and neuroradiological features of Usher syndrome type II. B-Ent 2008, 4, 201–206. [Google Scholar]
- Amore, G.; Spoto, G.; Ieni, A.; Vetri, L.; Quatrosi, G.; Di Rosa, G.; Nicotera, A.G. A Focus on the Cerebellum: From Embryogenesis to an Age-Related Clinical Perspective. Front. Syst. Neurosci. 2021, 15, 174–182. [Google Scholar] [CrossRef]
- Wang, D.; Zhou, J. The Kinocilia of Cochlear Hair Cells: Structures, Functions, and Diseases. Front. Cell. Dev. Biol. 2021, 9, 715037. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Et Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef] [Green Version]
- Mathur, P.D.; Yang, J. Usher syndrome and non-syndromic deafness: Functions of different whirlin isoforms in the cochlea, vestibular organs, and retina. Hear. Res. 2019, 375, 14–24. [Google Scholar] [CrossRef]
- Puffenberger, E.G.; Jinks, R.N.; Sougnez, C.; Cibulskis, K.; Willert, R.A.; Achilly, N.P.; Cassidy, R.P.; Fiorentini, C.J.; Heiken, K.F.; Lawrence, J.J.; et al. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE 2012, 7, e28936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Chen, Q.; Almishaal, A.; Mathur, P.D.; Zheng, T.; Tian, C.; Zheng, Q.Y.; Yang, J. The roles of USH1 proteins and PDZ domain-containing USH proteins in USH2 complex integrity in cochlear hair cells. Hum. Mol. Genet. 2017, 26, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Xu, K.; Ren, Y.; Xie, Y.; Zhang, X.; Tian, L.; Li, Y. Comprehensive Molecular Screening in Chinese Usher Syndrome Patients. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1229–1237. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, Z.M.; Frolenkov, G.I.; Riazuddin, S. Usher proteins in inner ear structure and function. Physiol. Genom. 2013, 45, 987–989. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, Z.M.; Jaworek, T.J.; Sarangdhar, G.N.; Zheng, L.; Gul, K.; Khan, S.N.; Friedman, T.B.; Sisk, R.A.; Bartles, J.R.; Riazuddin, S.; et al. Inframe deletion of human ESPN is associated with deafness, vestibulopathy and vision impairment. J. Med. Genet. 2018, 55, 479–488. [Google Scholar] [CrossRef]
- Smith, R.J.; Lee, E.C.; Kimberling, W.J.; Daiger, S.P.; Pelias, M.Z.; Keats, B.J.; Jay, M.; Bird, A.; Reardon, W.; Guest, M.; et al. Localization of two genes for Usher syndrome type I to chromosome 11. Genomics 1992, 14, 995–1002. [Google Scholar] [CrossRef]
- Weil, D.; Levy, G.; Sahly, I.; Levi-Acobas, F.; Blanchard, S.; El-Amraoui, A.; Crozet, F.; Philippe, H.; Abitbol, M.; Petit, C. Human myosin VIIA responsible for the Usher 1B syndrome: A predicted membrane-associated motor protein expressed in developing sensory epithelia. Proc. Natl. Acad. Sci. USA 1996, 93, 3232–3237. [Google Scholar] [CrossRef] [Green Version]
- Weil, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995, 374, 60–61. [Google Scholar] [CrossRef]
- Jaijo, T.; Aller, E.; Oltra, S.; Beneyto, M.; Najera, C.; Ayuso, C.; Baiget, M.; Carballo, M.; Antinolo, G.; Valverde, D.; et al. Mutation profile of the MYO7A gene in Spanish patients with Usher syndrome type I. Hum. Mutat. 2006, 27, 290–291. [Google Scholar] [CrossRef]
- Jaijo, T.; Aller, E.; Beneyto, M.; Najera, C.; Graziano, C.; Turchetti, D.; Seri, M.; Ayuso, C.; Baiget, M.; Moreno, F.; et al. MYO7A mutation screening in Usher syndrome type I patients from diverse origins. J. Med. Genet. 2007, 44, e71. [Google Scholar] [CrossRef] [Green Version]
- Galbis-Martinez, L.; Blanco-Kelly, F.; Garcia-Garcia, G.; Avila-Fernandez, A.; Jaijo, T.; Fuster-Garcia, C.; Perea-Romero, I.; Zurita-Munoz, O.; Jimenez-Rolando, B.; Carreno, E.; et al. Genotype-phenotype correlation in patients with Usher syndrome and pathogenic variants in MYO7A: Implications for future clinical trials. Acta Ophthalmol. 2021, 99, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Wafa, T.T.; Faridi, R.; King, K.A.; Zalewski, C.; Yousaf, R.; Schultz, J.M.; Morell, R.J.; Muskett, J.; Turriff, A.; Tsilou, E.; et al. Vestibular phenotype-genotype correlation in a cohort of 90 patients with Usher syndrome. Clin. Genet. 2021, 99, 226–235. [Google Scholar] [CrossRef]
- Davies, C.; Bergman, J.; Misztal, C.; Ramchandran, R.; Mittal, J.; Bulut, E.; Shah, V.; Mittal, R.; Eshraghi, A.A. The Outcomes of Cochlear Implantation in Usher Syndrome: A Systematic Review. J. Clin. Med. 2021, 10, 2915. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, A.A.; Polineni, S.P.; Davies, C.; Shahal, D.; Mittal, J.; Al-Zaghal, Z.; Sinha, R.; Jindal, U.; Mittal, R. Genotype-Phenotype Correlation for Predicting Cochlear Implant Outcome: Current Challenges and Opportunities. Front. Genet. 2020, 11, 678. [Google Scholar] [CrossRef]
- Liu, X.Z.; Angeli, S.I.; Rajput, K.; Yan, D.; Hodges, A.V.; Eshraghi, A.; Telischi, F.F.; Balkany, T.J. Cochlear implantation in individuals with Usher type 1 syndrome. Int. J. Pediatric Otorhinolaryngol. 2008, 72, 841–847. [Google Scholar] [CrossRef]
- Skilton, A.; Boswell, E.; Prince, K.; Francome-Wood, P.; Moosajee, M. Overcoming barriers to the involvement of deafblind people in conversations about research: Recommendations from individuals with Usher syndrome. Res. Involv. Engagem. 2018, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazmierczak, P.; Sakaguchi, H.; Tokita, J.; Wilson-Kubalek, E.M.; Milligan, R.A.; Muller, U.; Kachar, B. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 2007, 449, 87–91. [Google Scholar] [CrossRef]
- Alagramam, K.N.; Yuan, H.; Kuehn, M.H.; Murcia, C.L.; Wayne, S.; Srisailpathy, C.R.; Lowry, R.B.; Knaus, R.; Van Laer, L.; Bernier, F.P.; et al. Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum. Mol. Genet. 2001, 10, 1709–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownstein, Z.; Ben-Yosef, T.; Dagan, O.; Frydman, M.; Abeliovich, D.; Sagi, M.; Abraham, F.A.; Taitelbaum-Swead, R.; Shohat, M.; Hildesheimer, M.; et al. The R245X mutation of PCDH15 in Ashkenazi Jewish children diagnosed with nonsyndromic hearing loss foreshadows retinitis pigmentosa. Pediatric Res. 2004, 55, 995–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, Z.M.; Riazuddin, S.; Bernstein, S.L.; Ahmed, Z.; Khan, S.; Griffith, A.J.; Morell, R.J.; Friedman, T.B.; Riazuddin, S.; Wilcox, E.R. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am. J. Hum. Genet. 2001, 69, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Jaijo, T.; Oshima, A.; Aller, E.; Carney, C.; Usami, S.; Millan, J.M.; Kimberling, W.J. Mutation screening of the PCDH15 gene in Spanish patients with Usher syndrome type I. Mol. Vis. 2012, 18, 1719–1726. [Google Scholar]
- Wu, C.C.; Lin, Y.H.; Liu, T.C.; Lin, K.N.; Yang, W.S.; Hsu, C.J.; Chen, P.L.; Wu, C.M. Identifying Children With Poor Cochlear Implantation Outcomes Using Massively Parallel Sequencing. Medicine 2015, 94, e1073. [Google Scholar] [CrossRef]
- Nisenbaum, E.; Prentiss, S.; Yan, D.; Nourbakhsh, A.; Smeal, M.; Holcomb, M.; Cejas, I.; Telischi, F.; Liu, X.Z. Screening Strategies for Deafness Genes and Functional Outcomes in Cochlear Implant Patients. Otol. Neurotol. 2021, 42, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.M.; Xia, X.J.; Verpy, E.; Du, L.L.; Pandya, A.; Petit, C.; Balkany, T.; Nance, W.E.; Liu, X.Z. Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness. Hum. Genet. 2002, 111, 26–30. [Google Scholar] [CrossRef]
- Ebermann, I.; Lopez, I.; Bitner-Glindzicz, M.; Brown, C.; Koenekoop, R.K.; Bolz, H.J. Deafblindness in French Canadians from Quebec: A predominant founder mutation in the USH1C gene provides the first genetic link with the Acadian population. Genome Biol. 2007, 8, R47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebermann, I.; Koenekoop, R.K.; Lopez, I.; Bou-Khzam, L.; Pigeon, R.; Bolz, H.J. An USH2A founder mutation is the major cause of Usher syndrome type 2 in Canadians of French origin and confirms common roots of Quebecois and Acadians. Eur. J. Hum. Genet. 2009, 17, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Mustapha, M.; Chouery, E.; Torchard-Pagnez, D.; Nouaille, S.; Khrais, A.; Sayegh, F.N.; Megarbane, A.; Loiselet, J.; Lathrop, M.; Petit, C.; et al. A novel locus for Usher syndrome type I, USH1G, maps to chromosome 17q24-25. Hum. Genet. 2002, 110, 348–350. [Google Scholar] [CrossRef] [PubMed]
- DeWan, A.T.; Parrado, A.R.; Leal, S.M. A second kindred linked to DFNA20 (17q25.3) reduces the genetic interval. Clin. Genet. 2003, 63, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Weil, D.; El-Amraoui, A.; Masmoudi, S.; Mustapha, M.; Kikkawa, Y.; Laine, S.; Delmaghani, S.; Adato, A.; Nadifi, S.; Zina, Z.B.; et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum. Mol. Genet. 2003, 12, 463–471. [Google Scholar] [CrossRef]
- Riazuddin, S.; Belyantseva, I.A.; Giese, A.P.; Lee, K.; Indzhykulian, A.A.; Nandamuri, S.P.; Yousaf, R.; Sinha, G.P.; Lee, S.; Terrell, D.; et al. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat. Genet. 2012, 44, 1265–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaudy, F.; Zheng, L.; Ficarella, R.; Ballana, E.; Carella, M.; Melchionda, S.; Estivill, X.; Bartles, J.R.; Gasparini, P. Espin gene (ESPN) mutations associated with autosomal dominant hearing loss cause defects in microvillar elongation or organisation. J. Med. Genet. 2006, 43, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, E.; Pennings, R.J.; te Brinke, H.; Claassen, A.; Yntema, H.G.; Hoefsloot, L.H.; Cremers, F.P.; Cremers, C.W.; Kremer, H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am. J. Hum. Genet. 2004, 74, 738–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, L.; Maltese, P.E.; Castori, M.; El Shamieh, S.; Zeitz, C.; Audo, I.; Zulian, A.; Marinelli, C.; Benedetti, S.; Costantini, A.; et al. Molecular Epidemiology in 591 Italian Probands With Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Investig. Ophthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef]
- Dreyer, B.; Brox, V.; Tranebjaerg, L.; Rosenberg, T.; Sadeghi, A.M.; Moller, C.; Nilssen, O. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum. Mutat. 2008, 29, 451. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Chen, D.F.; Wang, L.; Wu, S.; Wei, X.; Li, H.; Jin, Z.B.; Sui, R. USH2A variants in Chinese patients with Usher syndrome type II and non-syndromic retinitis pigmentosa. Br. J. Ophthalmol. 2021, 105, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Vozzi, D.; Aaspollu, A.; Athanasakis, E.; Berto, A.; Fabretto, A.; Licastro, D.; Kulm, M.; Testa, F.; Trevisi, P.; Vahter, M.; et al. Molecular epidemiology of Usher syndrome in Italy. Mol. Vis. 2011, 17, 1662–1668. [Google Scholar]
- Ebermann, I.; Wiesen, M.H.; Zrenner, E.; Lopez, I.; Pigeon, R.; Kohl, S.; Lowenheim, H.; Koenekoop, R.K.; Bolz, H.J. GPR98 mutations cause Usher syndrome type 2 in males. J. Med. Genet. 2009, 46, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Hilgert, N.; Kahrizi, K.; Dieltjens, N.; Bazazzadegan, N.; Najmabadi, H.; Smith, R.J.; Van Camp, G. A large deletion in GPR98 causes type IIC Usher syndrome in male and female members of an Iranian family. J. Med. Genet. 2009, 46, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Hilgert, N. Novel human pathological mutations. Gene symbol: GPR98. Disease: Usher syndrome 2C. Hum. Genet. 2009, 125, 342. [Google Scholar]
- Lin, L.; Shi, Y.; Wang, M.; Wang, C.; Lu, Q.; Zhu, J.; Zhang, R. Phase separation-mediated condensation of Whirlin-Myo15-Eps8 stereocilia tip complex. Cell Rep. 2021, 34, 108770. [Google Scholar] [CrossRef] [PubMed]
- Mburu, P.; Mustapha, M.; Varela, A.; Weil, D.; El-Amraoui, A.; Holme, R.H.; Rump, A.; Hardisty, R.E.; Blanchard, S.; Coimbra, R.S.; et al. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat. Genet. 2003, 34, 421–428. [Google Scholar] [CrossRef]
- Ebermann, I.; Scholl, H.P.; Charbel Issa, P.; Becirovic, E.; Lamprecht, J.; Jurklies, B.; Millan, J.M.; Aller, E.; Mitter, D.; Bolz, H. A novel gene for Usher syndrome type 2: Mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum. Genet. 2007, 121, 203–211. [Google Scholar] [CrossRef]
- Ebermann, I.; Phillips, J.B.; Liebau, M.C.; Koenekoop, R.K.; Schermer, B.; Lopez, I.; Schafer, E.; Roux, A.F.; Dafinger, C.; Bernd, A.; et al. PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Investig. 2010, 120, 1812–1823. [Google Scholar] [CrossRef] [Green Version]
- Aller, E.; Jaijo, T.; van Wijk, E.; Ebermann, I.; Kersten, F.; Garcia-Garcia, G.; Voesenek, K.; Aparisi, M.J.; Hoefsloot, L.; Cremers, C.; et al. Sequence variants of the DFNB31 gene among Usher syndrome patients of diverse origin. Mol. Vis. 2010, 16, 495–500. [Google Scholar] [PubMed]
- Bonnet, C.; Riahi, Z.; Chantot-Bastaraud, S.; Smagghe, L.; Letexier, M.; Marcaillou, C.; Lefevre, G.M.; Hardelin, J.P.; El-Amraoui, A.; Singh-Estivalet, A.; et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 2016, 24, 1730–1738. [Google Scholar] [CrossRef] [Green Version]
- Booth, K.T.; Azaiez, H.; Kahrizi, K.; Simpson, A.C.; Tollefson, W.T.; Sloan, C.M.; Meyer, N.C.; Babanejad, M.; Ardalani, F.; Arzhangi, S.; et al. PDZD7 and hearing loss: More than just a modifier. Am. J. Med. Genet. 2015, 167, 2957–2965. [Google Scholar] [CrossRef] [Green Version]
- Vona, B.; Lechno, S.; Hofrichter, M.A.; Hopf, S.; Laig, A.K.; Haaf, T.; Keilmann, A.; Zechner, U.; Bartsch, O. Confirmation of PDZD7 as a Nonsyndromic Hearing Loss Gene. Ear Hear. 2016, 37, e238–e246. [Google Scholar] [CrossRef] [PubMed]
- Aparisi, M.J.; Aller, E.; Fuster-Garcia, C.; Garcia-Garcia, G.; Rodrigo, R.; Vazquez-Manrique, R.P.; Blanco-Kelly, F.; Ayuso, C.; Roux, A.F.; Jaijo, T.; et al. Targeted next generation sequencing for molecular diagnosis of Usher syndrome. Orphanet J. Rare Dis. 2014, 9, 168. [Google Scholar] [CrossRef] [Green Version]
- Geng, R.; Geller, S.F.; Hayashi, T.; Ray, C.A.; Reh, T.A.; Bermingham-McDonogh, O.; Jones, S.M.; Wright, C.G.; Melki, S.; Imanishi, Y.; et al. Usher syndrome IIIA gene clarin-1 is essential for hair cell function and associated neural activation. Hum. Mol. Genet. 2009, 18, 2748–2760. [Google Scholar] [CrossRef] [Green Version]
- Geng, R.; Melki, S.; Chen, D.H.; Tian, G.; Furness, D.N.; Oshima-Takago, T.; Neef, J.; Moser, T.; Askew, C.; Horwitz, G.; et al. The mechanosensory structure of the hair cell requires clarin-1, a protein encoded by Usher syndrome III causative gene. J. Neurosci. 2012, 32, 9485–9498. [Google Scholar] [CrossRef]
- Herrera, W.; Aleman, T.S.; Cideciyan, A.V.; Roman, A.J.; Banin, E.; Ben-Yosef, T.; Gardner, L.M.; Sumaroka, A.; Windsor, E.A.; Schwartz, S.B.; et al. Retinal disease in Usher syndrome III caused by mutations in the clarin-1 gene. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2651–2660. [Google Scholar] [CrossRef]
- Isosomppi, J.; Vastinsalo, H.; Geller, S.F.; Heon, E.; Flannery, J.G.; Sankila, E.M. Disease-causing mutations in the CLRN1 gene alter normal CLRN1 protein trafficking to the plasma membrane. Mol. Vis. 2009, 15, 1806–1818. [Google Scholar] [PubMed]
- Jouret, G.; Poirsier, C.; Spodenkiewicz, M.; Jaquin, C.; Gouy, E.; Arndt, C.; Labrousse, M.; Gaillard, D.; Doco-Fenzy, M.; Lebre, A.S. Genetics of Usher Syndrome: New Insights From a Meta-analysis. Otol. Neurotol. 2019, 40, 121–129. [Google Scholar] [CrossRef]
- Vastinsalo, H.; Jalkanen, R.; Bergmann, C.; Neuhaus, C.; Kleemola, L.; Jauhola, L.; Bolz, H.J.; Sankila, E.M. Extended mutation spectrum of Usher syndrome in Finland. Acta Ophthalmol. 2013, 91, 325–334. [Google Scholar] [CrossRef]
- Adato, A.; Vreugde, S.; Joensuu, T.; Avidan, N.; Hamalainen, R.; Belenkiy, O.; Olender, T.; Bonne-Tamir, B.; Ben-Asher, E.; Espinos, C.; et al. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur. J. Hum. Genet. 2002, 10, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Adato, A.; Kalinski, H.; Weil, D.; Chaib, H.; Korostishevsky, M.; Bonne-Tamir, B. Possible interaction between USH1B and USH3 gene products as implied by apparent digenic deafness inheritance. Am. J. Hum. Genet. 1999, 65, 261–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joensuu, T.; Hämäläinen, R.; Yuan, B.; Johnson, C.; Tegelberg, S.; Gasparini, P.; Zelante, L.; Pirvola, U.; Pakarinen, L.; Lehesjoki, A.E.; et al. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am. J. Hum. Genet. 2001, 69, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Pietola, L.; Aarnisalo, A.A.; Abdel-Rahman, A.; Vastinsalo, H.; Isosomppi, J.; Lopponen, H.; Kentala, E.; Johansson, R.; Valtonen, H.; Vasama, J.P.; et al. Speech recognition and communication outcomes with cochlear implantation in Usher syndrome type 3. Otol. Neurotol. 2012, 33, 38–41. [Google Scholar] [CrossRef]
- Eisenberger, T.; Slim, R.; Mansour, A.; Nauck, M.; Nurnberg, G.; Nurnberg, P.; Decker, C.; Dafinger, C.; Ebermann, I.; Bergmann, C.; et al. Targeted next-generation sequencing identifies a homozygous nonsense mutation in ABHD12, the gene underlying PHARC, in a family clinically diagnosed with Usher syndrome type 3. Orphanet J. Rare Dis. 2012, 7, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, S.L.; Pelletier, L. Centriolar satellite biogenesis and function in vertebrate cells. J. Cell Sci. 2020, 133, jcs239566. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Khateb, S.; Zelinger, L.; Mizrahi-Meissonnier, L.; Ayuso, C.; Koenekoop, R.K.; Laxer, U.; Gross, M.; Banin, E.; Sharon, D. A homozygous nonsense CEP250 mutation combined with a heterozygous nonsense C2orf71 mutation is associated with atypical Usher syndrome. J. Med. Genet. 2014, 51, 460–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikopoulos, K.; Farinelli, P.; Giangreco, B.; Tsika, C.; Royer-Bertrand, B.; Mbefo, M.K.; Bedoni, N.; Kjellstrom, U.; El Zaoui, I.; Di Gioia, S.A.; et al. Mutations in CEP78 Cause Cone-Rod Dystrophy and Hearing Loss Associated with Primary-Cilia Defects. Am. J. Hum. Genet. 2016, 99, 770–776. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Xu, M.; Chen, X.; Sheng, X.; Yuan, Z.; Liu, Y.; Li, H.; Sun, Z.; Li, H.; Yang, L.; et al. CEP78 is mutated in a distinct type of Usher syndrome. J. Med. Genet. 2017, 54, 190–195. [Google Scholar] [CrossRef]
- Namburi, P.; Ratnapriya, R.; Khateb, S.; Lazar, C.H.; Kinarty, Y.; Obolensky, A.; Erdinest, I.; Marks-Ohana, D.; Pras, E.; Ben-Yosef, T.; et al. Bi-allelic Truncating Mutations in CEP78, Encoding Centrosomal Protein 78, Cause Cone-Rod Degeneration with Sensorineural Hearing Loss. Am. J. Hum. Genet. 2016, 99, 1222–1223. [Google Scholar] [CrossRef]
- Khateb, S.; Kowalewski, B.; Bedoni, N.; Damme, M.; Pollack, N.; Saada, A.; Obolensky, A.; Ben-Yosef, T.; Gross, M.; Dierks, T.; et al. A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical Usher syndrome in humans. Genet. Med. 2018, 20, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Nolen, R.M.; Hufnagel, R.B.; Friedman, T.B.; Turriff, A.E.; Brewer, C.C.; Zalewski, C.K.; King, K.A.; Wafa, T.T.; Griffith, A.J.; Brooks, B.P.; et al. Atypical and ultra-rare Usher syndrome: A review. Ophthalmic Genet. 2020, 41, 401–412. [Google Scholar] [CrossRef]
- Abad-Morales, V.; Navarro, R.; Bures-Jelstrup, A.; Pomares, E. Identification of a novel homozygous ARSG mutation as the second cause of Usher syndrome type 4. Am. J. Ophthalmol. Cas. Rep. 2020, 19, 100736. [Google Scholar] [CrossRef]
- Peter, V.G.; Quinodoz, M.; Sadio, S.; Held, S.; Rodrigues, M.; Soares, M.; Sousa, A.B.; Coutinho Santos, L.; Damme, M.; Rivolta, C. New clinical and molecular evidence linking mutations in ARSG to Usher syndrome type IV. Hum. Mutat. 2021, 42, 261–271. [Google Scholar] [CrossRef]
- Espinos, C.; Millan, J.M.; Beneyto, M.; Najera, C. Epidemiology of Usher syndrome in Valencia and Spain. Community Genet 1998, 1, 223–228. [Google Scholar] [CrossRef]
- Kimberling, W.J.; Hildebrand, M.S.; Shearer, A.E.; Jensen, M.L.; Halder, J.A.; Trzupek, K.; Cohn, E.S.; Weleber, R.G.; Stone, E.M.; Smith, R.J. Frequency of Usher syndrome in two pediatric populations: Implications for genetic screening of deaf and hard of hearing children. Genet. Med. 2010, 12, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi, M.; Kimberling, W.; Tranebjœrg, L.; Möller, C. The prevalence of Usher Syndrome in Sweden: A nationwide epidemiological and clinical survey. Audiol. Med. 2004, 2, 220–228. [Google Scholar] [CrossRef]
- Ness, S.L.; Ben-Yosef, T.; Bar-Lev, A.; Madeo, A.C.; Brewer, C.C.; Avraham, K.B.; Kornreich, R.; Desnick, R.J.; Willner, J.P.; Friedman, T.B.; et al. Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J. Med. Genet. 2003, 40, 767–772. [Google Scholar] [CrossRef] [Green Version]
- Malm, E.; Ponjavic, V.; Moller, C.; Kimberling, W.J.; Stone, E.S.; Andreasson, S. Alteration of rod and cone function in children with Usher syndrome. Eur. J. Ophthalmol. 2011, 21, 30–38. [Google Scholar] [CrossRef]
- Kimberling, W.J.; Moller, C. Clinical and molecular genetics of Usher syndrome. J. Am. Acad. Audiol. 1995, 6, 63–72. [Google Scholar]
- Sadeghi, A.M.; Eriksson, K.; Kimberling, W.J.; Sjostrom, A.; Moller, C. Longterm visual prognosis in Usher syndrome types 1 and 2. Acta Ophthalmol. Scand. 2006, 84, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Busi, M.; Rosignoli, M.; Castiglione, A.; Minazzi, F.; Trevisi, P.; Aimoni, C.; Calzolari, F.; Granieri, E.; Martini, A. Cochlear Implant Outcomes and Genetic Mutations in Children with Ear and Brain Anomalies. BioMed Res. Int. 2015, 2015, 696281. [Google Scholar] [CrossRef] [Green Version]
- Hartel, B.P.; Lofgren, M.; Huygen, P.L.; Guchelaar, I.; Lo, A.N.K.N.; Sadeghi, A.M.; van Wijk, E.; Tranebjaerg, L.; Kremer, H.; Kimberling, W.J.; et al. A combination of two truncating mutations in USH2A causes more severe and progressive hearing impairment in Usher syndrome type IIa. Hear. Res. 2016, 339, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, A.M.; Cohn, E.S.; Kimberling, W.J.; Halvarsson, G.; Moller, C. Expressivity of hearing loss in cases with Usher syndrome type IIA. Int. J. Audiol. 2013, 52, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Nisenbaum, E.; Thielhelm, T.P.; Nourbakhsh, A.; Yan, D.; Blanton, S.H.; Shu, Y.; Koehler, K.R.; El-Amraoui, A.; Chen, Z.; Lam, B.L.; et al. Review of Genotype-Phenotype Correlations in Usher Syndrome. Ear Hear. 2021, 43, 1–8. [Google Scholar] [CrossRef]
- Sadeghi, M.; Cohn, E.S.; Kimberling, W.J.; Tranebjaerg, L.; Moller, C. Audiological and vestibular features in affected subjects with USH3: A genotype/phenotype correlation. Int. J. Audiol. 2005, 44, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Ehn, M.; Wahlqvist, M.; Danermark, B.; Dahlstrom, O.; Moller, C. Health, work, social trust, and financial situation in persons with Usher syndrome type 1. Work 2018, 60, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlqvist, M.; Moller, C.; Moller, K.; Danermark, B. Similarities and Differences in Health, Social Trust, and Financial Situation in People With Usher Syndrome, a Bio-Psychosocial Perspective. Front. Psychol. 2020, 11, 1760. [Google Scholar] [CrossRef] [PubMed]
- French, L.S.; Mellough, C.B.; Chen, F.K.; Carvalho, L.S. A Review of Gene, Drug and Cell-Based Therapies for Usher Syndrome. Front. Cell Neurosci. 2020, 14, 183. [Google Scholar] [CrossRef]
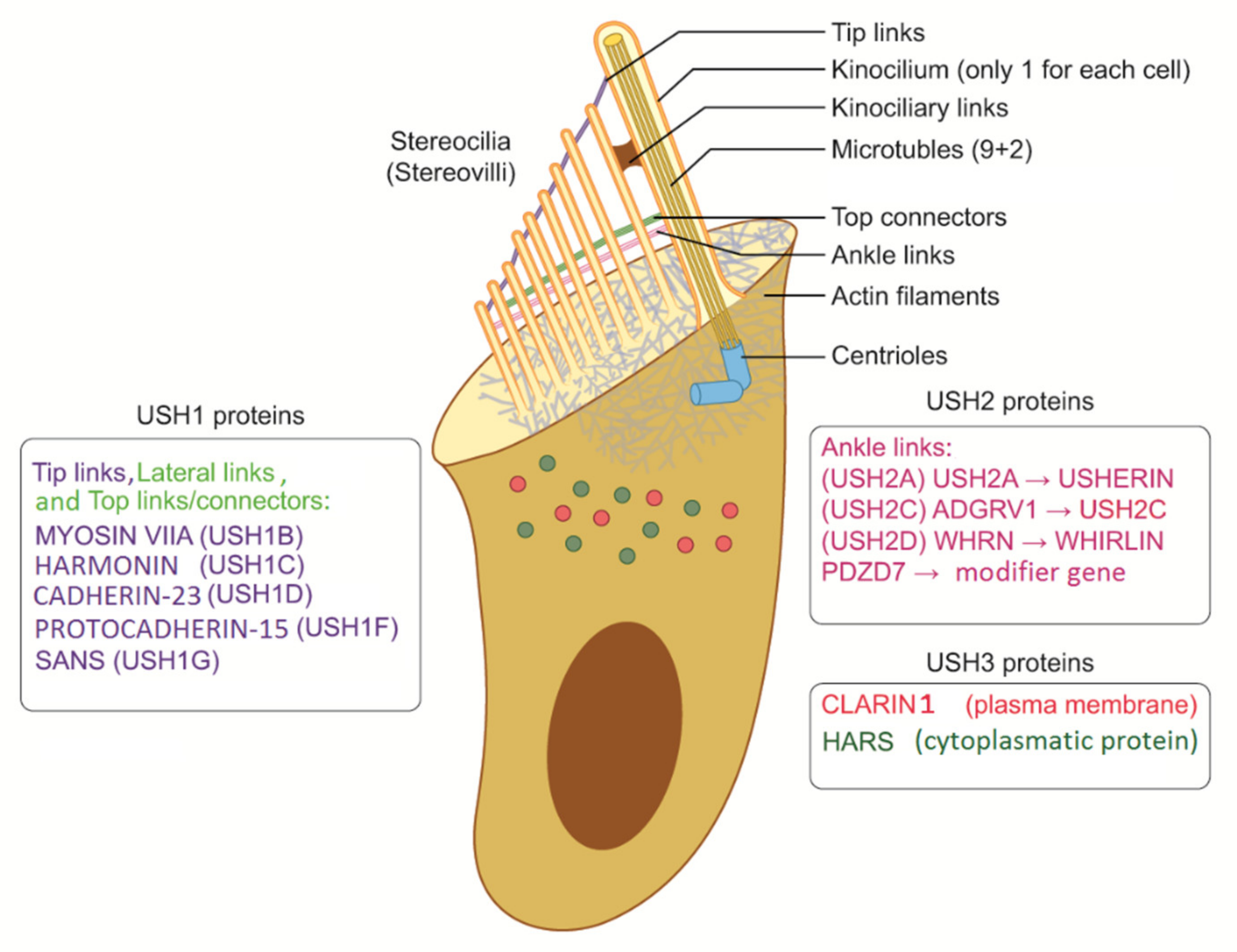




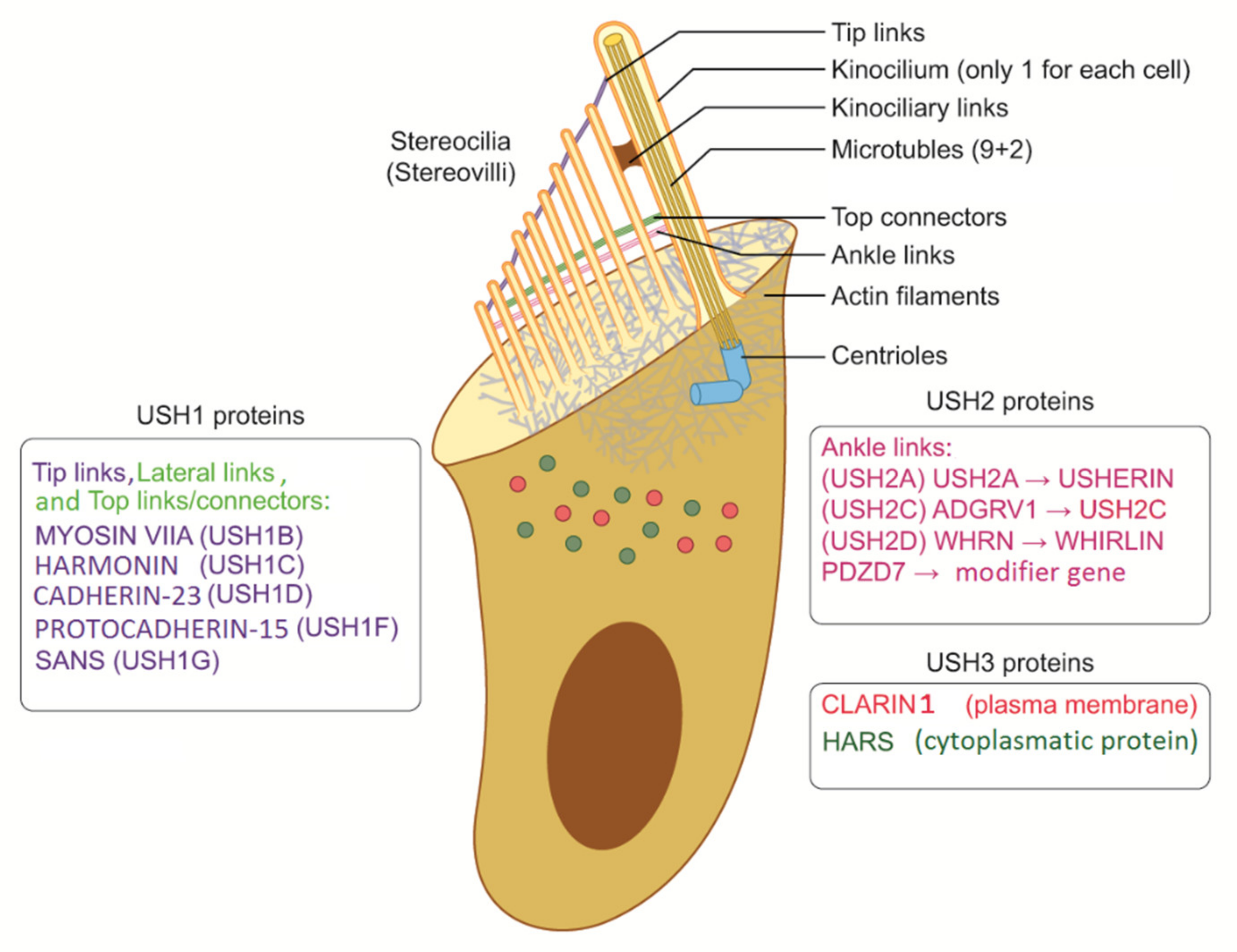{kind=link}
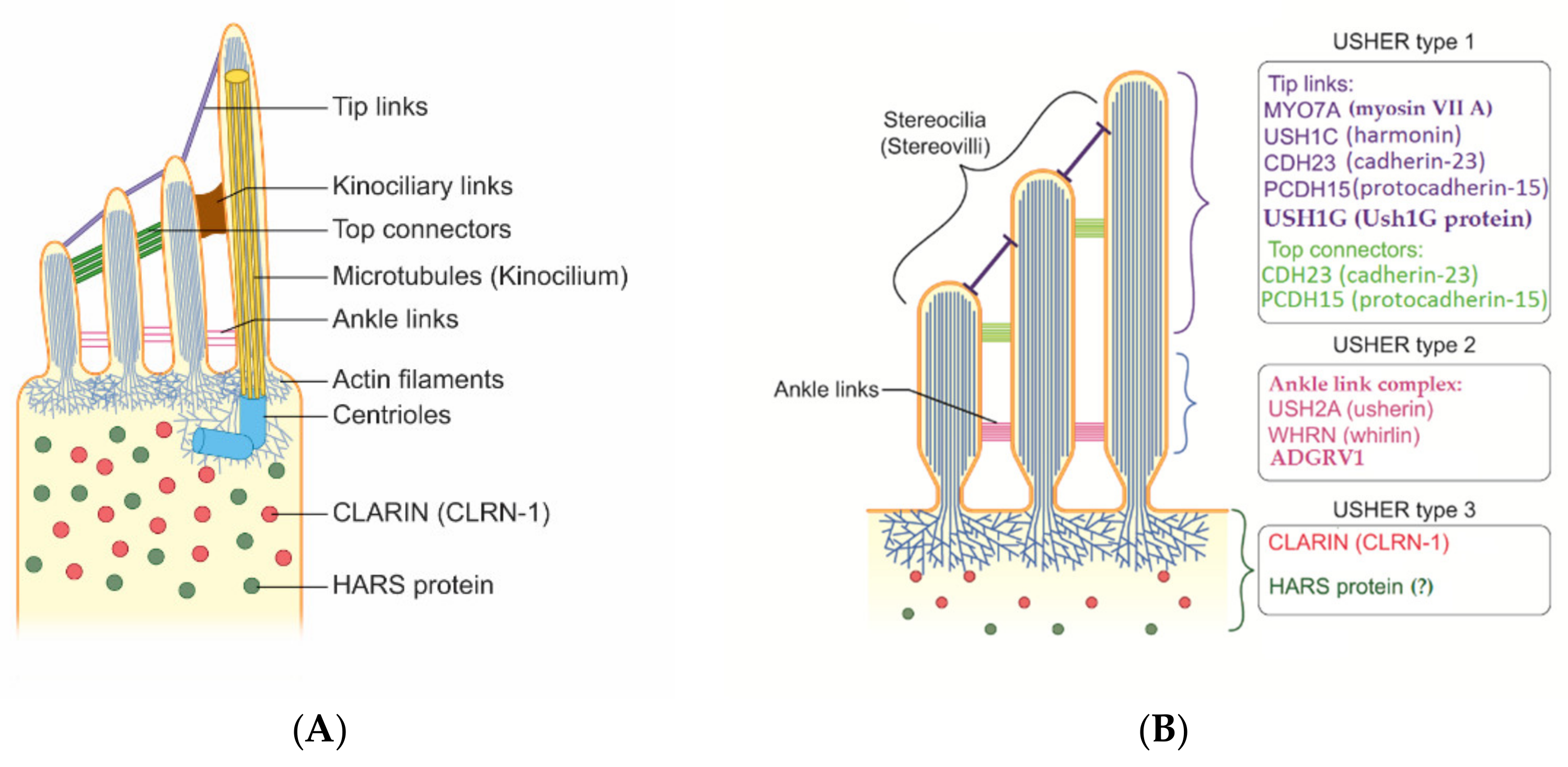{kind=link}
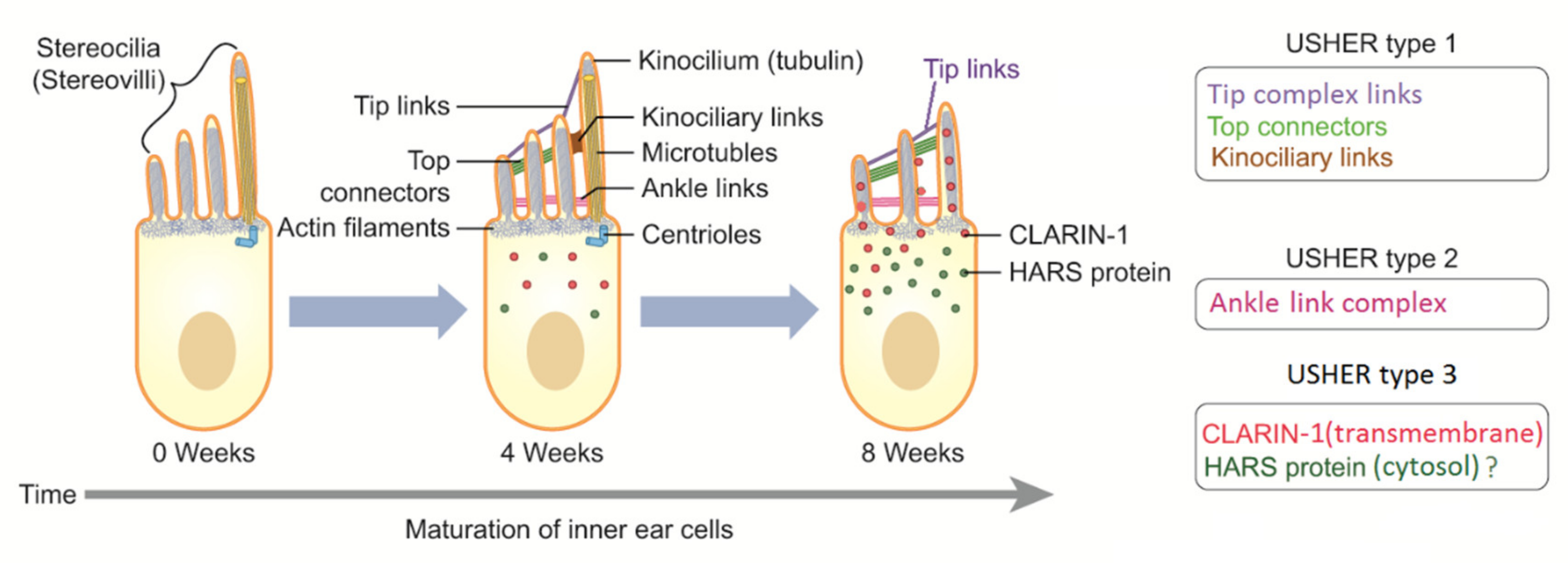{kind=link}
{kind=link}
| Gene | Chr. (Loci) | Protein | Role | Expression | Effect When Mutated | Usher Type | Grade of Evidence |
|---|---|---|---|---|---|---|---|
| ABHD12 | 20p11.21 | Lysophosphatidylserine lipase ABHD12 (abhydrolase domain containing 12) | Functional protein (enzyme involved: endocannabinoid system and neurotransmission) | Nervous system, thyroid, retina, skin, nasal epithelium | Neurodegeneration | USH3? | To be confirmed |
| ADGRV1 | 5q14.3 | Adhesion G protein-coupled receptor V1 | Functional protein G-protein transmembrane receptor. Usher protein complex 2. | Nervous system, eye, and ear. | Ciliary dysfunction. Vision impairment. Familial febrile seizures | USH2C | Confirmed and widely reported |
| ARSG | 17q24.2 | Arylsulfatase G | Functional protein, Enzyme (sulfatase) | Lysosomes | Dysregulation of hormone biosynthesis | USH4? | To be confirmed |
| CDH23 | 10q22.1 | Cadherin-23 | Structural protein (adhesion protein) | Retina and cochlea; Usher protein complex 1. | Poor intercellular adhesion | USH1D and DFNB12 | Confirmed and widely reported |
| CEP250 | 20q11.22 | Centrosome-associated protein CEP250 | Protein required for centriole-centriole cohesion during interphase of the cell cycle | Ubiquitous, centrosome and cilia body | Centrosomal dysfunctional activity and ciliopathies | atypical USH? | To be confirmed |
| CEP78 | 9q21.2 | Centrosomal protein of 78 kDa | Protein required for centriole-centriole cohesion during interphase of the cell cycle | Ubiquitous, centrosome and cilia body | Centrosomal dysfunctional activity and ciliopathies | atypical USH? | To be confirmed |
| CIB2 | 15q25.1 | Calcium and integrin-binding family member 2 | Intracellular calcium homeostasis | Ubiquitous | Dysfunction in cellular activities | USH1J? | No longer considered an Usher gene |
| CLRN1 | 3q25.1 | Clarin-1 | Transmembrane protein | Synapses, inner ear, and retina | Interruption of the signal transmission | USH3A | Confirmed |
| WHRN | 9q32 | Whirlin | Gene product is a PDZ scaffold protein expressed in hair cells and photoreceptors | Usher protein complex 2 | Profound prelingual deafness; rare cause of recessive deafness and RP | USH2D and DFNB31 | Confirmed |
| ESPN | 1p36.31 | Espin | The gene product is an actin-bundling protein that plays a role in transduction in mechanosensory and chemosensory cells | Tip complex | Possible USH with or without vestibular symptoms | USH1M? and DFNB36 | To be confirmed |
| HARS1 | 5q31.3 | Histidine--tRNA ligase, cytoplasmic | Protein-coding gene | Cytoplasmic enzyme that belongs to the class II family of aminoacyl-tRNA synthetases | Possible USH as recessive pattern and Charcot–Marie– Tooth disease as dominant pattern | USH3B? | Its role must be confirmed |
| MYO7A | 11q13.5 | Unconventional myosin-VIIa | Myosin, a structural component of cilia and microvilli, found in several tissues, including inner ear hair cells, photoreceptors, and RPE | Ubiquitous; Usher protein complex 1. | USH and DFNB2 | USH1B | Confirmed |
| PCDH15 | 10q21.1 | Protocadherin-15 | Structural protein involved in tip links (USH1 complex). | Stereocilia in inner ear hair cells and photoreceptors. Usher protein complex 1. | USH and DFNB23 | USH1F | Confirmed |
| PDZD7 | 10q24.31 | PDZ domain-containing protein 7 | It is considered a modifier gene for Usher syndrome. | Usher protein complex 2 | It is responsible for autosomal recessive hearing loss DFNB57 | USH2? | To be confirmed |
| USH1C | 11p15.1 | Harmonin | Structural protein | Usher protein complex 1 | USH and DFNB18 | USH1C | Confirmed |
| USH1G (SANS) | 17q25.1 | Usher syndrome type-1G protein | Structural scaffold protein | Usher protein complex 1 | Usher | USH1G | Confirmed |
| USH2A | 1q41 | Usherin | Structural protein | Usher protein complex 2 | Usher | USH2A | Confirmed |
| ? | USH1E * | 21q21 | ? | mapping linkage | |||
| ? | USH1H * | 15q22-q23 | ? | mapping linkage | |||
| ? | USH1K * | 10p11.21-q21.1 | ? | mapping linkage |
| Type | Gene | Chr. (Locus) | Protein | Epidemiology * (% Mutations a) | Year of Identification |
|---|---|---|---|---|---|
| Usher type I (35–40%) | |||||
| IB | MYO7A | 11q13.5 | Myosin Vlla | 50–70% | 1995 |
| IC | USH1C | 11p14.3 | Harmonin | 6–20% | 2000 |
| ID | CDH23 | 10q22.1 | Cadherin 23 | 10–20% | 2001 |
| IE | Unknown | 21q21.3 | Unknown | Unknown | 1997 |
| IF | PCDH15 | 10q21.1 | Protocadherin 15 | 5–10% | 2001 |
| IG | USH1G (SANS) | 17q25.1 | Usher syndrome type 1G protein | 0–5% | 2003 |
| IH | Unknown | 15q22-q23 | Unknown | 2009 | |
| IJ | CIB2 (?) | 15q25.1 | CIB2 | No longer USH gene | 2012 |
| IK | Unknown | 10p11.21-q21.1 | Unknown | Unknown | ? |
| Usher type II (60–65%) | |||||
| IIA | USH2A | 1q41 | Usherin | 50–80% | 1998 |
| IIC | ADGRV1 (GPR98) | 5q14.3 | 5–20% | 2004 | |
| IID | WHRN (DFNB31) | 9q32 | Whirlin | 0–10% | 2007 |
| Usher type III (0–5%) | |||||
| IIIA | CLRN1 | 3q25.1 | Clarin-1 transcript variant | 90–95% | 2001 |
| IIIB | HARS | 5q31.3 | Cytoplasmic histidine--tRNA ligase | 5–10%, however its role must be confirmed | ? |
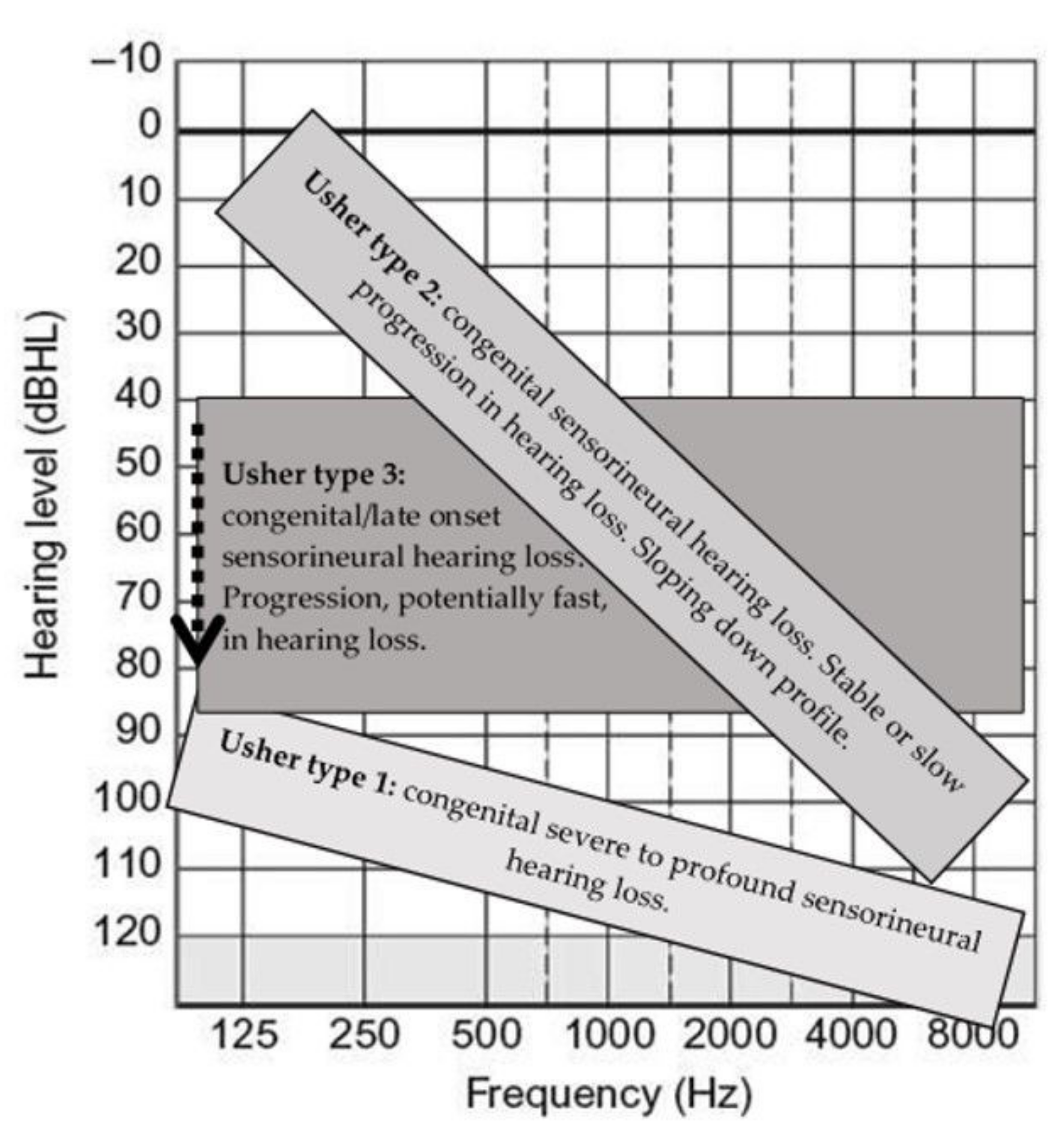
| Type | Hearing Loss | Vestibular Function | Retinitis Pigmentosa (RP) | Hearing Rehabilitation and Communication |
|---|---|---|---|---|
| USH1 | Congenital, severe-to-profound sensorineural hearing loss; however, exceptions with moderate hearing loss are reported. | Absent or abnormal vestibular function; however, exceptions are widely reported, from moderate to normal function | Early onset | Bilateral. Good outcomes with early cochlear implantations. Some exceptions have been reported for patients with PCDH15 mutations. However, further studies are required to confirm this statement. Tactile signing and sign language may be necessary in selected cases. |
| USH2 | Congenital/prelingual sloping down, moderate-to-severe sensorineural hearing loss. | Normal | Late onset, but usually in young/adult age. | Outcomes can be related to the hearing loss onset. Patients with stable hearing loss may benefit from hearing aids. Cochlear implantations have also shown good outcomes. Bilateral. Bimodal rehabilitation should be considered. They may benefit from sign language. |
| USH3 | Variable onset of moderate-to-severe hearing loss potentially fast progressive. | Variable | Variable onset, but essentially during early adulthood | Hearing aids till surgery if indicated. Progression of hearing loss should carefully be monitored. Bilateral rehabilitation. Due to progression, if indicated, bilateral cochlear implantation is a valid option, but bimodal stimulation can also be effective. Efficacy of sign language in selected cases depends on onset and progression. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castiglione, A.; Möller, C. Usher Syndrome. Audiol. Res. 2022, 12, 42-65. https://doi.org/10.3390/audiolres12010005
Castiglione A, Möller C. Usher Syndrome. Audiology Research. 2022; 12(1):42-65. https://doi.org/10.3390/audiolres12010005
Chicago/Turabian StyleCastiglione, Alessandro, and Claes Möller. 2022. "Usher Syndrome" Audiology Research 12, no. 1: 42-65. https://doi.org/10.3390/audiolres12010005
APA StyleCastiglione, A., & Möller, C. (2022). Usher Syndrome. Audiology Research, 12(1), 42-65. https://doi.org/10.3390/audiolres12010005