

Synthesis, DFT Studies, Molecular Docking and Biological Activity Evaluation of Thiazole-Sulfonamide Derivatives as Potent Alzheimer’s Inhibitors
 , , , , , , , and
, , , , , , , and
Abstract
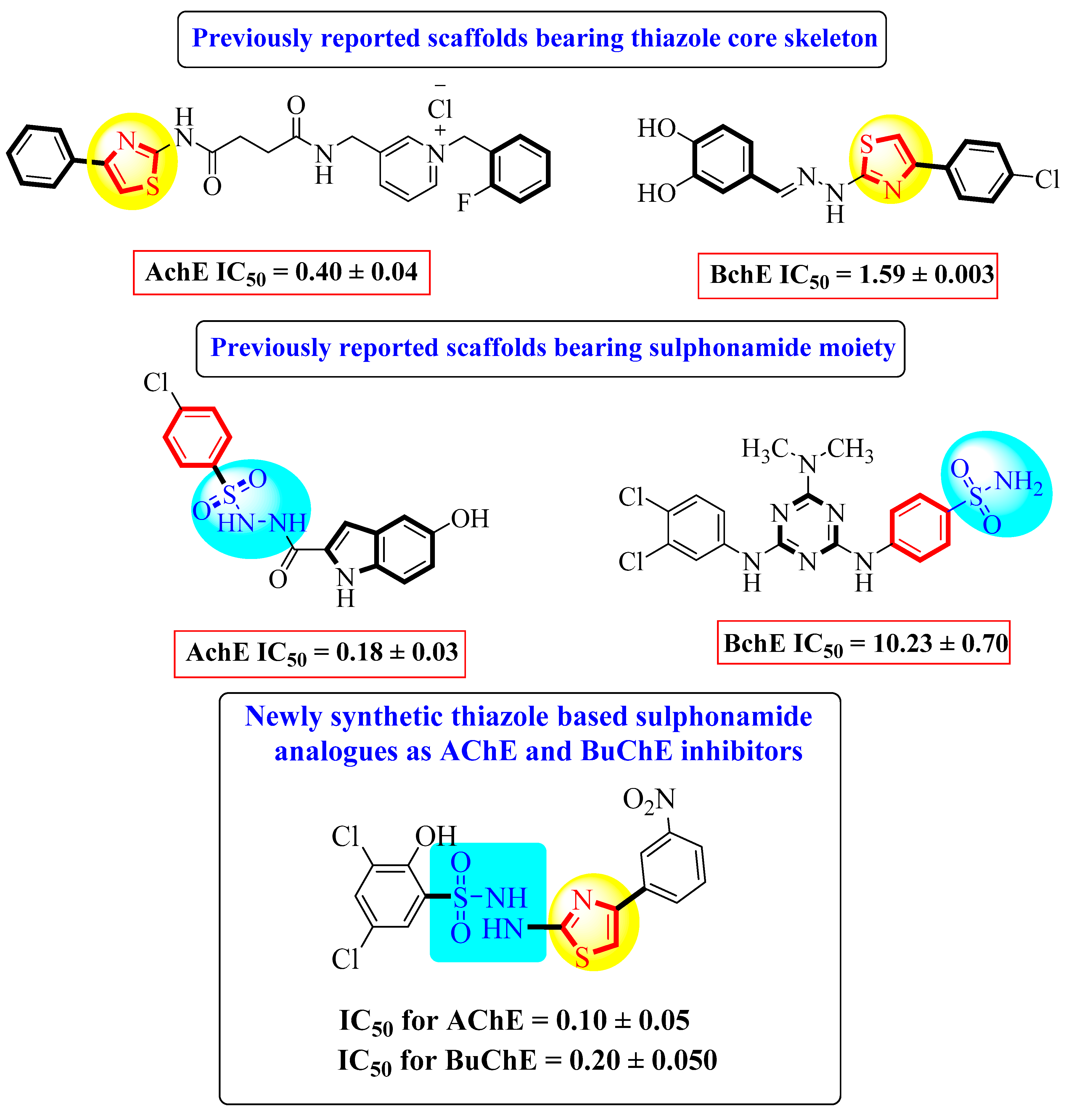
:1. Introduction
2. Results and Discussion
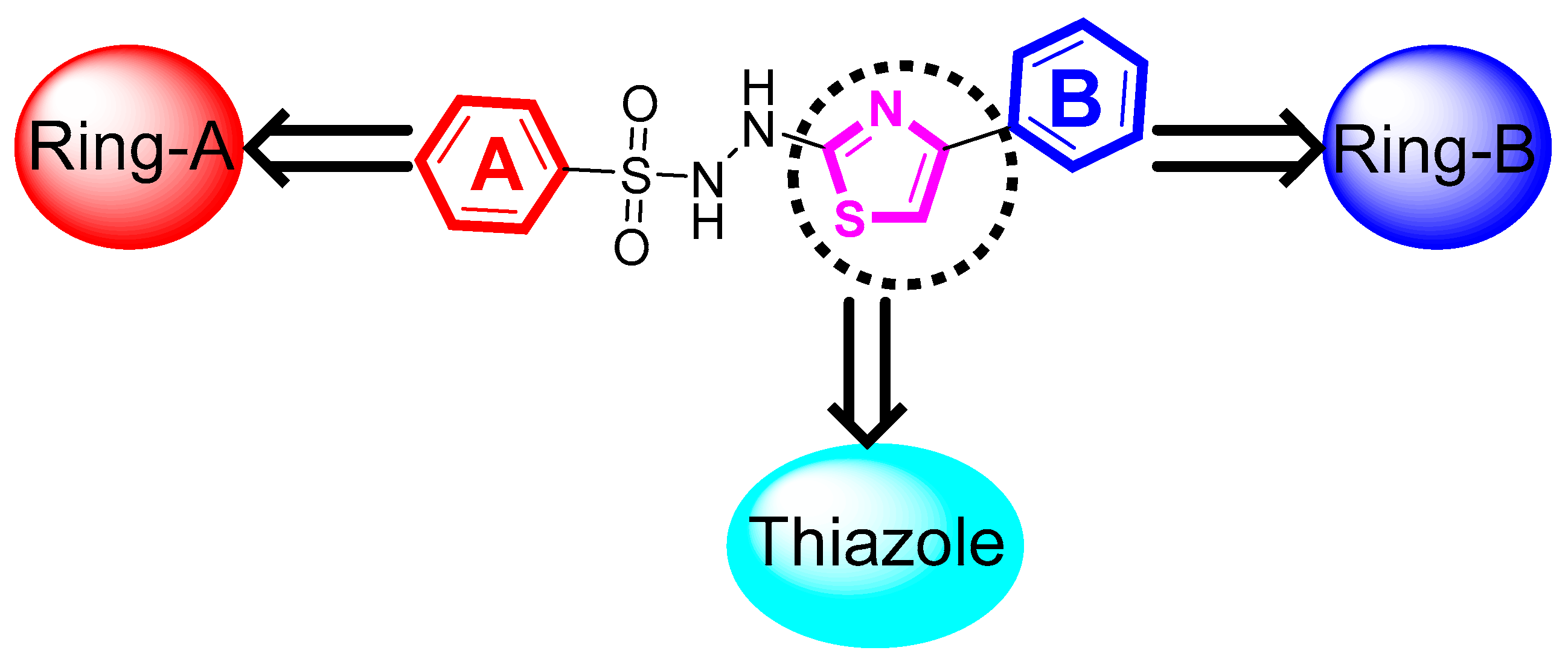
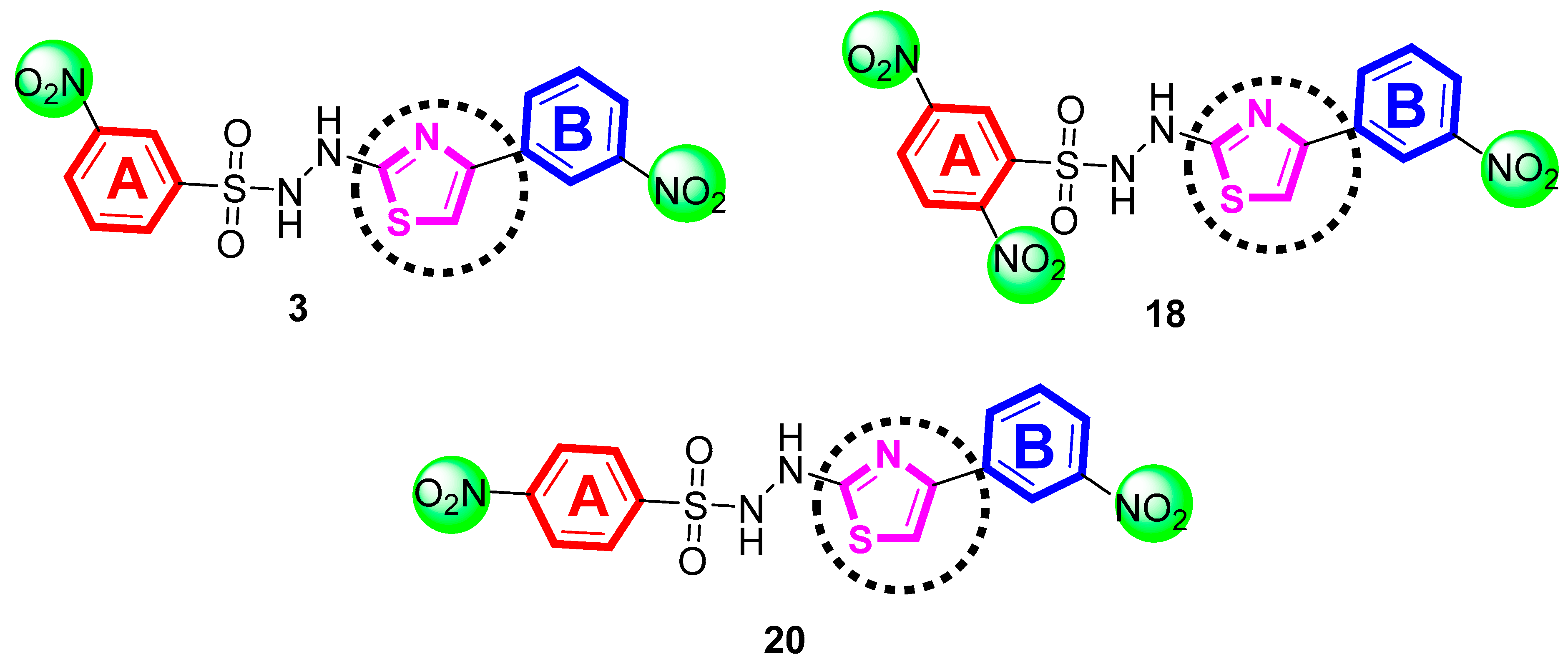
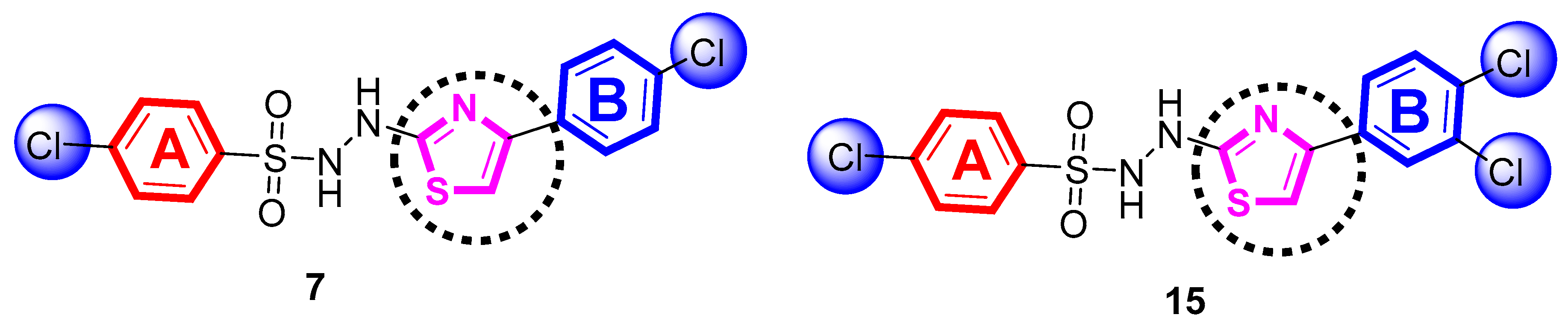
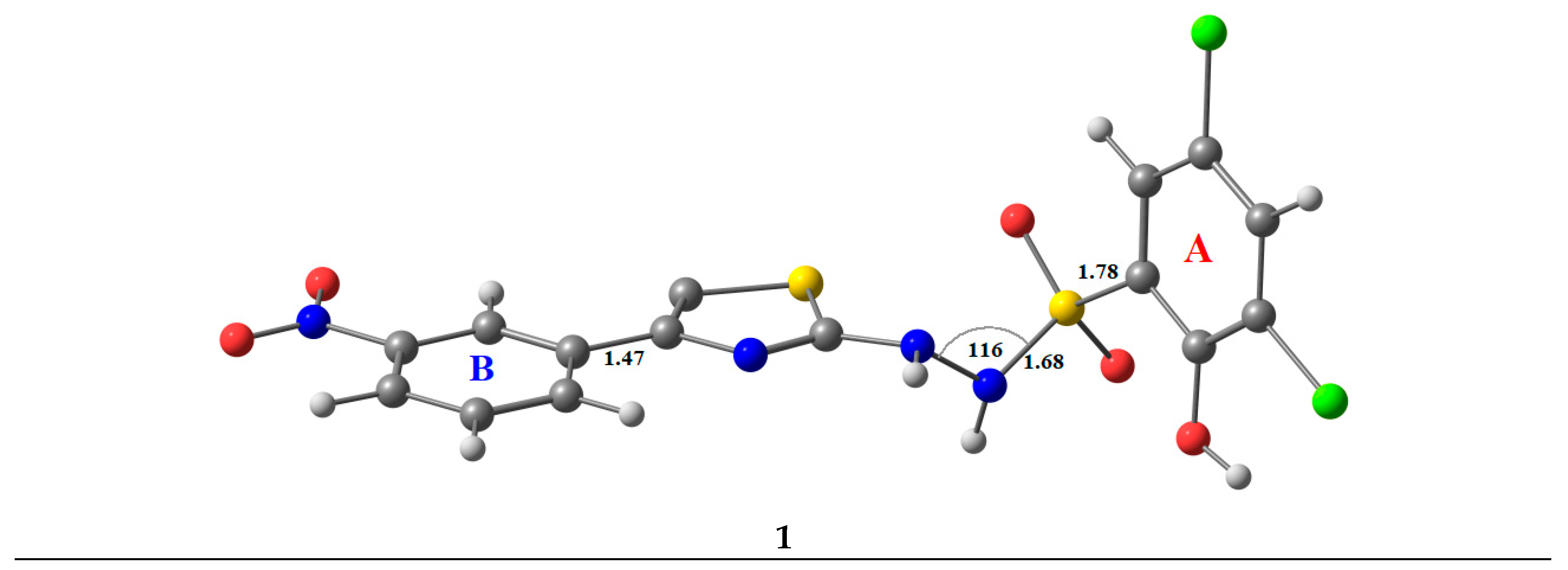
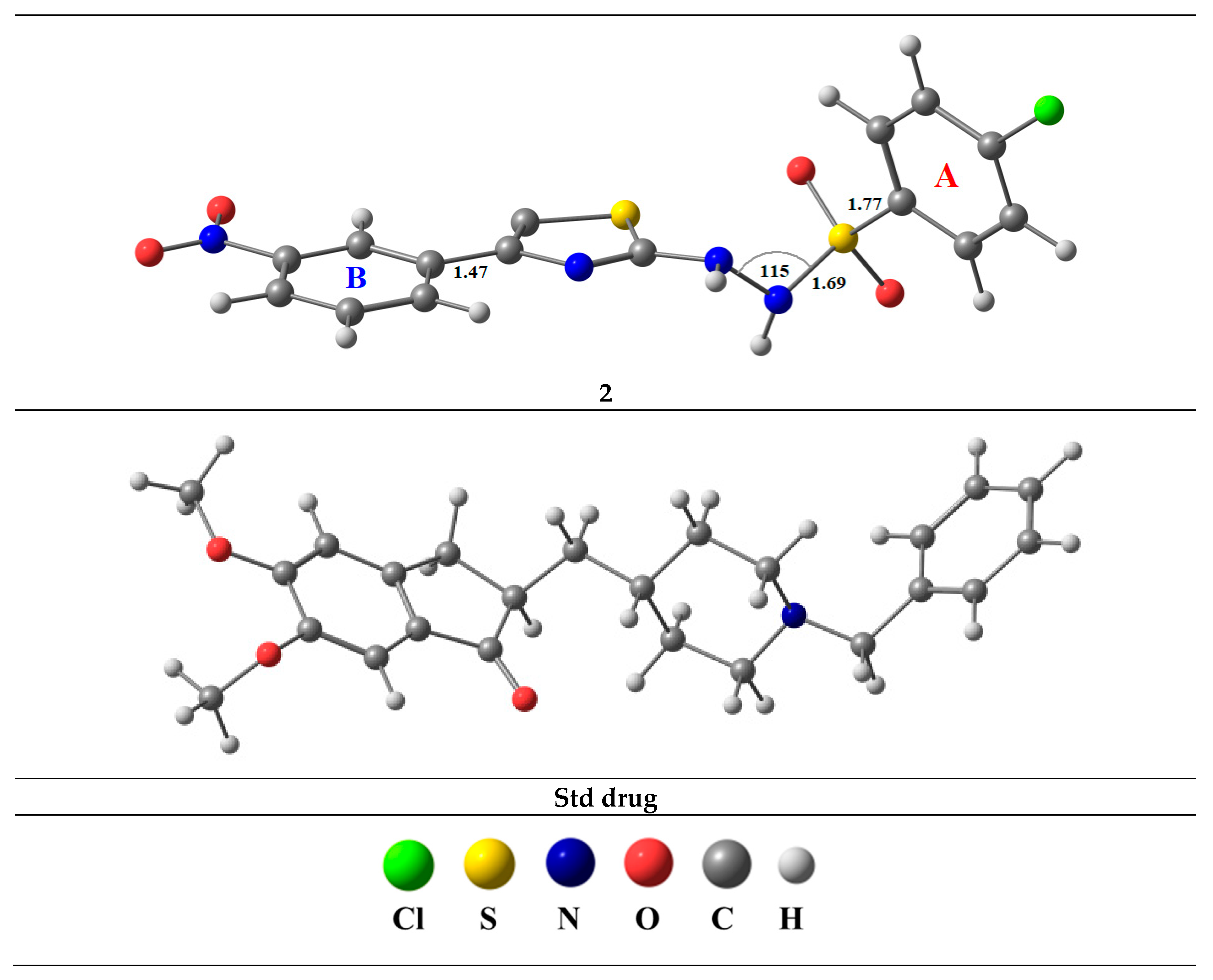
2.1. Chemistry
2.2. Biological Activities
2.2.1. In Vitro Acetylcholinesterase (AchE) Inhibitory Activity
2.2.2. In Vitro Butyrylcholinesterase (BchE) Inhibitory Activity
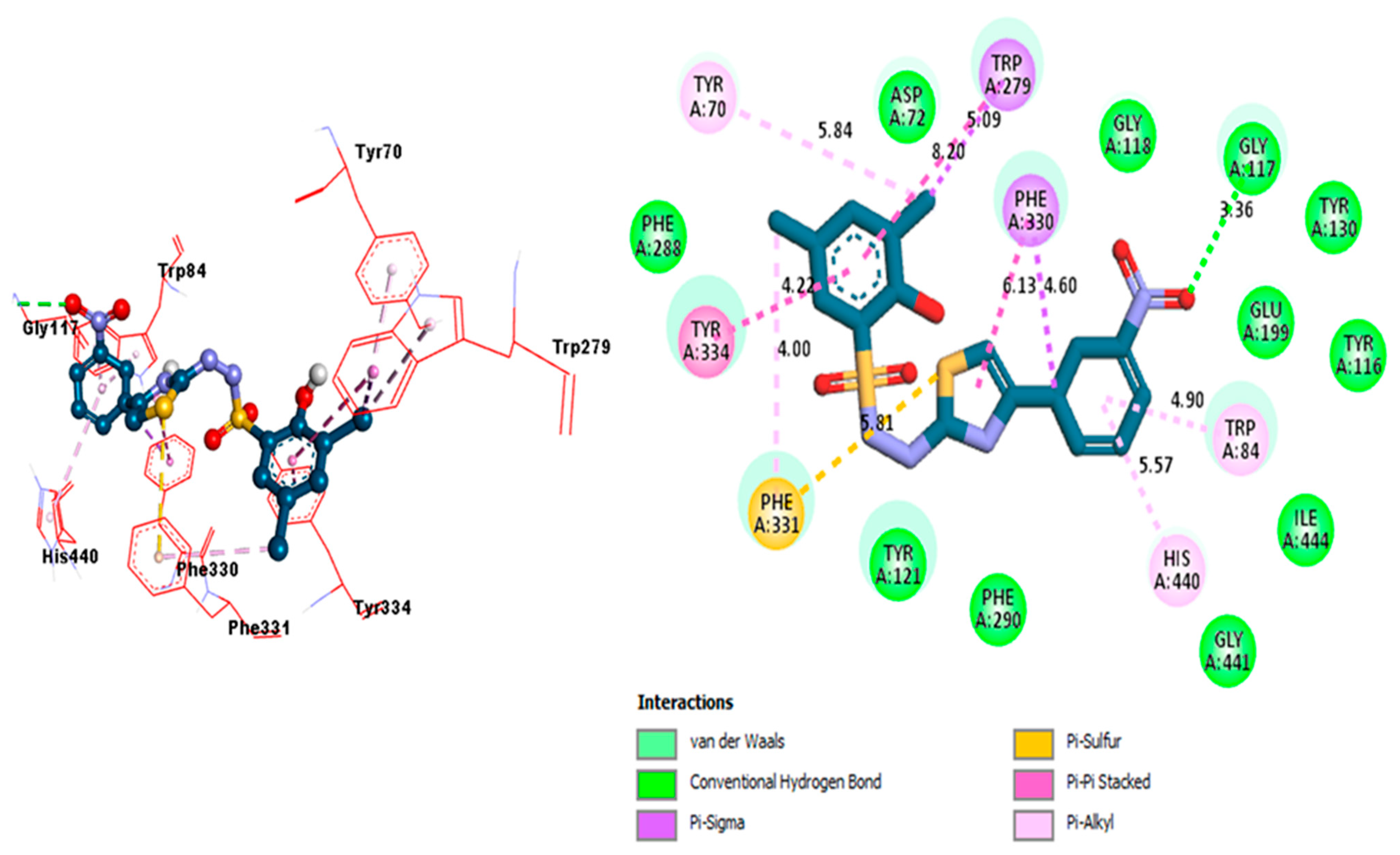
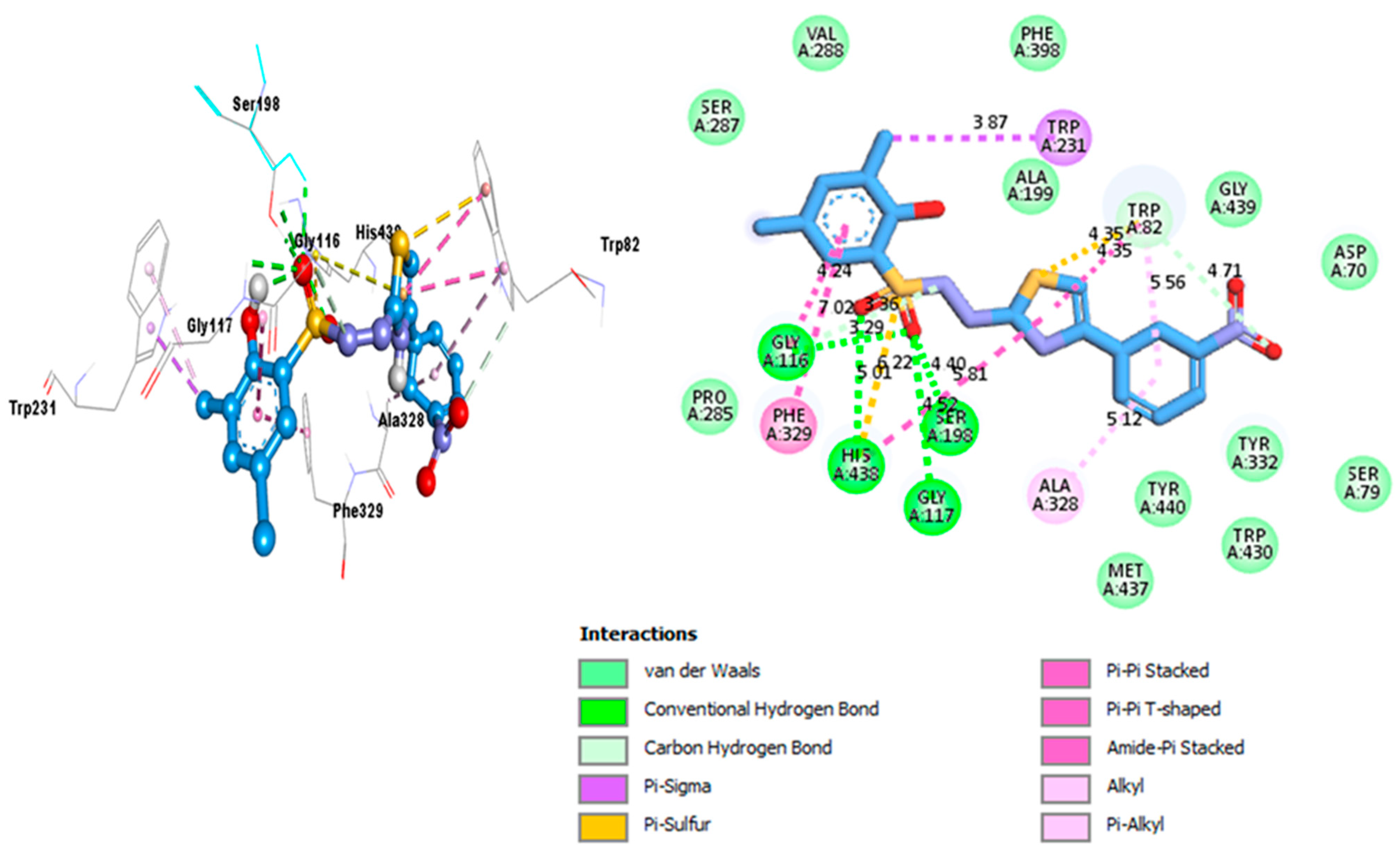
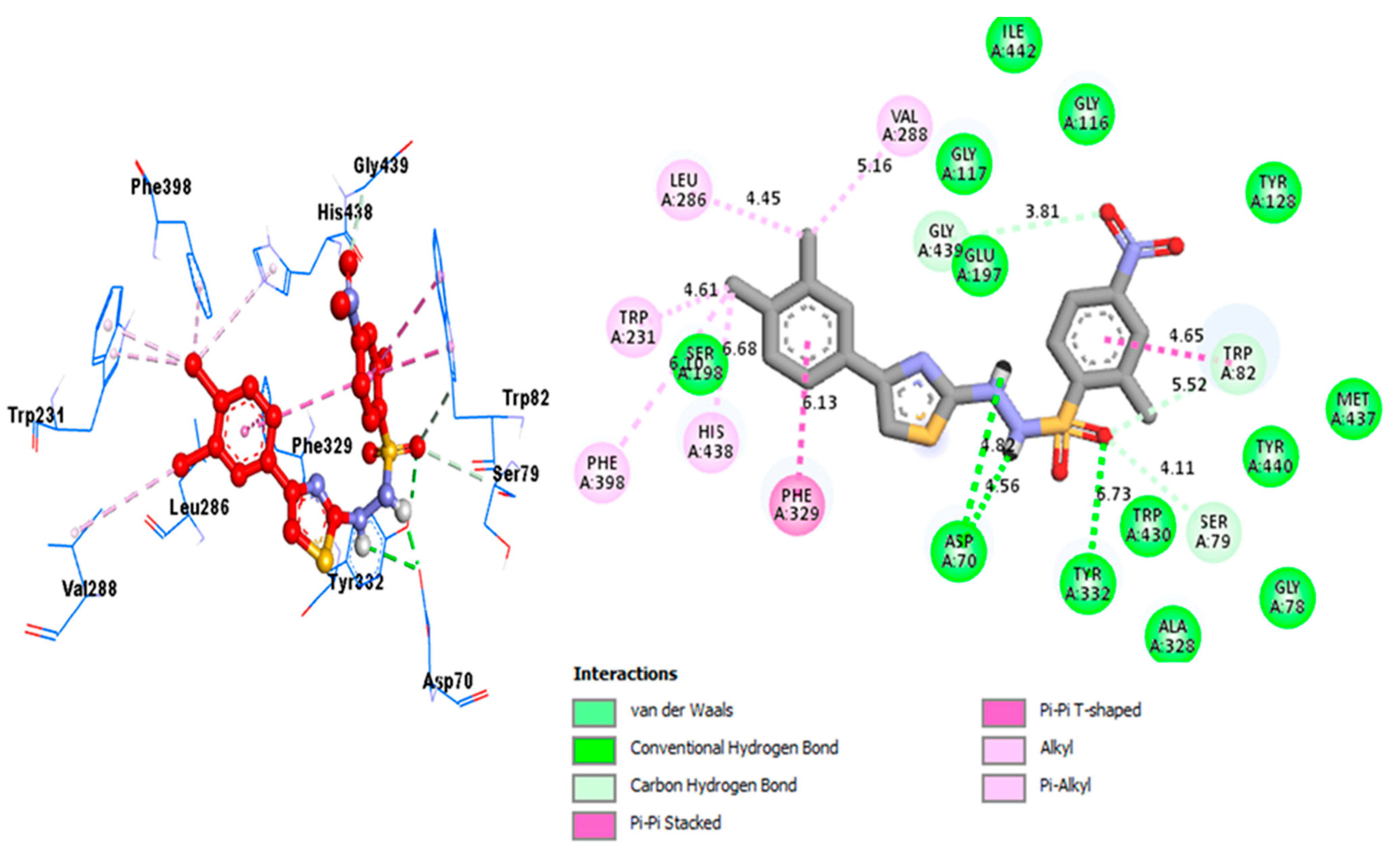
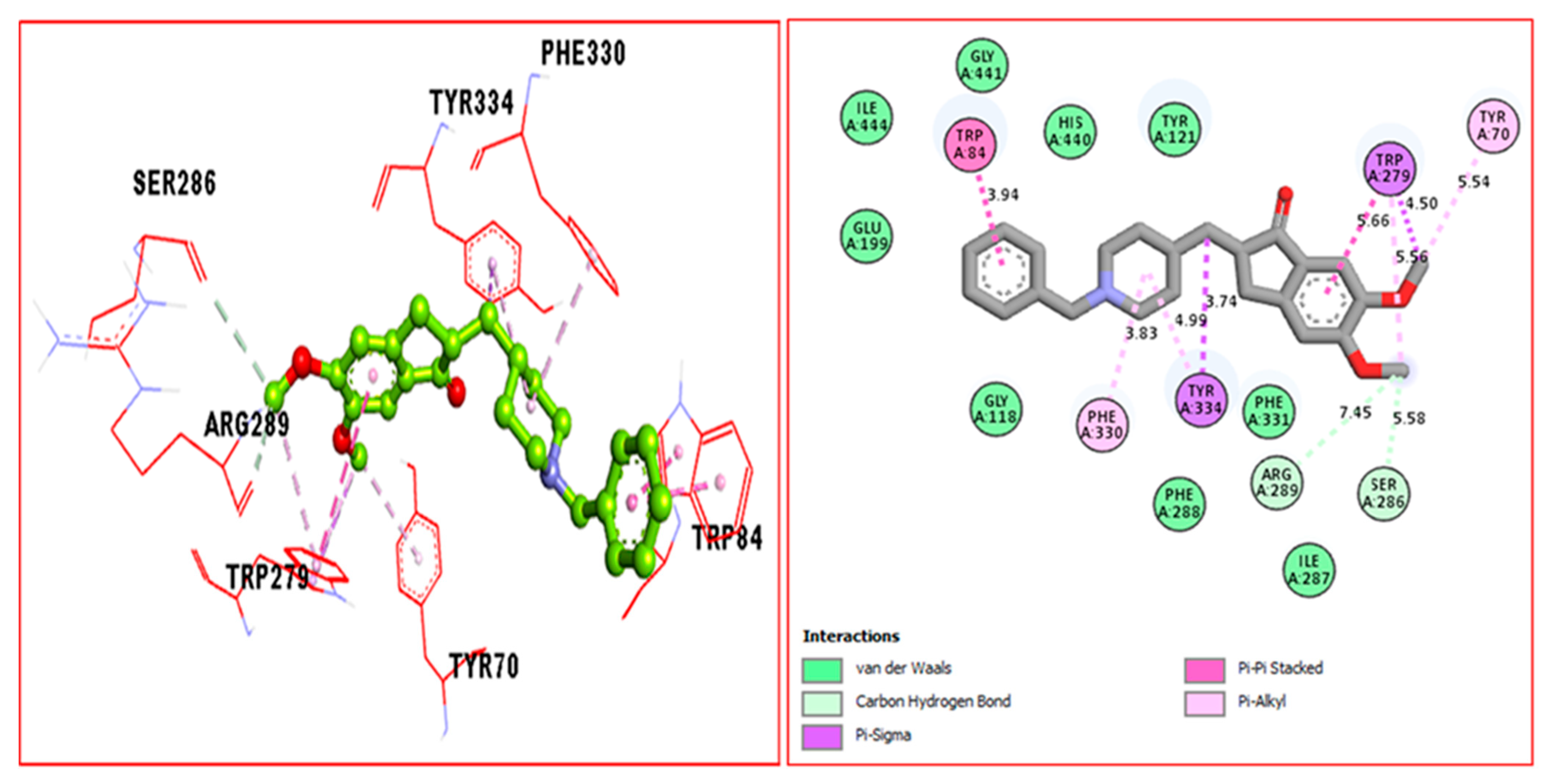
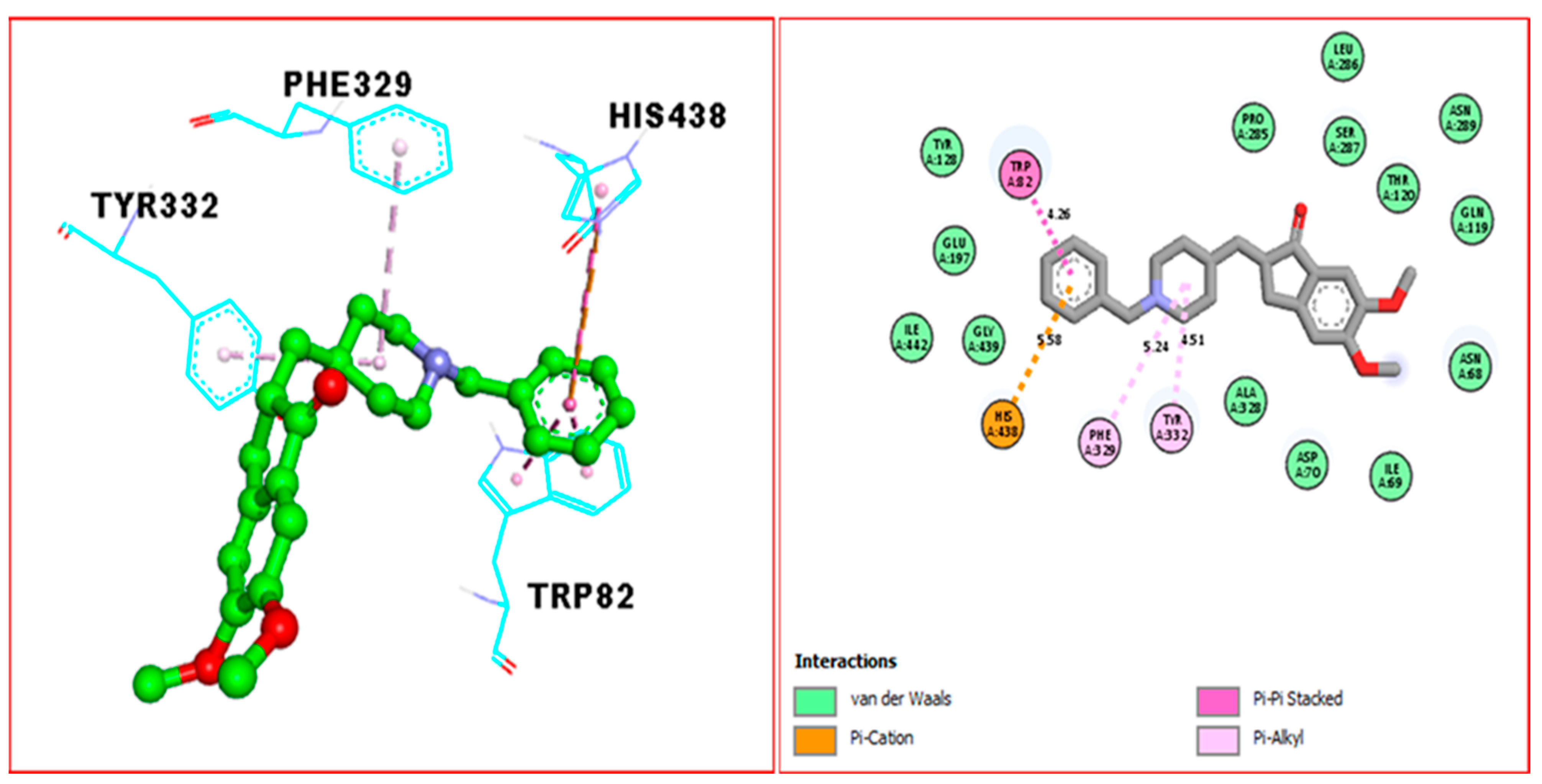
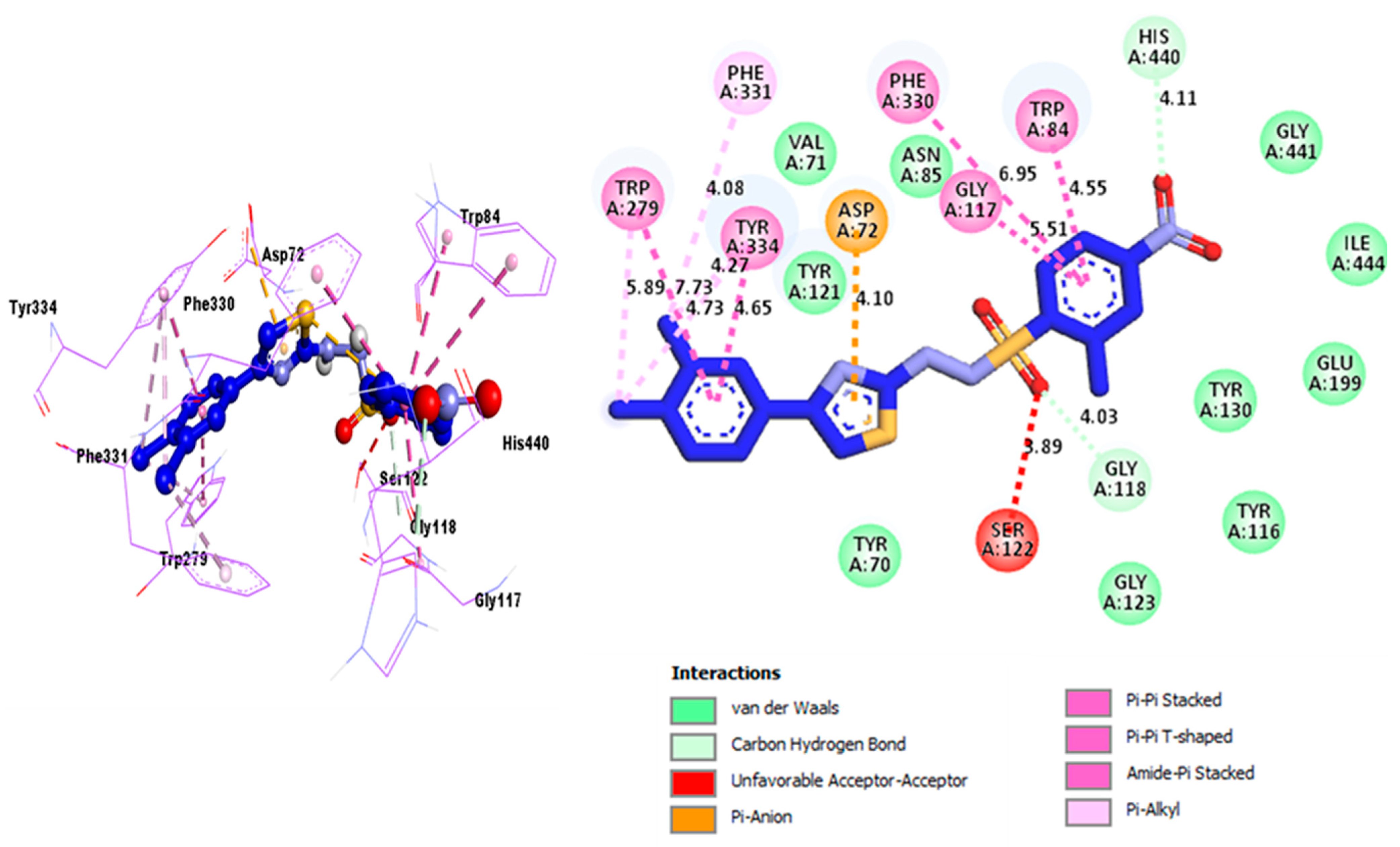
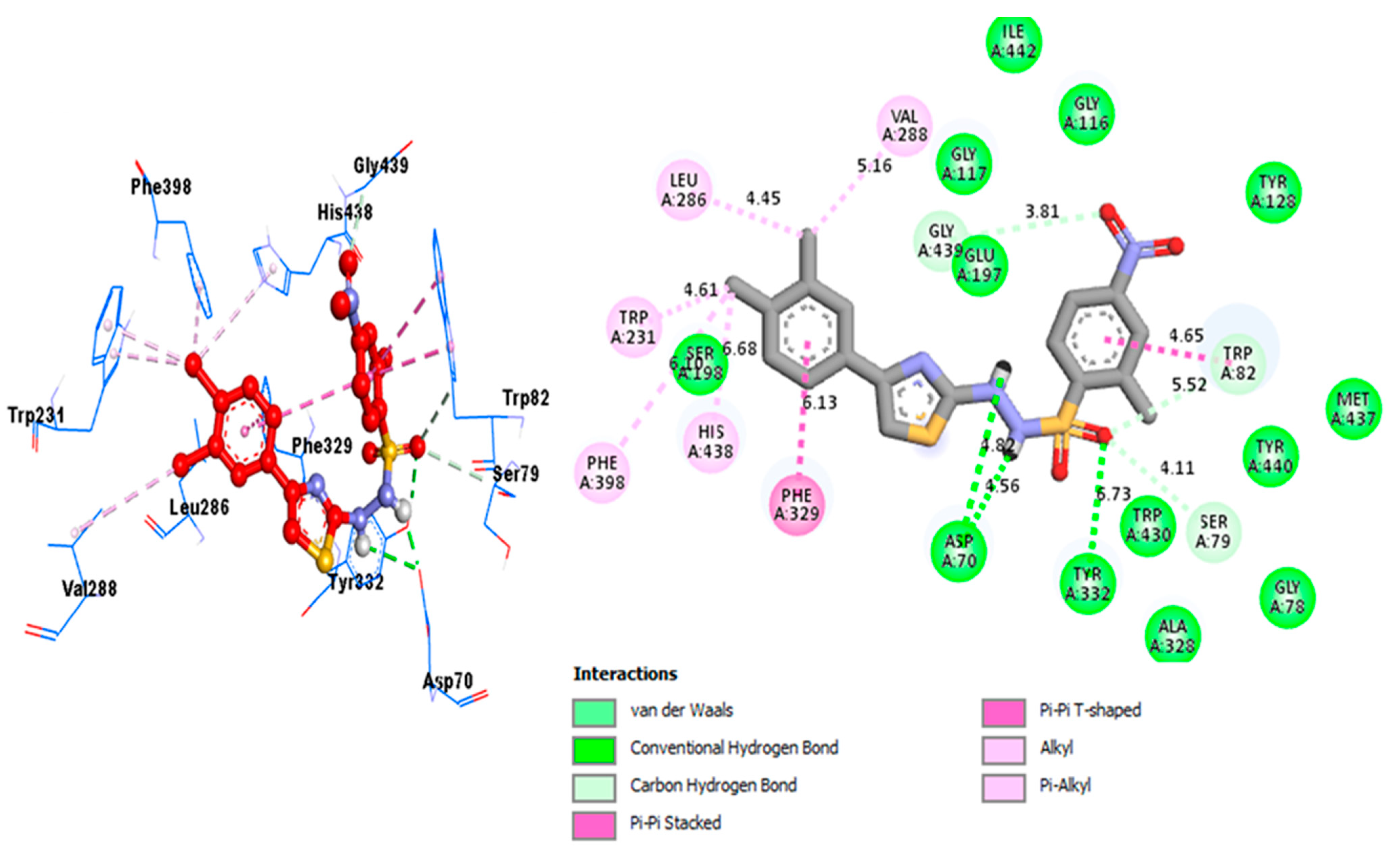
2.3. Docking Study
2.4. Computational Details
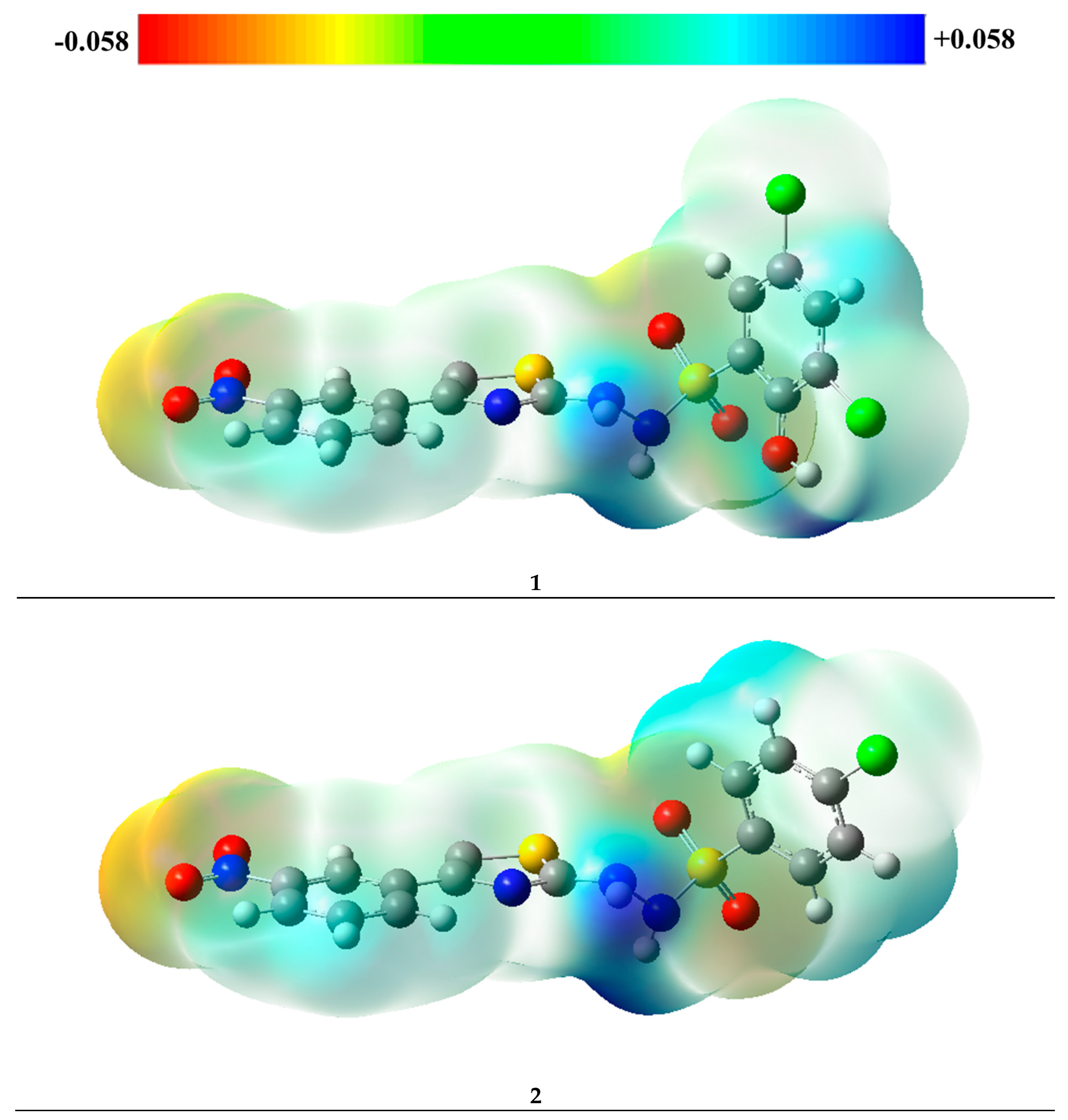
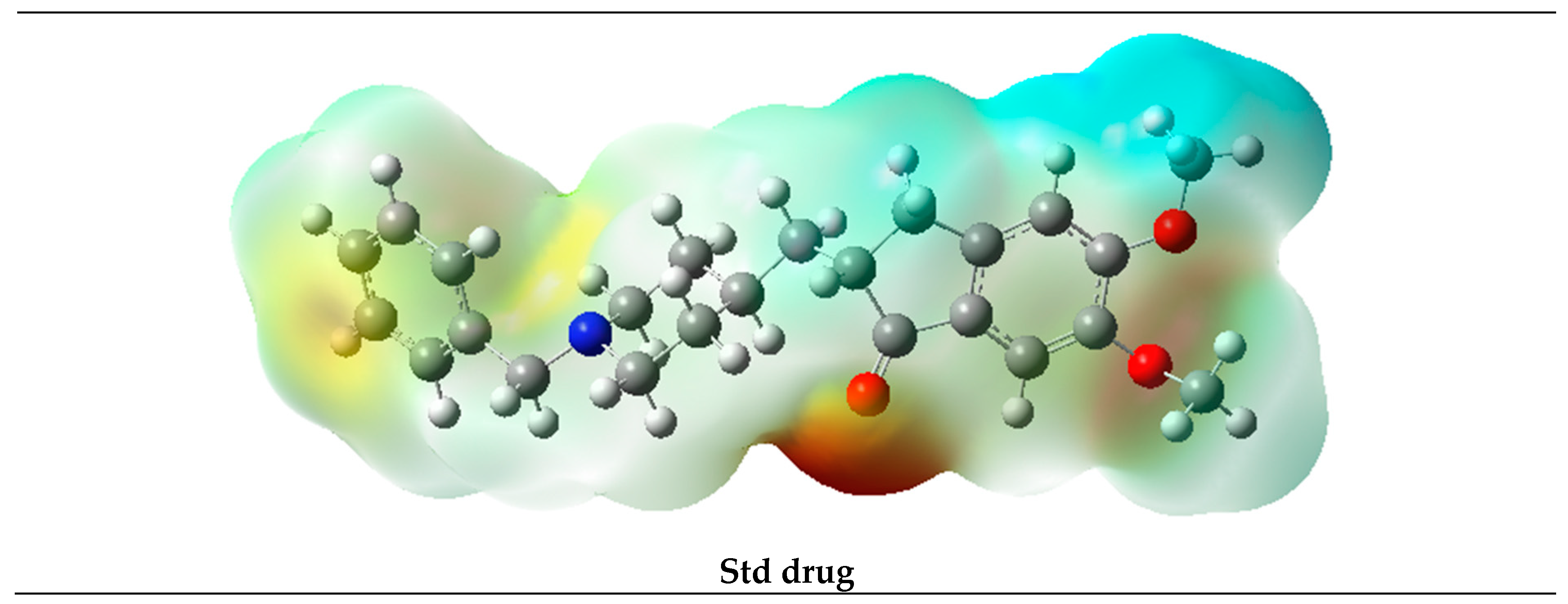
2.5. Molecular Electrostatic Potential
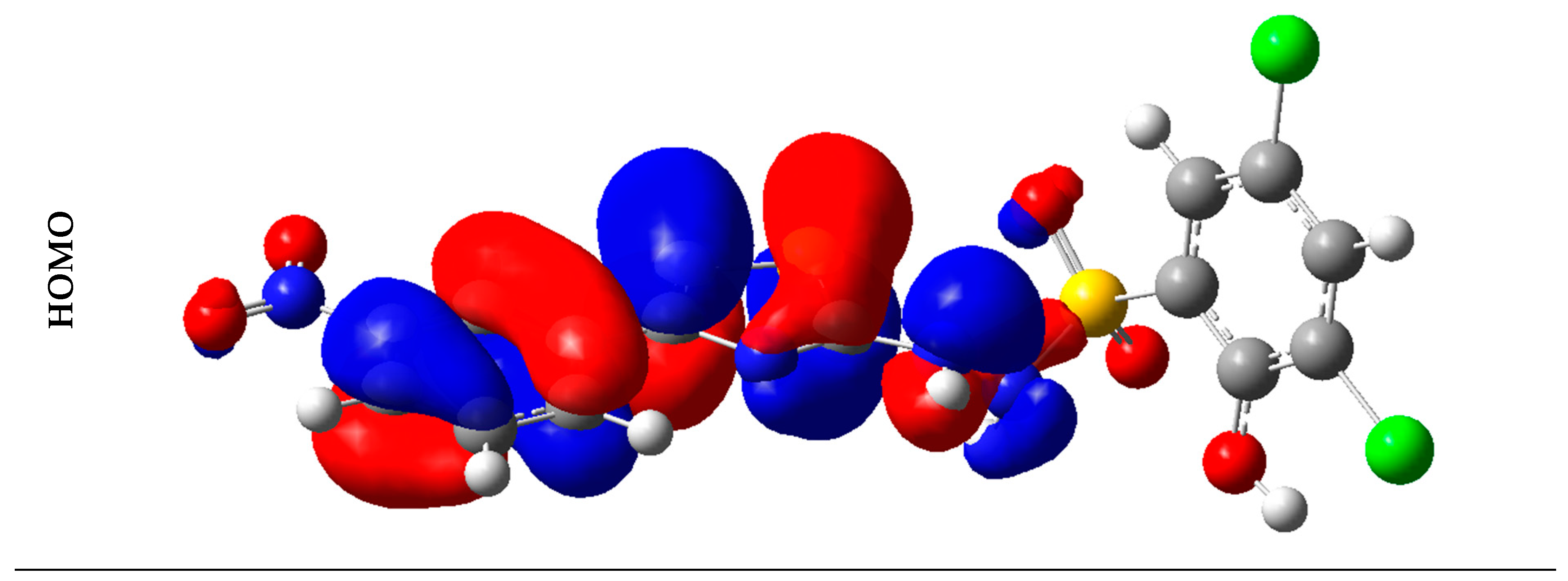
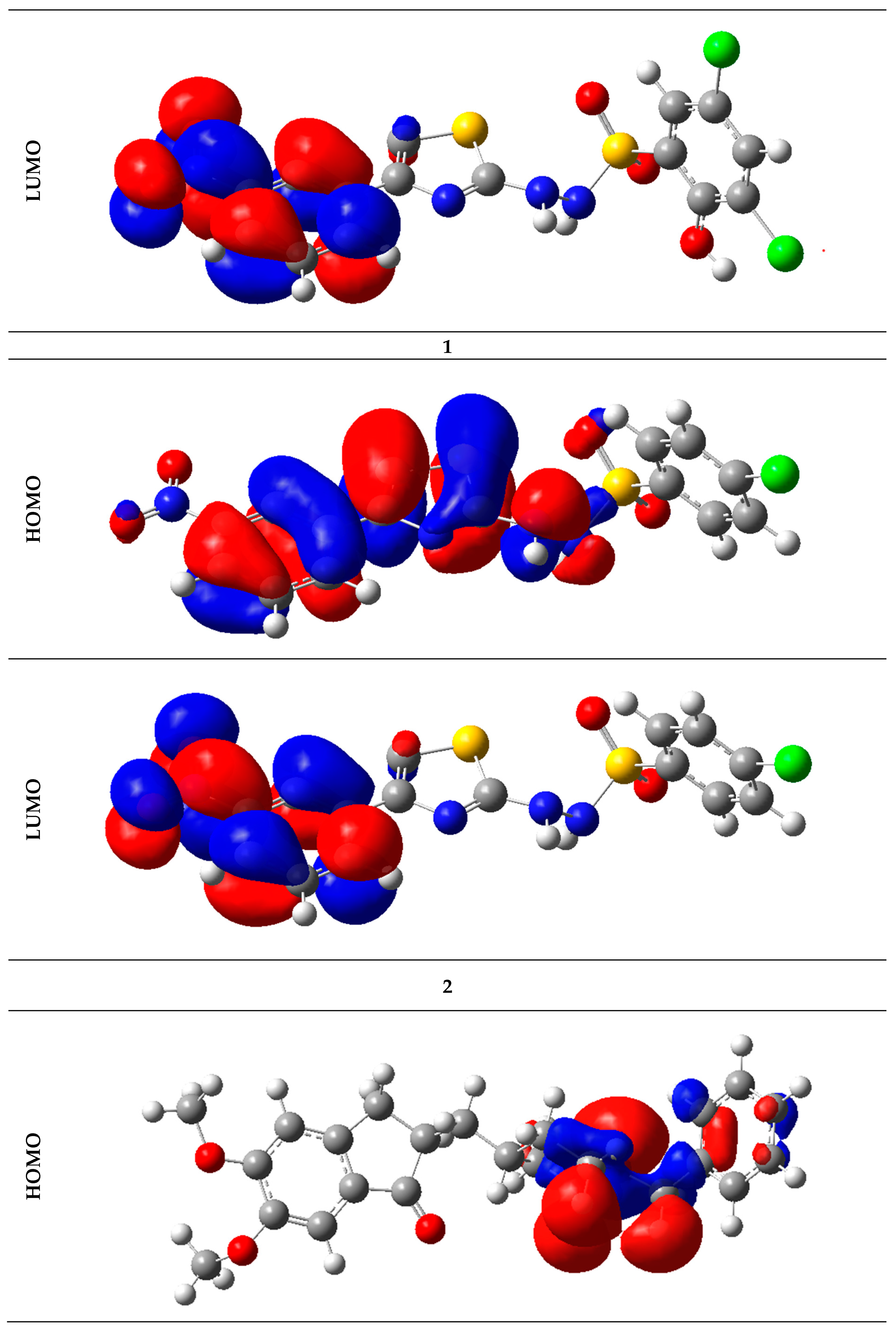
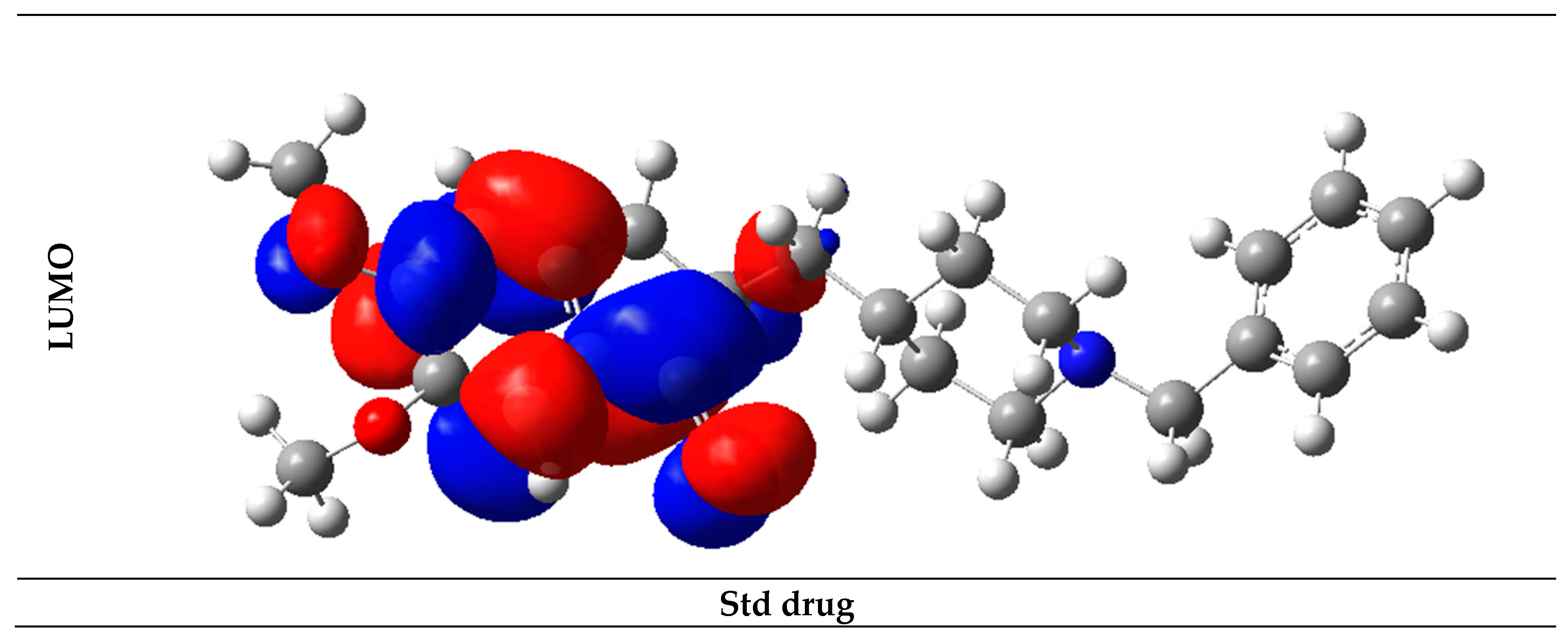
2.6. Frontier Molecular Orbital Analysis
3. Conclusions
4. Experimental
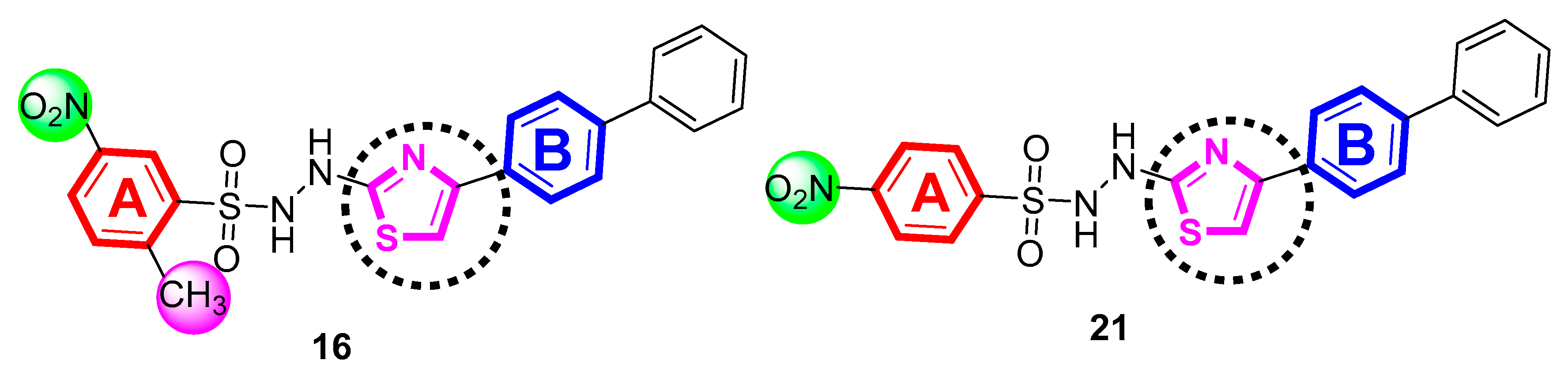
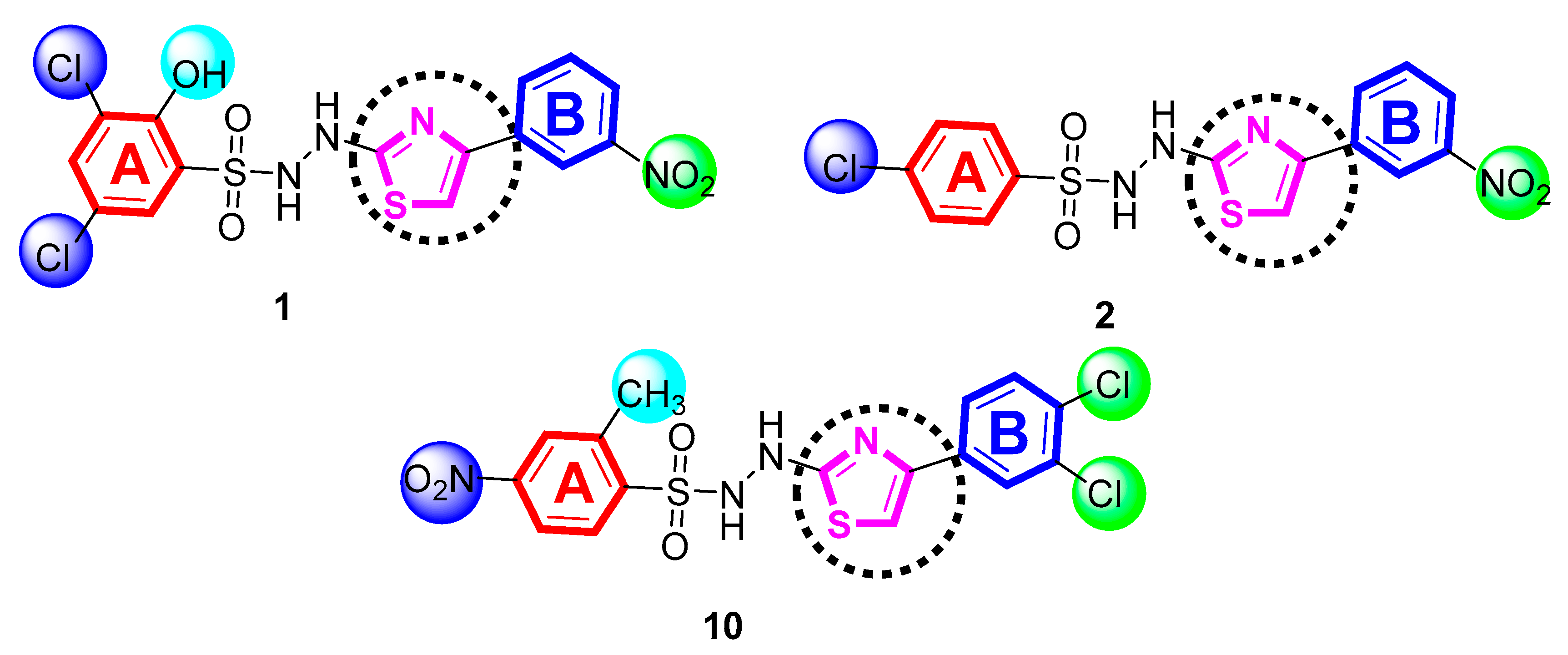
4.1. General Procedure for the Synthesis of Thiazole-Bearing Sulfonamide Analogs (1–21)
4.2. Spectral Analysis
4.2.1. 3,5-Dichlro-2-hydroxy-N′-(4-(3-nitrophenyl)thiazol-2-yl)benzenesulfonohydrazide (1)
4.2.2. 4-Chloro-N′-(4-(3-nitrophenyl)thiazol-2-yl)benzenesulfonohydrazide (2)
4.2.3. 3-Nitro-N′-(4-(3-nitrophenyl)thiazol-2-yl)benzenesulfonohydrazide (3)
4.2.4. 2-Methyl-5-nitro-N′-(4-(3-nitrophenyl)thiazol-2-yl)benzenesulfonohydrazide (4)
4.2.5. 4-Bromo-N′-(4-phenylthiazol-2-yl)benzenesulfonohydrazide (5)
4.2.6. 4-Nitro-N′-(4-phenylthiazol-2-yl)benzenesulfonohydrazide (6)
4.2.7. 4-Chloro-N′-(4-(4-chlorophenyl)thiazol-2-yl)benzenesulfonohydrazide (7)
4.2.8. N′-(4-([1,1′-biphenyl]-4-yl)thiazol-2-yl)-4-chlorobenzenesulfonohydrazide (8)
4.2.9. 3,5-Dichloro-N′-(4-(4-chlorophenyl)thiazol-2-yl)-2-hydroxybenzenesulfonohydrazide (9)
4.2.10. N′-(4-(3,4-dichlorophenyl)thiazol-2-yl)-2-methyl-5-nitrobenzenesulfonohydrazide (10)
4.2.11. N′-(4-(4-chlorophenyl)thiazol-2-yl)-2-methyl-5-nitrobenzenesulfonohydrazide (11)
4.2.12. N′-(4-(4-bromophenyl)thiazol-2-yl)-2-methyl-5-nitrobenzenesulfonohydrazide (12)
4.2.13. N′-(4-(3,4-dichlorophenyl)thiazol-2-yl)-3-nitrobenzenesulfonohydrazide (13)
4.2.14. N′-(4-(4-chlorophenyl)thiazol-2-yl)-3-nitrobenzenesulfonohydrazide (14)
4.2.15. 4-Chloro-N′-(4-(3,4-dichlorophenyl)thiazol-2-yl)benzenesulfonohydrazide (15)
4.2.16. N′-(4-([1,1′-biphenyl]-4-yl)thiazol-2-yl)-2-methyl-5-nitrobenzenesulfonohydrazide (16)
4.2.17. N′-(4-([1,1′-biphenyl]-4-yl)thiazol-2-yl)-4-chlorobenzenesulfonohydrazide (17)
4.2.18. 2,5-Dinitro-N′-(4-(3-nitrophenyl)thiazol-2-yl)benzenesulfonohydrazide (18)
4.2.19. N′-(4-([1,1′-biphenyl]-4-yl)thiazol-2-yl)-2,5-dinitrobenzenesulfonohydrazide (19)
4.2.20. 4-Nitro-N′-(4-(3-nitrophenyl)thiazol-2-yl)benzenesulfonohydrazide (20)
4.2.21. N′-(4-([1,1′-biphenyl]-4-yl)thiazol-2-yl)-4-nitrobenzenesulfonohydrazide (21)
4.3. Molecular Docking Study Assay
4.4. Acetylcholinesterase and Butyrylcholinesterase Activity Assay Protocol
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahmad, S.; Iftikhar, F.; Ullah, F.; Sadiq, A.; Rashid, U. Rational design and synthesis of dihydropyrimidine based dual binding site acetylcholinesterase inhibitors. Bioorg. Chem. 2016, 69, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Auld, D.S.; Kornecook, T.J.; Bastianetto, S.; Quirion, R. Alzheimer’s disease and the basal forebrain cholinergic system: Relations to β-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 2002, 68, 209–245. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.L.; Craig, P.L.; Parsons, O.A. Neuropsychology of dementia. Neurol. Clin. 1986, 4, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Davis, K.L. The search for disease-modifying treatment for Alzheimer’s disease. Neurology 1997, 48, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Jann, M.W. Preclinical pharmacology of metrifonate. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1998, 18, 55–67. [Google Scholar]
- Massoulié, J.; Pezzementi, L.; Bon, S.; Krejci, E.; Vallette, F. Molecular and cellular biology of cholinesterases. Prog. Neurobiol. 1993, 41, 31–91. [Google Scholar] [CrossRef]
- Mushtaq, G.; Greig, N.H.; Khan, J.; Kamal, M.A. Status of Acetylcholinesterase and Butyrylcholinesterase in Alzheimer’s Disease and Type 2 Diabetes Mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1432–1439. [Google Scholar] [CrossRef]
- Ecobichon, D.J.; Comeau, A.M. Pseudocholinesterases of mammalian plasma: Physicochemical properties and organophosphate inhibition in eleven species. Toxicol. Appl. Pharmacol. 1973, 24, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Rahim, F.; Javed, M.T.; Ullah, H.; Wadood, A.; Taha, M.; Ashraf, M.; Ain, Q.U.; Khan, M.A.; Khan, F.; Mirza, S.; et al. Synthesis, Molecular Docking, Acetylcholinesterase and Butyrylcholinesterase Inhibitory Potential of Thiazole Analogs as New Inhibitors for Alzheimer Disease. Bioorg. Chem. 2015, 62, 106–116. [Google Scholar] [CrossRef]
- Rahim, F.; Ullah, H.; Taha, M.; Wadood, A.; Javed, M.T.; Rehman, W.; Nawaz, M.; Ashraf, M.; Ali, M.; Sajid, M.; et al. Synthesis and in vitro acetylcholinesterase and butyrylcholinesterase inhibitory potential of hydrazide based Schiff bases. Bioorg. Chem. 2016, 68, 30–40. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, K.; Mintzer, J.; Truyen, L.; Wessel, T.; Wilkinson, D. Effects of a flexible galantamine dose in Alzheimer’s disease: A randomised, controlled trial. J. Neurol. Neurosurg. Psychiatry 2001, 71, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesulam, M.; Guillozet, A.; Shaw, P.; Quinn, B. Widely spread butyrylcholinesterase can hydrolyze acetylcholine in the normal and Alzheimer brain. Neurobiol. Dis. 2002, 9, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greig, N.H.; Utsuki, T.; Yu, Q.S.; Zhu, X.; Holloway, H.W.; Perry, T.; Lee, B.; Ingram, D.K.; Lahiri, D.K. A new therapeutic target in Alzheimer’s disease treatment: Attention to butyrylcholinesterase. Curr. Med. Res. Opin. 2001, 17, 159–165. [Google Scholar] [CrossRef]
- Gabr, M.T.; Abdel-Raziq, M.S. Design and synthesis of donepezil analogues as dual AChE and BACE-1 inhibitors. Bioorg. Chem. 2018, 80, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Melzer, D. New drug treatment for Alzheimer’s disease: Lessons for healthcare policy. Brit. Med. J. 1998, 316, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.; Arshad, M.F.; Ahsan, W.; Alam, M.S. Thiazoles: A valuable insight into the recent advances and biological activities. Int. J. Pharm. Sci. Drug Res. 2009, 1, 136–143. [Google Scholar]
- Mishra, C.B.; Kumari, S.; Tiwari, M. Thiazole: A promising heterocycle for the development of potent CNS active agents. Eur. J. Med. Chem. 2015, 92, 1–34. [Google Scholar] [CrossRef]
- Pasqualotto, A.C.; Thiele, K.O.; Goldani, L.Z. Novel triazole antifungal drugs: Focus on isavuconazole, ravuconazole and albaconazole. Curr. Opin. Investig. Drugs 2010, 11, 165–174. [Google Scholar]
- Das, J.; Chen, P.; Norris, D.; Padmanabha, R.; Lin, J.; Moquin, R.V.; Behnia, K.; Schieven, G.L.; Wityak, J.; Barrish, J.C. 2-Aminothiazole as a Novel Kinase Inhibitor Template. Structure− Activity Relationship Studies toward the Discovery of N-(2-Chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl)]-2-methyl-4-pyrimidinyl] amino)]-1, 3-thiazole-5-carboxamide (Dasatinib, BMS-354825) as a Potent pan-Src Kinase Inhibitor. J. Med. Chem. 2006, 49, 6819–6832. [Google Scholar]
- Siddiqui, H.L.; Zia-Ur-Rehman, M.; Ahmad, N.; Weaver, G.W.; Lucas, P.D. Synthesis and Antibacterial Activity of Bis [2-amino-4-phenyl-5-thiazolyl] Disulfides. Chem. Pharm. Bull. 2007, 55, 1014–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knadler, M.P.; Bergstrom, R.F.; Callaghan, L.T.; Rubin, A.L.A.N. Nizatidine, an H2-blocker. Its metabolism and disposition in man. Drug Metab. Dispos. 1986, 14, 175–182. [Google Scholar] [PubMed]
- Riaz, S.; Khan, I.U.; Bajda, M.; Ashraf, M.; Shaukat, A.; Rehman, T.U.; Mutahir, S.; Hussain, S.; Mustafa, G.; Yar, M. Pyridine sul-fonamide as a small key organic molecule for the potential treatment of type-II diabetes mellitus and alzheimer’s disease in vitro studies against yeast α-glucosidase, acetylcholinesterase and butyrylcholinesterase. Bioorg. Chem. 2015, 63, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Zajdel, P.; Partyka, A.; Marciniec, K.; Bojarski, A.J.; Pawlowski, M.; Wesolowska, A. Quinoline- and isoquinoline-sulfonamide analogs of aripiprazole: Novel antipsychotic agents? Futur. Med. Chem. 2014, 6, 57–75. [Google Scholar] [CrossRef]
- Mutahir, S.; Jończyk, J.; Bajda, M.; Khan, I.U.; Khan, M.A.; Ullah, N.; Ashraf, M.; Ain, Q.U.; Riaz, S.; Hussain, S.; et al. Novel biphenyl bis -sulfonamides as acetyl and butyrylcholinesterase inhibitors: Synthesis, biological evaluation and molecular modeling studies. Bioorg. Chem. 2016, 64, 13–20. [Google Scholar] [CrossRef]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau-Post-Translational Modifications in Wild-Type and Human Amyloid Precursor Protein Transgenic Mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef]
- Gul, H.I.; Yamali, C.; Sakagami, H.; Angeli, A.; Leitans, J.; Kazaks, A.; Tars, K.; Ozgun, D.O.; Supuran, C.T. New anticancer drug candidates sulfonamides as selective hCA IX or hCA XII inhibitors. Bioorg. Chem. 2018, 77, 411–419. [Google Scholar] [CrossRef]
- Ullah, H.; Uddin, I.; Rahim, F.; Khan, F.; Sobia; Taha, M.; Khan, M.U.; Hayat, S.; Ullah, M.; Gul, Z.; et al. In vitro α-glucosidase and α-amylase inhibitory potential and molecular docking studies of benzohydrazide based imines and thiazolidine-4-one derivatives. J. Mol. Struct. 2021, 1251, 132058. [Google Scholar] [CrossRef]
- Ullah, H.; Ahmad, S.; Khan, F.; Taha, M.; Rahim, F.; Sarfraz, M.; Aziz, A.; Wadood, A. Synthesis, in-vitro and in-silico studies of triazinoindole bearing bis-Schiff base as β-glucuronidase inhibitors. J. Mol. Struct. 2021, 1244, 131003. [Google Scholar] [CrossRef]
- Uddin, I.; Ullah, H.; Bibi, A.; Taha, M.; Khan, F.; Rahim, F.; Wadood, A.; Ahmad, N.; Khan, A.A.; Ahmad, F.; et al. Synthesis, in vitro alpha glucosidase, urease activities and molecular docking study of bis-indole bearing Schiff base analogs. Chem. Data Collect. 2020, 28, 100396. [Google Scholar] [CrossRef]
- Taha, M.; Imran, S.; Ismail, N.H.; Selvaraj, M.; Rahim, F.; Chigurupati, S.; Ullah, H.; Khan, F.; Salar, U.; Javid, M.T.; et al. Biology-oriented drug synthesis (BIODS) of 2-(2-methyl-5-nitro-1Himidazol-1-yl)ethyl aryl ether derivatives, in vitro α-amylase inhibitory activity and in silico studies. Bioorg. Chem. 2017, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Rahim, F.; Ullah, H.; Wadood, A.; Farooq, R.K.; Shah, S.A.A.; Nawaz, M.; Zakaria, Z.A. Synthesis, in vitro urease inhibitory potential and molecular docking study of benzofuran-based-thiazoldinone analogues. Sci. Rep. 2020, 10, 10673. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Ismail, N.H.; Imran, S.; Rahim, F.; Wadood, A.; Khan, H.; Ullah, H.; Salar, U.; Khan, K.M. Synthesis, β-Glucuronidase Inhibition and Molecular Docking Studies of Hybrid Bisindole-Thiosemicarbazides Analogs. Bioorg. Chem. 2016, 68, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Javid, M.T.; Imran, S.; Selvaraj, M.; Ullah, H.; Rahim, F.; Khan, F.; Mohammad, J.I.; Khan, K.M. Synthesis and study of the a-amylase inhibitory potential of thiadiazole quinoline derivatives. Bioorg. Chem. 2017, 74, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Ullah, H.; Ullah, H.; Taha, M.; Khan, F.; Rahim, F.; Uddin, I.; Sarfraz, M.; Shah, S.A.A.; Aziz, A.; Mubeen, S. Synthesis, in vitro α-amylase activity and molecular docking study of new benzimidazole analogs. Russ. J. Org. Chem. 2021, 57, 968–975. [Google Scholar] [CrossRef]
- Taha, M.; Rahim, F.; Imran, S.; Ismail, N.H.; Ullah, H.; Selvaraj, M.; Javid, M.T.; Salar, U.; Ali, M.; Khan, K.M. Synthesis, α-glucosidase inhibitory activity and in silico study of tris-indole hybrid scaffold with oxadiazole ring: As potential leads for the management of type-II diabetes mellitus. Bioorg. Chem. 2017, 74, 30–40. [Google Scholar] [CrossRef]
- Rahim, F.; Ullah, H.; Wadood, A.; Khan, F.; Javid, M.T.; Taha, M.; Rehman, W.; Rehman, A.U.; Khan, K.M. Isatin based Schiff bases as inhibitors of α-glucosidase: Synthesis, characterization, in vitro evaluation and molecular docking studies. Bioorg. Chem. 2015, 60, 42–48. [Google Scholar] [CrossRef]
- Ahmat, N.; Zawawi, N.K.N.A.; Taha, M.; Wadood, A.; Rahim, F.; Ullah, H. Biscoumarin analogs: Synthesis, α-glucosidase inhibitory potential and molecular docking study. Malays. J. Chem. 2020, 22, 111–119. [Google Scholar]
- Taha, M.; Ullah, H.; Khan, M.N.; Rahim, F.; Ahmat, N.; Ali, M.; Perveen, S. Synthesis of bis-indolylmethanes as new potential inhibitors of β-glucuronidase and their molecular docking studies. Eur. J. Med. Chem. 2018, 143, 1757–1767. [Google Scholar] [CrossRef]
- Rahim, F.; Zaman, K.; Ullah, H.; Taha, M.; Ashraf, M.; Uddin, R.; Uddin, I.; Asghar, H.; Khan, A.A.; Khan, K.M. Synthesis of 4-thiazolidinone Analogs as potent in vitro Anti-Urease Agents. Bioorg. Chem. 2015, 63, 123–131. [Google Scholar] [CrossRef]
- Ullah, H.; Rahim, F.; Taha, M.; Hussain, R.; Wadood, A.; Nawaz, M.; Wahab, Z.; Khan, K.M. Synthesis, in vitro α-glucosidase Inhibitory Potential and Molecular Docking Studies of 2-Amino-1,3,4-Oxadiazole Derivatives. Med. Chem. 2020, 16, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Rahim, F.; Ullah, K.; Ullah, H.; Wadood, A.; Taha, M.; Ashraf, M.; Shaukat, A.; Rehman, W.; Hussain, S.; Khan, K.M. Triazinoindole analogs as potent inhibitors of α-glucosidase: Synthesis, biological evaluation and molecular docking studies. Bioorg. Chem. 2015, 58, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Ghotbi, G.; Mahdavi, M.; Najafi, Z.; Moghadam, F.H.; Hamzeh-Mivehroud, M.; Davaran, S.; Dastmalchi, S. Design, synthesis, biological evaluation, and docking study of novel dual-acting thiazole-pyridiniums inhibiting acetylcholinesterase and β-amyloid aggregation for Alzheimer’s disease. Bioorg. Chem. 2020, 103, 104186. [Google Scholar] [CrossRef] [PubMed]
- Zada, H.; Ullah, H.; Hayat, S.; Rahim, F.; Khan, F.; Wadood, A. Synthesis of triazinoindole bearing sulfonamide derivatives, in vitro α-amylase activity and their molecular docking study. Chem. Data Collect. 2022, 39, 100875. [Google Scholar] [CrossRef]
- Rahim, F.; Ullah, H.; Javid, M.T.; Wadood, A.; Taha, M.; Ashraf, M.; Shaukat, A.; Junaid, M.; Hussain, S.; Rehman, W.; et al. Synthesis, in vitro evaluation and molecular docking studies of thiazole derivatives as new inhibitors of α-glucosidase. Bioorg. Chem. 2015, 62, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Oguz, M.; Kalay, E.; Akocak, S.; Nocentini, A.; Lolak, N.; Boga, M.; Yilmaz, M.; Supuran, C.T.J. Synthesis of calix [4] azacrown substituted sulphonamides with antioxidant, acetylcholinesterase, butyrylcholinesterase, tyrosinase and carbonic anhydrase inhibitory action, Enzyme inhib. Med. Chem. 2020, 35, 1215–1223. [Google Scholar]
- Channar, P.A.; Saeed, A.; Albericio, F.; Larik, F.A.; Abbas, Q.; Hassan, M.; Raza, H.; Seo, S.-Y. Sulfonamide-Linked Ciprofloxacin, Sulfadiazine and Amantadine Derivatives as a Novel Class of Inhibitors of Jack Bean Urease; Synthesis, Kinetic Mechanism and Molecular Docking. Molecules 2017, 22, 1352. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.; Clemente, F. Gaussian 09, Revision A. 01; Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., et al., Eds.; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Khan, A.U.; Muhammad, S.; Khera, R.A.; Shehzad, R.A.; Ayub, K.; Iqbal, J. DFT study of superhalogen (AlF4) doped boron nitride for tuning their nonlinear optical properties. Optik 2021, 231, 166464. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Saeidi, N. DFT calculations on the catalytic oxidation of CO over Si-doped (6,0) boron nitride nanotubes. Struct. Chem. 2015, 27, 595–604. [Google Scholar] [CrossRef]
- Khan, S.; Yar, M.; Kosar, N.; Ayub, K.; Arshad, M.; Zahid, M.N.; Mahmood, T. First-principles study for exploring the adsorption behavior of G-series nerve agents on graphdyine surface. Comput. Theor. Chem. 2020, 1191, 113043. [Google Scholar] [CrossRef]
- Prasad, O.; Sinha, L.; Kumar, N. Theoretical Raman and IR spectra of tegafur and comparison of molecular electrostatic potential surfaces, polarizability and hyerpolarizability of tegafur with 5-fluoro-uracil by density functional theory. J. At. Mol. Sci. 2010, 1, 201–214. [Google Scholar] [CrossRef]
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci. 2020, 21, 3922. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-B.; Gao, W.-W.; Tangadanchu, V.K.R.; Zhou, C.-H.; Geng, R.-X. Novel aminopyrimidinyl benzimidazoles as potentially antimicrobial agents: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2018, 143, 66–84. [Google Scholar] [CrossRef] [PubMed]
- Marinho, E.S.; Marinho, M.M. A DFT study of synthetic drug topiroxostat: MEP, HOMO, LUMO. Int. J. Sci. Eng. Res. 2016, 7, 1264–1270. [Google Scholar]
- Özcan, M.; Dehri, I.; Erbil, M. Organic sulphur-containing compounds as corrosion inhibitors for mild steel in acidic media: Correlation between inhibition efficiency and chemical structure. Appl. Surf. Sci. 2004, 236, 155–164. [Google Scholar] [CrossRef]
- Rehman, A.U.; Zhen, G.; Zhong, B.; Ni, D.; Li, J.; Nasir, A.; Gabr, M.T.; Rafiq, H.; Wadood, A.; Lu, S.; et al. Mechanism of zinc ejection by disulfiram in nonstructural protein 5A. J. Phys. Chem. Chem. Phys. 2021, 23, 12204–12215. [Google Scholar] [CrossRef]
- Riaz, M.; Rehman, A.U.; Shah, S.A.; Rafiq, H.; Lu, S.; Qiu, Y.; Wadood, A. Predicting Multi-Interfacial Binding Mechanisms of NLRP3 and ASC Pyrin Domains in Inflammasome Activation. ACS Chem. Neurosci. 2021, 12, 603–612. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholin-esterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/No | R1 | R2 | AchE IC50 (µM) | BuChE IC50 (µM) |
|---|---|---|---|---|
| 1 | 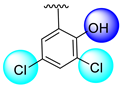 | 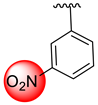 | 0.10 ± 0.05 | 0.20 ± 0.050 |
| 2 |  |  | 1.90 ± 0.10 | 2.70 ± 0.10 |
| 3 | 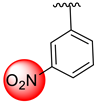 | 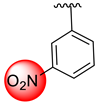 | 2.90 ± 0.10 | 3.80 ± 0.10 |
| 4 |  | 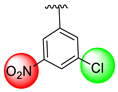 | 3.20 ± 0.10 | 6.10 ± 0.20 |
| 5 |  |  | N.A. | N.A. |
| 6 | 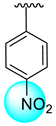 | 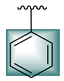 | 11.40 ± 0.20 | 14.30 ± 0.30 |
| 7 | 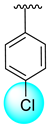 | 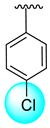 | 2.60 ± 0.10 | 4.20 ± 0.10 |
| 8 | 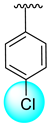 |  | 7.30 ± 0.10 | 9.20 ± 0.10 |
| 9 | 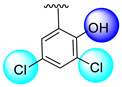 | 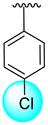 | 0.40 ± 0.050 | 0.70 ± 0.05 |
| 10 | 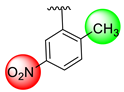 |  | 0.20 ± 0.050 | 0.60 ± 0.050 |
| 11 | 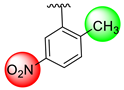 | 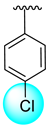 | 3.10 ± 0.10 | 4.40 ± 0.10 |
| 12 | 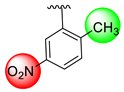 |  | N.A. | N.A. |
| 13 | 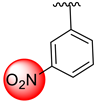 | 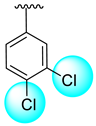 | 0.30 ± 0.10 | 1.40 ± 0.10 |
| 14 | 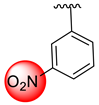 | 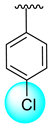 | 1.20 ± 0.10 | 2.70 ± 0.10 |
| 15 |  | 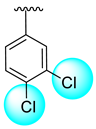 | 0.40 ± 0.10 | 0.90 ± 0.10 |
| 16 | 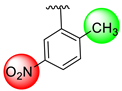 |  | 0.30 ± 0.050 | 0.30 ± 0.050 |
| 17 | 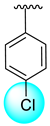 | 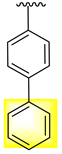 | N.A. | N.A. |
| 18 | 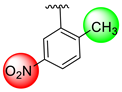 | 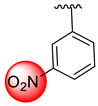 | 0.80 ± 0.050 | 1.20 ± 0.050 |
| 19 |  | 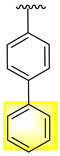 | 3.10 ± 0.10 | 4.30 ± 0.10 |
| 20 | 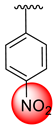 |  | 3.50 ± 0.10 | 5.50 ± 0.10 |
| 21 | 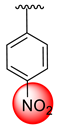 | 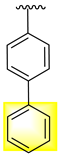 | 3.20 ± 0.10 | 5.40 ± 0.10 |
| Standard drug donepezil | 2.16 ± 0.12 | 4.5 ± 0.11 | ||
| Compound | Receptor | Distance | Interaction | Binding Affinity |
|---|---|---|---|---|
| Compound Analog 1 (A) against AchE | TYR-A-334 | Pi-Pi Stacked | 4.22A° | −10.9 |
| PHE-A-331 | Pi-Sulfur | 5.81A° | ||
| PHE-A-331 | Pi-Alkyl | 4.00A° | ||
| HIS-A-440 | Pi-Alkyl | 5.57A° | ||
| TRP-A-84 | Pi-Alkyl | 4.90A° | ||
| GLY-A-117 | H-B | 3.36A° | ||
| PHE-A-330 | Pi-Sigma | 4.60A° | ||
| PHE-A-330 | Pi-Pi Stacked | 6.13A° | ||
| TRP-A-279 | Pi-Sigma | 5.09A° | ||
| TRP-A-279 | Pi-Pi Stacked | 8.20A° | ||
| TYR-A-70 | Pi-Alkyl | 5.84A° | ||
| Analog 1 (B) against BuChE | GLY-A-116 | Pi-Pi Stacked | 4.24A° | −10.3 |
| GLY-A-116 | H-B | 3.29A° | ||
| PHE-A-329 | Pi-Pi T-Shaped | 7.02A° | ||
| HIS-A-438 | H-B | 5.01A° | ||
| HIS-A-438 | Pi-Sulfur | 3.36A° | ||
| HIS-A-438 | Pi-Pi Stacked | 5.81A° | ||
| GLY-A-117 | H-B | 4.52A° | ||
| ALA-A-328 | Alkyl | 5.12A° | ||
| TRP-A-82 | C-HB | 4.71A° | ||
| TRP-A-82 | Alkyl | 5.56A° | ||
| TRP-A-82 | Pi-Pi T-shaped | 4.35A° | ||
| TRP-A-82 | Pi-Sulfur | 4.35A° | ||
| TRP-A-231 | Pi-Sigma | 3.87A° | ||
| LEU-A-286 | Alkyl | 3.01A° | ||
| Analog 10 (C) against AchE | SER-A-122 | A-A | 3.89A° | −11.8 |
| GLY-A-118 | C-HB | 4.03A° | ||
| HIS-A-440 | C-HB | 4.11A° | ||
| TRP-A-84 Amide | Pi-Stacked | 4.55A° | ||
| PHE-A-330 | Pi-Pi Stacked | 6.95A° | ||
| GLY-A-117 | Pi-Pi T-shaped | 5.51A° | ||
| ASP-A-72 | P-Anion | 4.10A° | ||
| TYR-A-334 | Amide Pi-Stacked | 4.65A° | ||
| TYR-A-334 | Pi-Alkyl | 4.73A° | ||
| TYR-A-334 | Pi-Alkyl | 4.27A° | ||
| PHE-A-331 | Pi-Alkyl | 4.08A° | ||
| TRP-A-279 | Pi-Pi Stacked | 7.73A° | ||
| TRP-A-279 | Pi-Alkyl | 5.89A° | ||
| Analog 10 (D) against BuChE | TRP-A-231 | Pi-Alkyl | 4.61A° | −10.1 |
| PHE-A-398 | Alkyl | 6.19A° | ||
| HIS-A-438 | Pi-Alkyl | 6.68A° | ||
| PHE-A-334 | Pi-Pi T-shaped | 6.13A° | ||
| ASP-A-70 | H-B | 4.82A° | ||
| ASP-A-70 | H-B | 4.56A° | ||
| TYR-A-332 | H-B | 6.73A° | ||
| SER-A-79 | C-HB | 4.11A° | ||
| TRP-A-82 | C-HB | 5.52A° | ||
| TRP-A-82 | Pi-Pi Stacked | 4.65A° | ||
| GLY-A-439 | C-HB | 3.81A° | ||
| VAL-A-288 | Alkyl | 5.16A° | ||
| LEU-A-286 | Pi-Alkyl | 4.45A° | ||
| Donepezil (E) against AchE | TRP-A 84 | 3.94A° | Pi-Pi Stacked | −9.80 |
| TRP-A 279 | 5.66A° | Pi-Sigma | ||
| TYR-A 70 | 5.54A° | Pi-alkyl | ||
| SER-A 286 | 5.5.8A° | C-H bond | ||
| ARG-A289 | 7.45A° | C-H bond | ||
| TYR-A 334 | 3.74A° | Pi-Sigma | ||
| PHE-A 330 | 3.83A° | Pi-alkyl | ||
| Donepezil (F) against BuChE | TRP-A 82 | 4.26A° | Pi-Pi Stacked | −7.30 |
| HIS-A 438 | 5.58A° | Pi-Cation | ||
| PHE-A 329 | 5.24A° | Pi-alkyl | ||
| TYR-A 332 | 4.51A° | Pi-alkyl |
| Analogs | HOMO | LUMO | Egap | Analogs | HOMO | LUMO | Egap |
|---|---|---|---|---|---|---|---|
| 1 | −8.36 | −0.46 | −7.91 | 12 | −8.13 | −0.92 | −7.20 |
| 2 | −8.41 | −0.49 | −7.92 | 13 | −8.30 | −1.06 | −7.24 |
| 3 | −8.53 | −1.10 | −7.43 | 14 | −8.14 | −1.04 | −7.11 |
| 4 | −8.74 | −0.99 | −7.75 | 15 | −8.20 | 0.18 | −8.38 |
| 5 | −7.94 | 0.27 | −8.21 | 16 | −7.81 | −0.85 | −6.97 |
| 6 | −8.07 | −1.17 | −6.90 | 17 | −7.70 | 0.28 | −7.98 |
| 7 | −8.04 | 0.22 | −8.26 | 18 | −8.58 | −0.96 | −7.62 |
| 8 | −7.70 | 0.28 | −7.98 | 19 | −7.74 | −1.51 | −6.23 |
| 9 | −7.83 | 0.05 | −7.88 | 20 | −8.54 | −1.29 | −7.26 |
| 10 | −8.33 | −0.93 | −7.40 | 21 | −7.79 | −1.17 | −6.62 |
| 11 | −8.18 | −0.91 | −7.27 | Std drug | −7.77 | 0.64 | −8.41 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.; Ullah, H.; Taha, M.; Rahim, F.; Sarfraz, M.; Iqbal, R.; Iqbal, N.; Hussain, R.; Ali Shah, S.A.; Ayub, K.; et al. Synthesis, DFT Studies, Molecular Docking and Biological Activity Evaluation of Thiazole-Sulfonamide Derivatives as Potent Alzheimer’s Inhibitors. Molecules 2023, 28, 559. https://doi.org/10.3390/molecules28020559
Khan S, Ullah H, Taha M, Rahim F, Sarfraz M, Iqbal R, Iqbal N, Hussain R, Ali Shah SA, Ayub K, et al. Synthesis, DFT Studies, Molecular Docking and Biological Activity Evaluation of Thiazole-Sulfonamide Derivatives as Potent Alzheimer’s Inhibitors. Molecules. 2023; 28(2):559. https://doi.org/10.3390/molecules28020559
Chicago/Turabian StyleKhan, Shoaib, Hayat Ullah, Muhammad Taha, Fazal Rahim, Maliha Sarfraz, Rashid Iqbal, Naveed Iqbal, Rafaqat Hussain, Syed Adnan Ali Shah, Khurshid Ayub, and et al. 2023. "Synthesis, DFT Studies, Molecular Docking and Biological Activity Evaluation of Thiazole-Sulfonamide Derivatives as Potent Alzheimer’s Inhibitors" Molecules 28, no. 2: 559. https://doi.org/10.3390/molecules28020559
APA StyleKhan, S., Ullah, H., Taha, M., Rahim, F., Sarfraz, M., Iqbal, R., Iqbal, N., Hussain, R., Ali Shah, S. A., Ayub, K., Albalawi, M. A., Abdelaziz, M. A., Alatawi, F. S., & Khan, K. M. (2023). Synthesis, DFT Studies, Molecular Docking and Biological Activity Evaluation of Thiazole-Sulfonamide Derivatives as Potent Alzheimer’s Inhibitors. Molecules, 28(2), 559. https://doi.org/10.3390/molecules28020559