Poisonous Vapor Adsorption on Pure and Modified Aluminum Nitride Nanosheet for Environmental Safety: A DFT Exploration




Abstract
:1. Introduction
2. Methods
3. Results and Discussion
3.1. Interaction of m-DNB Molecule with Pure AlN Nanosheet
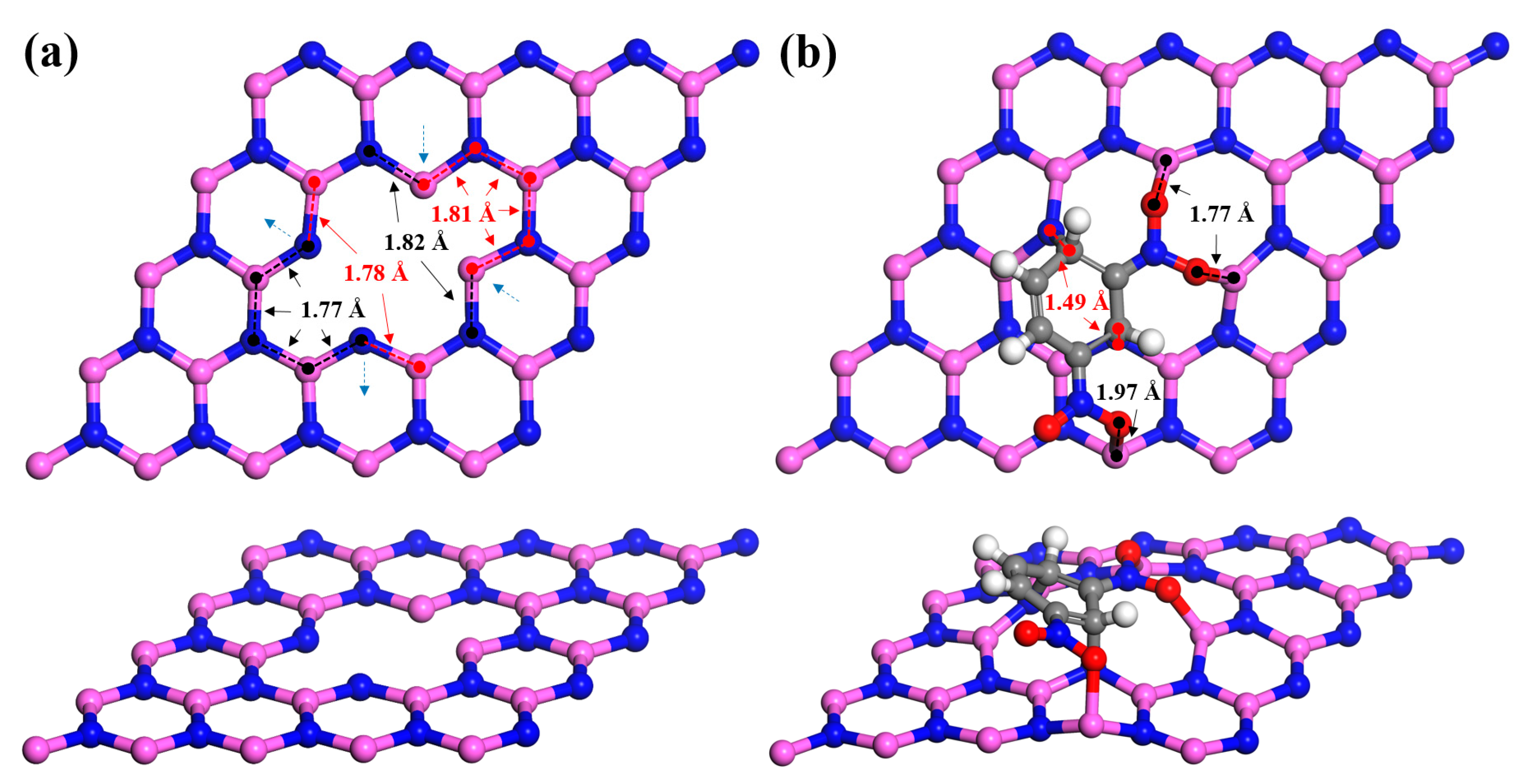
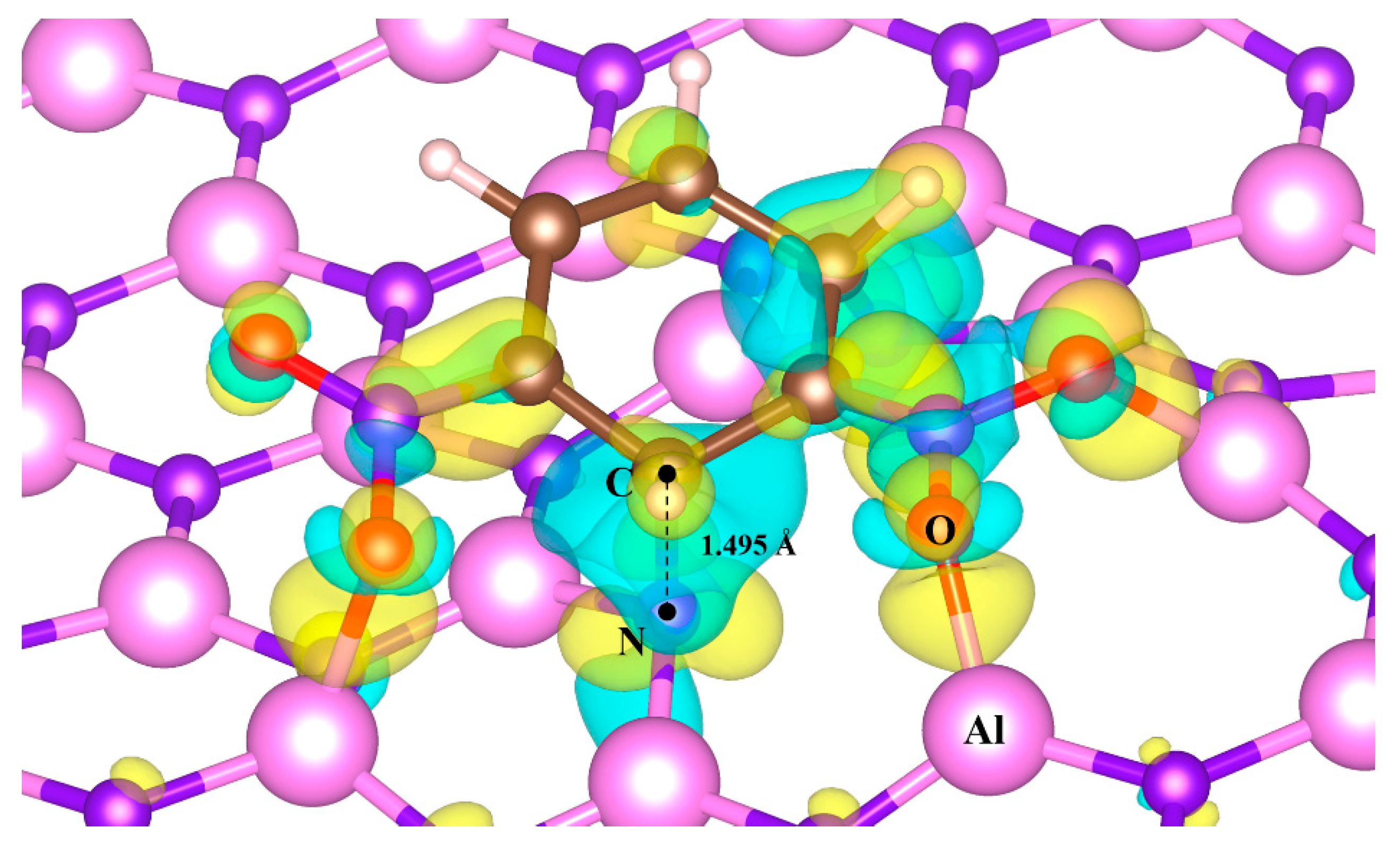
3.2. Adsorption of m-DNB Molecule on AlN Nanosheet with Al-N Vacancies
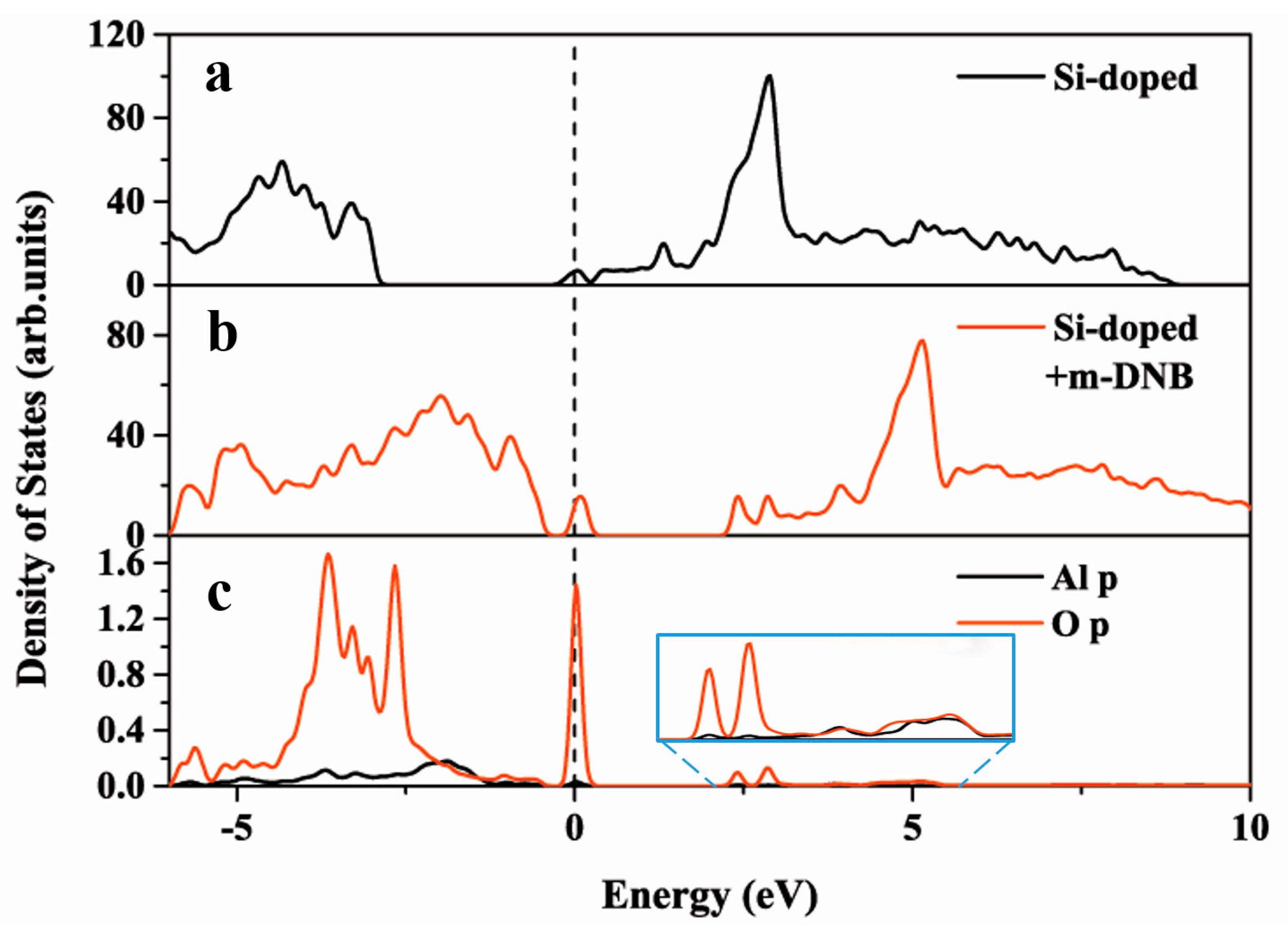
3.3. Interaction of m-DNB Molecule with Si-doped AlN Nanosheet
4. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Stankovich, S.; Dikin, D.A.; Piner, R.D.; Kohlhaas, K.A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S.T.; Ruoff, R.S. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 2007, 45, 1558–1565. [Google Scholar] [CrossRef]
- Prasongkit, J.; Amorim, R.G.; Chakraborty, S.; Ahuja, R.; Scheicher, R.H.; Amornkitbamrung, V. Highly sensitive and selective gas detection based on silicene. J. Phys. Chem. C 2015, 119, 16934–16940. [Google Scholar] [CrossRef]
- Liu, C.; Feng, W.; Yao, Y. Quantum spin hall effect in silicene and two-dimensional germanium. Phys. Rev. Lett. 2011, 107, 076802. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Neal, A.T.; Zhu, Z.; Luo, Z.; Xu, X.; Tomanek, D.; Ye, P.D. Phosphorene: An unexplored 2D semiconductor with a high hole mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Ci, L.; Lu, H.; Sorokin, P.B.; Jin, C.; Ni, J.; Kvashnin, A.G.; Kvashnin, D.G.; Lou, J.; Yakobson, B.I.; et al. Large scale growth and characterization of atomic hexagonal boron nitride layers. Nano Lett. 2010, 10, 3209–3215. [Google Scholar] [CrossRef]
- Golberg, D.; Bando, Y.; Huang, Y.; Terao, T.; Mitome, M.; Tang, C.; Zhi, C. Boron nitride nanotubes and nanosheets. ACS Nano 2010, 4, 2979–2993. [Google Scholar] [CrossRef]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef]
- Ganji, M.D.; Jameh-Bozorgi, S.; Rezvani, M. A comparative study of structural and electronic properties of formaldehyde molecule on monolayer honeycomb structures based on vdW-DF prospective. Appl. Surf. Sci. 2016, 384, 175–181. [Google Scholar] [CrossRef]
- Schedin, F.; Geim, A.K.; Morozov, S.V.; Hill, E.W.; Blake, P.; Katsnelson, M.I.; Novoselov, K.S. Detection of individual gas molecules adsorbed on graphene. Nat. Mater. 2007, 6, 652–655. [Google Scholar] [CrossRef]
- Safari, L.; Vessally, E.; Bekhradnia, A.; Hosseinian, A.; Edjlali, L. A density functional theory study of the sensitivity of two-dimensional BN nanosheet to nerve agents cyclosarin and tabun. Thin Solid Film. 2017, 623, 157–163. [Google Scholar] [CrossRef]
- Kou, L.; Frauenheim, T.; Chen, C. Phosphorene as a superior gas sensor: Selective adsorption and distinct I–V response. J. Phys. Chem. Lett. 2014, 5, 2675–2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, T.; Kaewmaraya, T.; Chakraborty, S.; Ahuja, R. Defect and substitution-induced silicene sensor to probe toxic gases. J. Phys. Chem. C 2016, 120, 25256–25262. [Google Scholar] [CrossRef]
- Yuan, W.; Shi, G. Graphene-based gas sensors. J. Mater. Chem. A 2013, 1, 10078–10091. [Google Scholar] [CrossRef]
- Dai, J.; Yuan, J.; Giannozzi, P. Gas adsorption on graphene doped with B, N, Al, and S: A theoretical study. Appl. Phys. Lett. 2009, 95, 232105. [Google Scholar] [CrossRef]
- Lopez-Corral, I.; Piriz, S.; Faccio, R.; Juan, A.; Avena, M. A van der waals DFT study of PtH2 systems absorbed on pristine and defective graphene. Appl. Surf. Sci. 2016, 382, 80–87. [Google Scholar] [CrossRef]
- Lopez-Corral, I.; German, E.; Juan, A.; Volpe, M.A.; Brizuela, G.P. Hydrogen adsorption on palladium dimer decorated graphene: A bonding study. Int. J. Hydrog. Energy 2012, 37, 6653–6665. [Google Scholar] [CrossRef]
- Basu, S.; Bhattacharyya, P. Recent developments on graphene and graphene oxide based solid state gas sensors. Sens. Actuators B Chem. 2012, 173, 1–21. [Google Scholar] [CrossRef]
- Varghese, S.S.; Lonkar, S.; Singh, K.K.; Swaminathan, S.; Abdala, A. Recent advances in graphene based gas sensors. Sens. Actuators B Chem. 2015, 218, 160–183. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Z.; Hark, S. Synthesis and optical characterization of single-crystalline AlN nanosheets. Solid State Commun. 2007, 143, 317–320. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Z.; Chen, Z.; Zhou, Z. Computational investigation on structural and physical properties of ain nanosheets and nanoribbons. J. Nanosci. Nanotechnol. 2010, 10, 7200–7203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zheng, F. First-principles prediction on electronic and magnetic properties of hydrogenated AlN nanosheets. J. Comput. Chem. 2011, 32, 3122–3128. [Google Scholar] [CrossRef] [PubMed]
- Valedbagi, S.; Fathalian, A.; Elahi, S.M. Electronic and optical properties of AlN nanosheet: An ab initio study. Opt. Commun. 2013, 309, 153–157. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, P.; Li, P.; Zheng, F. First-principles study on electronic structures and magnetic properties of AlN nanosheets and nanoribbons. J. Appl. Phys. 2012, 111, 094304. [Google Scholar] [CrossRef]
- Ouyang, T.; Qian, Z.; Ahuja, R.; Liu, X. First-principles investigation of CO adsorption on pristine, C-doped and N-vacancy defected hexagonal AlN nanosheets. Appl. Surf. Sci. 2018, 439, 196–201. [Google Scholar] [CrossRef]
- Du, W.; Zhao, C.; Liu, K.; Li, H.; Chen, Y.; Bai, Y.; Ahuja, R.; Qian, Z. Defective and doped aluminum nitride monolayers for NO adsorption: Physical insight. Chem. Phys. Lett. 2020, 753, 137592. [Google Scholar] [CrossRef]
- Rastegar, S.F.; Hadipour, N.L.; Soleymanabadi, H. Theoretical investigation on the selective detection of SO2 molecule by AlN nanosheets. J. Mol. Model. 2014, 20, 2439. [Google Scholar] [CrossRef]
- Rastegar, S.F.; Peyghan, A.A.; Ghenaatian, H.R.; Hadipour, N.L. NO2 detection by nanosized AlN sheet in the presence of NH3: DFT studies. Appl. Surf. Sci. 2013, 274, 217–220. [Google Scholar] [CrossRef]
- Rad, A.S.; Shabestari, S.S.; Jafari, S.A.; Zardoost, M.R.; Mirabi, A. N-doped graphene as a nanostructure adsorbent for carbon monoxide: DFT calculations. Mol. Phys. 2016, 114, 1756–1762. [Google Scholar] [CrossRef]
- Nasehnia, F.; Seifi, M. Adsorption of molecular oxygen on VIIIB transition metal-doped graphene: A DFT study. Mod. Phys. Lett. B 2014, 28, 1450237. [Google Scholar] [CrossRef]
- Li, F.; Shi, C. NO-sensing performance of vacancy defective monolayer MoS2 predicted by density function theory. Appl. Surf. Sci. 2018, 434, 294–306. [Google Scholar] [CrossRef]
- Ouyang, T.; Qian, Z.; Hao, X.; Ahuja, R.; Liu, X. Effect of defects on adsorption characteristics of AlN monolayer towards SO2 and NO2: Ab initio exposure. Appl. Surf. Sci. 2018, 462, 615–622. [Google Scholar] [CrossRef]
- Chen, X.; Chen, B. Macroscopic and spectroscopic investigations of the adsorption of nitroaromatic compounds on graphene oxide, reduced graphene oxide, and graphene nanosheets. Environ. Sci. Technol. 2015, 49, 6181–6189. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, V.; Chandiramouli, R. Investigation on probing explosive nitroaromatic compound vapors using graphyne nanosheet: A first-principle study. Struct. Chem. 2019, 30, 657–667. [Google Scholar] [CrossRef]
- Bhuvaneswari, R.; Nagarajan, V.; Chandiramouli, R. Arsenene nanotube as a chemical sensor to detect the presence of explosive vapors: A first-principles insight. J. Inorg. Organomet. Polym. Mater. 2018, 28, 2844–2853. [Google Scholar] [CrossRef]
- Zhang, J.R.; Yue, Y.Y.; Luo, H.Q.; Li, N.B. Supersensitive and selective detection of picric acid explosive by fluorescent Ag nanoclusters. Analyst 2016, 141, 1091–1097. [Google Scholar] [CrossRef]
- Chandiramouli, R. Antimonene nanosheet device for detection of explosive vapors-A first-principles inspection. Chem. Phys. Lett. 2018, 708, 130–137. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab-initio molecular-dynamics for open-shell transition-metals. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab-initio molecular-dynamics simulation of the liquid-metal amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Beiranvand, R.; Valedbagi, S. Electronic and optical properties of advance semiconductor materials: BN, AlN and GaN nanosheets from first principles. Optik 2016, 127, 1553–1560. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Configuration | a | b | c | d | e | f | g | h | i |
|---|---|---|---|---|---|---|---|---|---|
| Ead (eV) | −0.806 | −0.799 | −0.742 | −0.717 | −0.901 | −0.903 | −0.649 | −0.646 | −0.829 |
| d (Å) | 3.270 | 3.212 | 3.254 | 3.340 | 3.029 | 2.924 | 2.602 | 2.962 | 2.083 |
| Systems | Ead (eV) | Adsorption Distance (Å) | Band Gap Change (eV) |
|---|---|---|---|
| Pure | −0.903 | 2.924 | −0.092 |
| Al-vacancy | −2.693 | 1.463 | −0.420 |
| N-vacancy | −3.790 | 1.870 | 0.210 |
| Al-N-vacancies | −6.957 | 1.495 | −0.690 |
| C-doped | −2.623 | 1.895 | −0.452 |
| Mg-doped | −1.083 | 2.399 | −0.151 |
| P-doped | −2.633 | 2.590 | 0.690 |
| Si-doped | −3.360 | 2.019 | −0.691 |
| Ti-doped | −3.310 | 2.012 | −0.180 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Du, W.; Zhao, T.; Ahuja, R.; Qian, Z. Poisonous Vapor Adsorption on Pure and Modified Aluminum Nitride Nanosheet for Environmental Safety: A DFT Exploration. Sustainability 2020, 12, 10097. https://doi.org/10.3390/su122310097
Zhang H, Du W, Zhao T, Ahuja R, Qian Z. Poisonous Vapor Adsorption on Pure and Modified Aluminum Nitride Nanosheet for Environmental Safety: A DFT Exploration. Sustainability. 2020; 12(23):10097. https://doi.org/10.3390/su122310097
Chicago/Turabian StyleZhang, Hongni, Wenzheng Du, Tong Zhao, Rajeev Ahuja, and Zhao Qian. 2020. "Poisonous Vapor Adsorption on Pure and Modified Aluminum Nitride Nanosheet for Environmental Safety: A DFT Exploration" Sustainability 12, no. 23: 10097. https://doi.org/10.3390/su122310097
APA StyleZhang, H., Du, W., Zhao, T., Ahuja, R., & Qian, Z. (2020). Poisonous Vapor Adsorption on Pure and Modified Aluminum Nitride Nanosheet for Environmental Safety: A DFT Exploration. Sustainability, 12(23), 10097. https://doi.org/10.3390/su122310097