
Thermodynamic and Kinetic Description of the Main Effects Related to the Memory Effect during Carbon Dioxide Hydrates Formation in a Confined Environment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
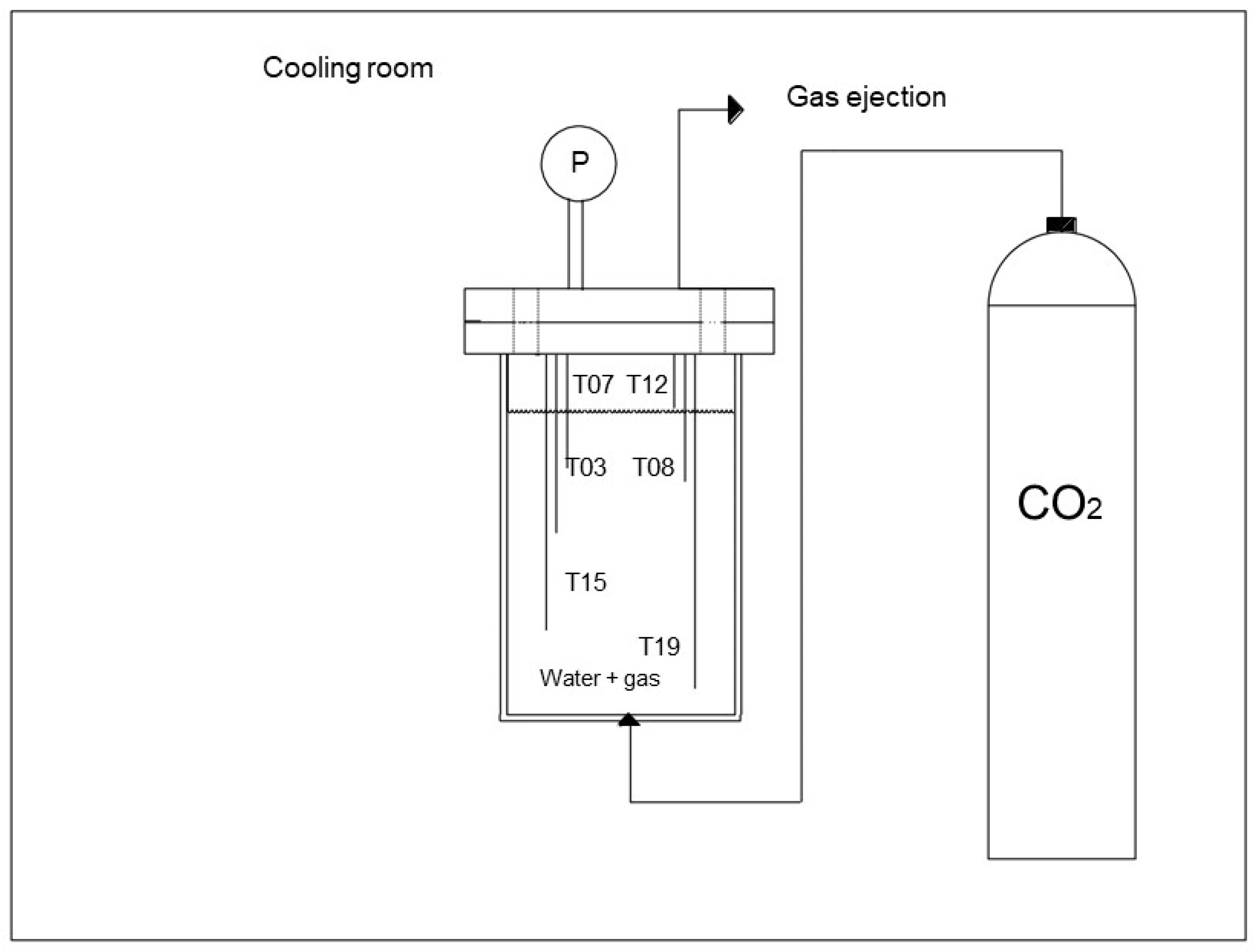
2.1. Experimental Apparatus
2.2. Materials
2.3. Methods
The Role of Porous Sand on the Distribution of Hydrates within the Reactor
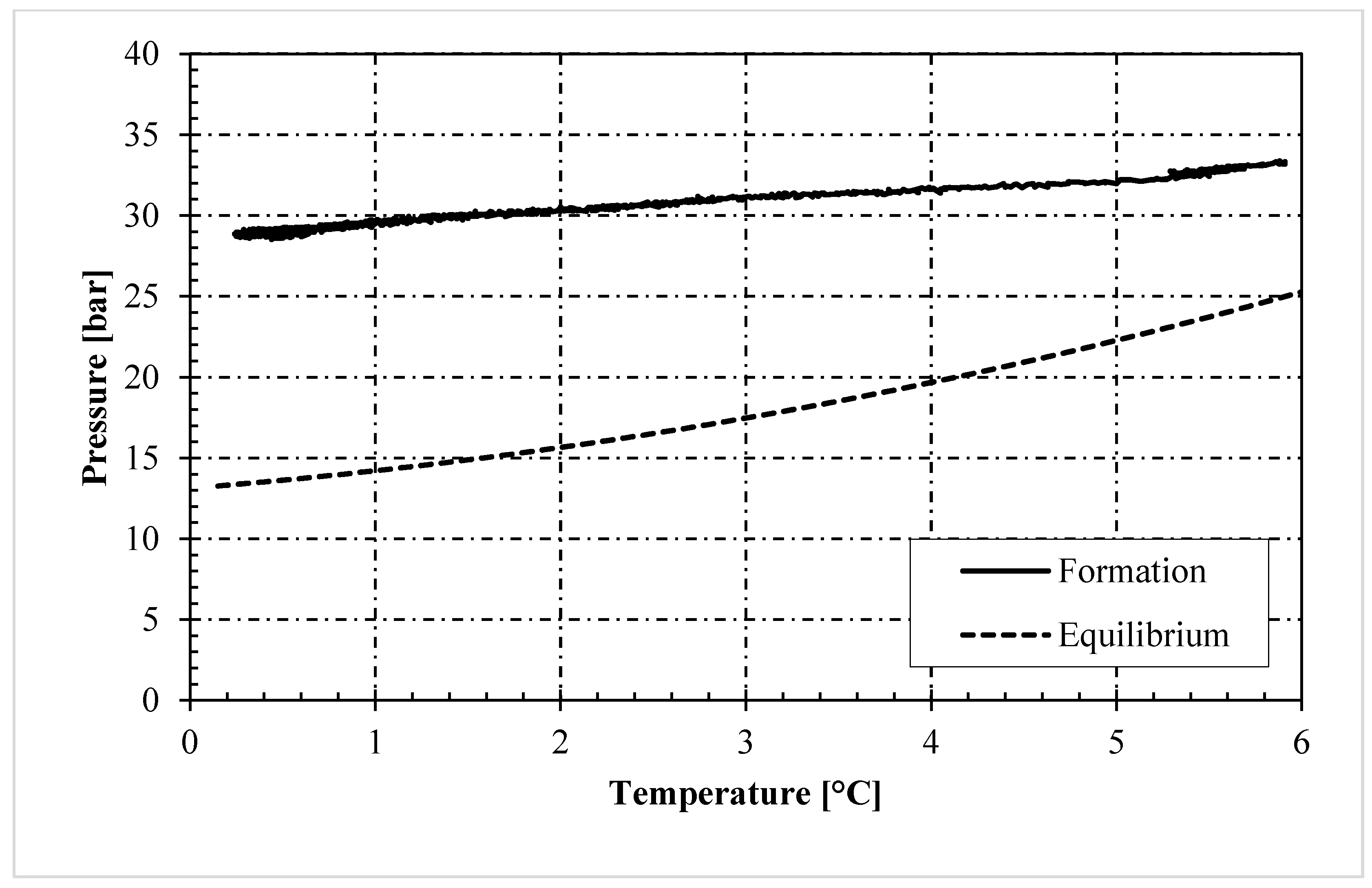
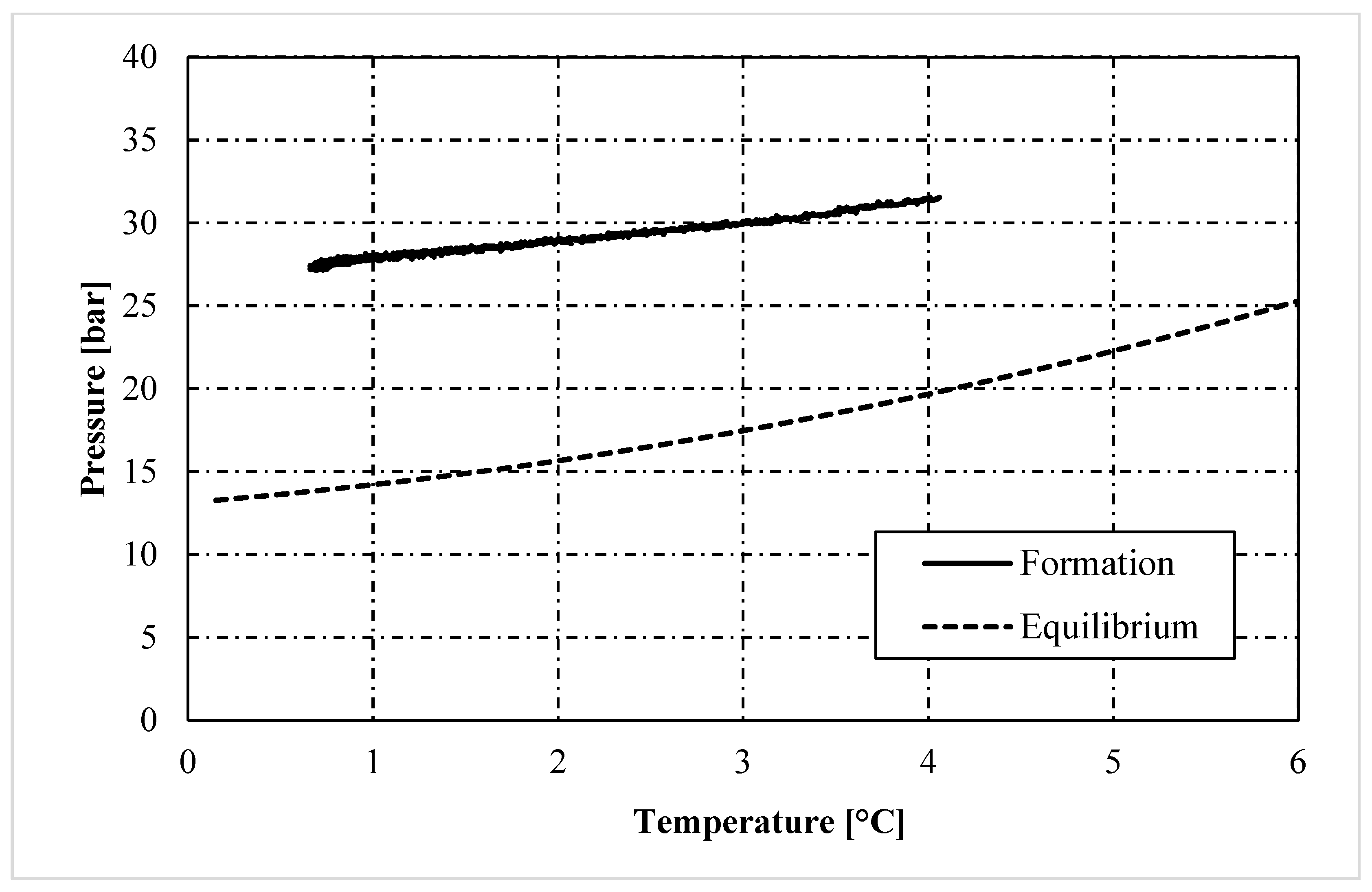
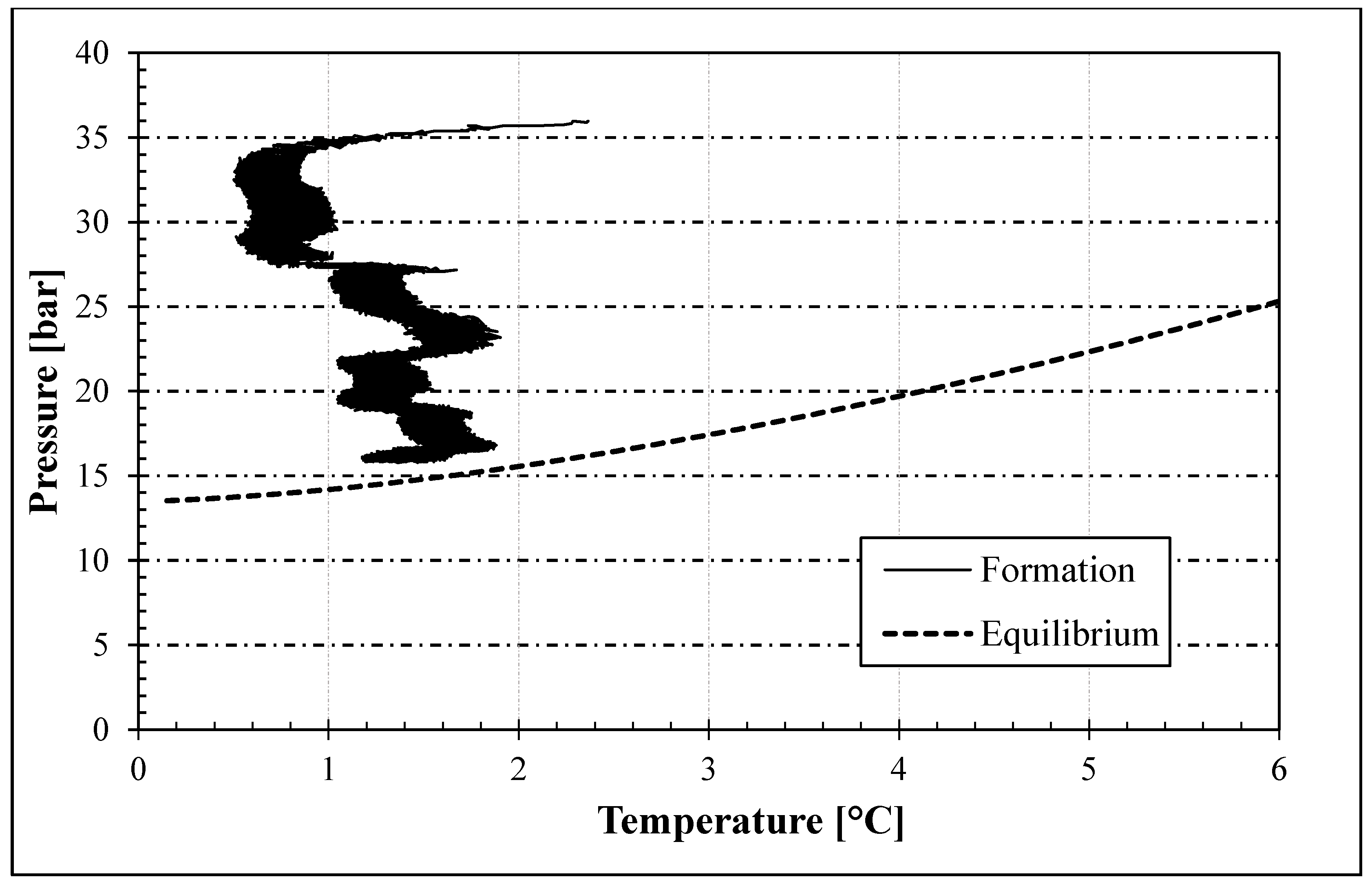
3. Results and Discussion
4. Conclusions
- In the presence of memory effect, the formation trend was almost linear and well approximated the phase boundary equilibrium. The distance between the two curves mainly depended on the presence of the porous medium, with its specific characteristics, and on the process variables which always characterize the formation phase (equilibrium lines are defined during dissociation). Conversely, in tests without memory, the process started only after the system entered widely within the stability zone; thus, it occurred at lower temperatures and its trend was completely different from the equilibrium curve.
- Time required for the process completion was drastically higher in tests without memory, up to four times higher.
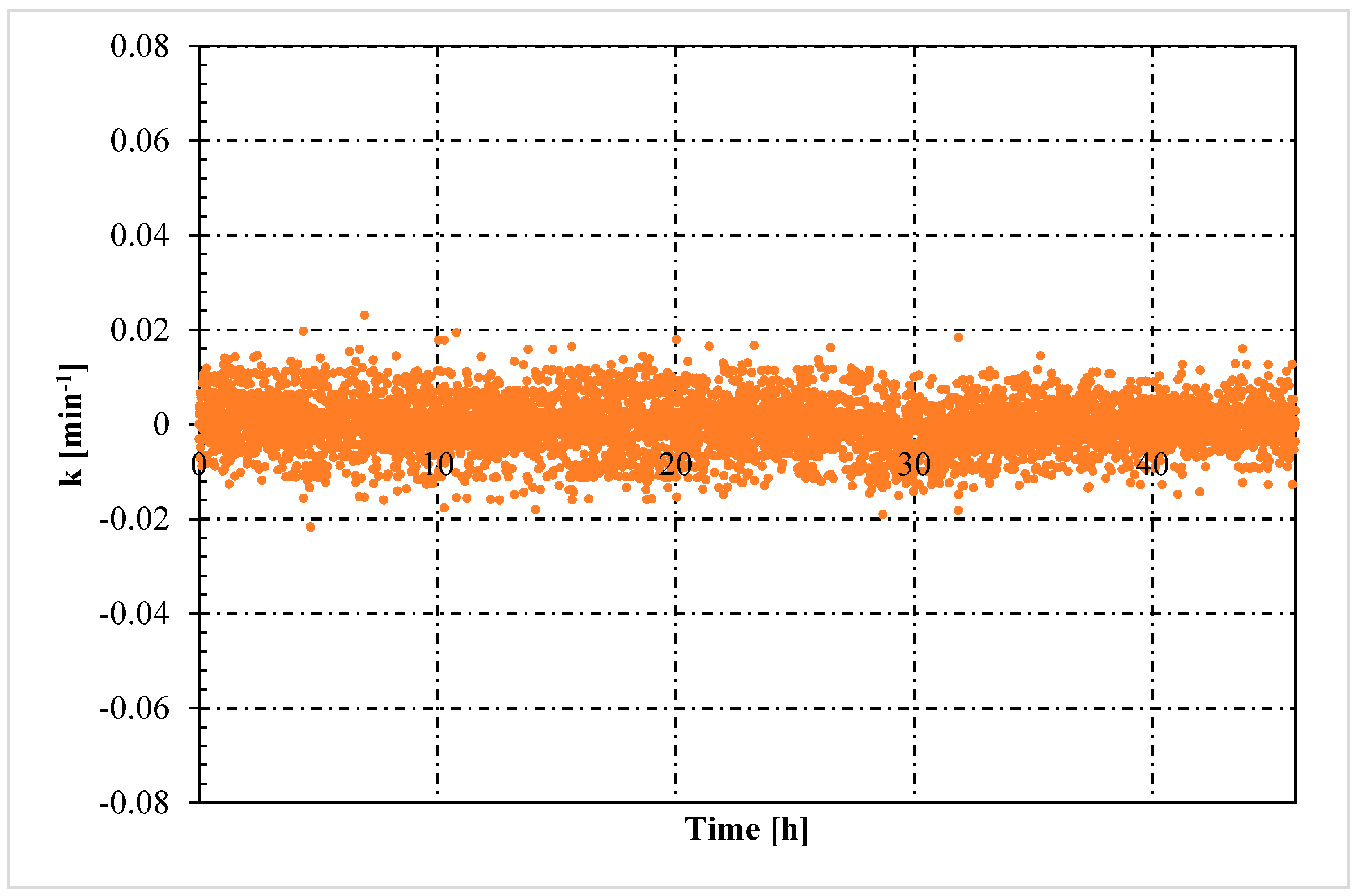
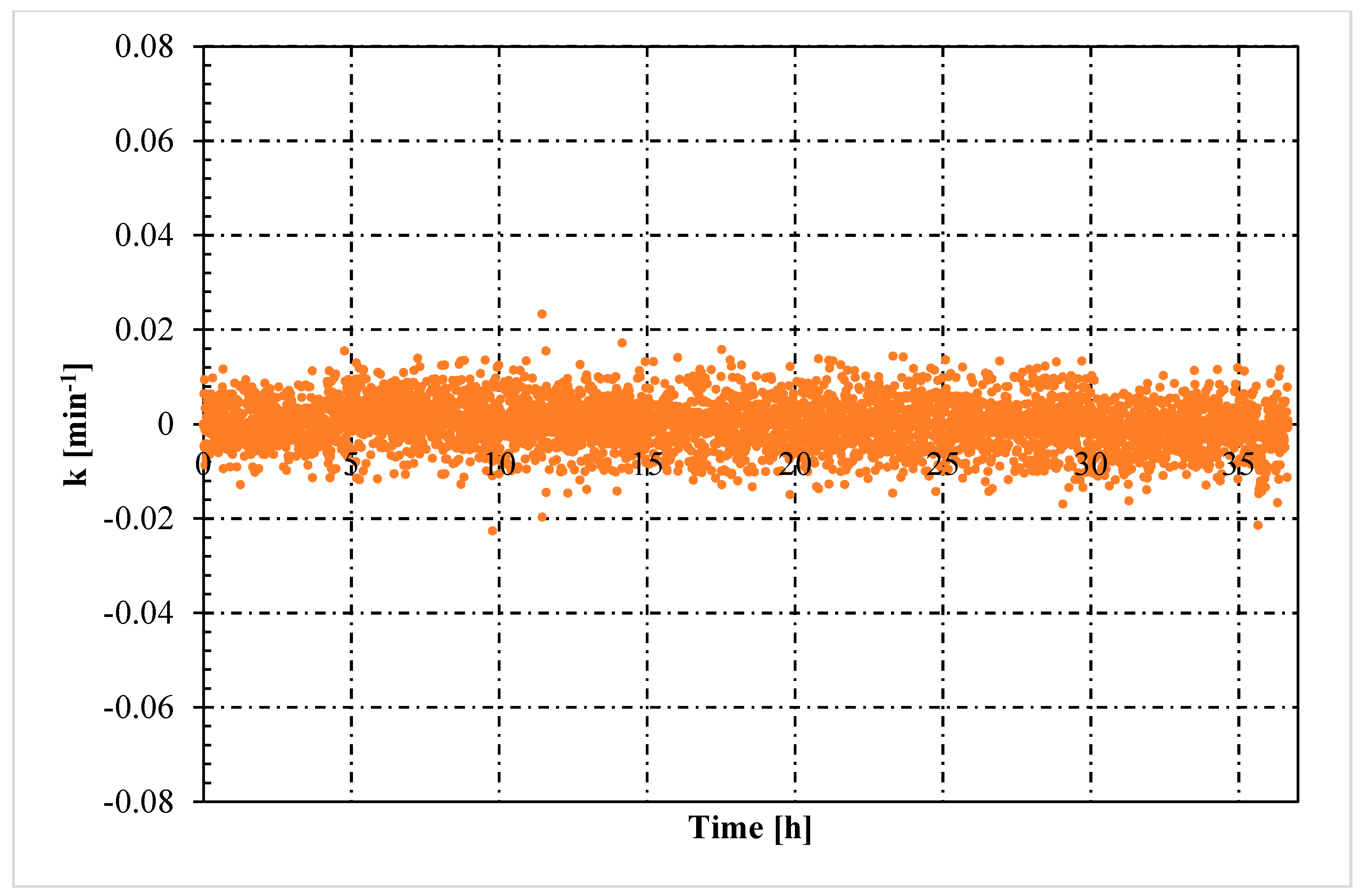
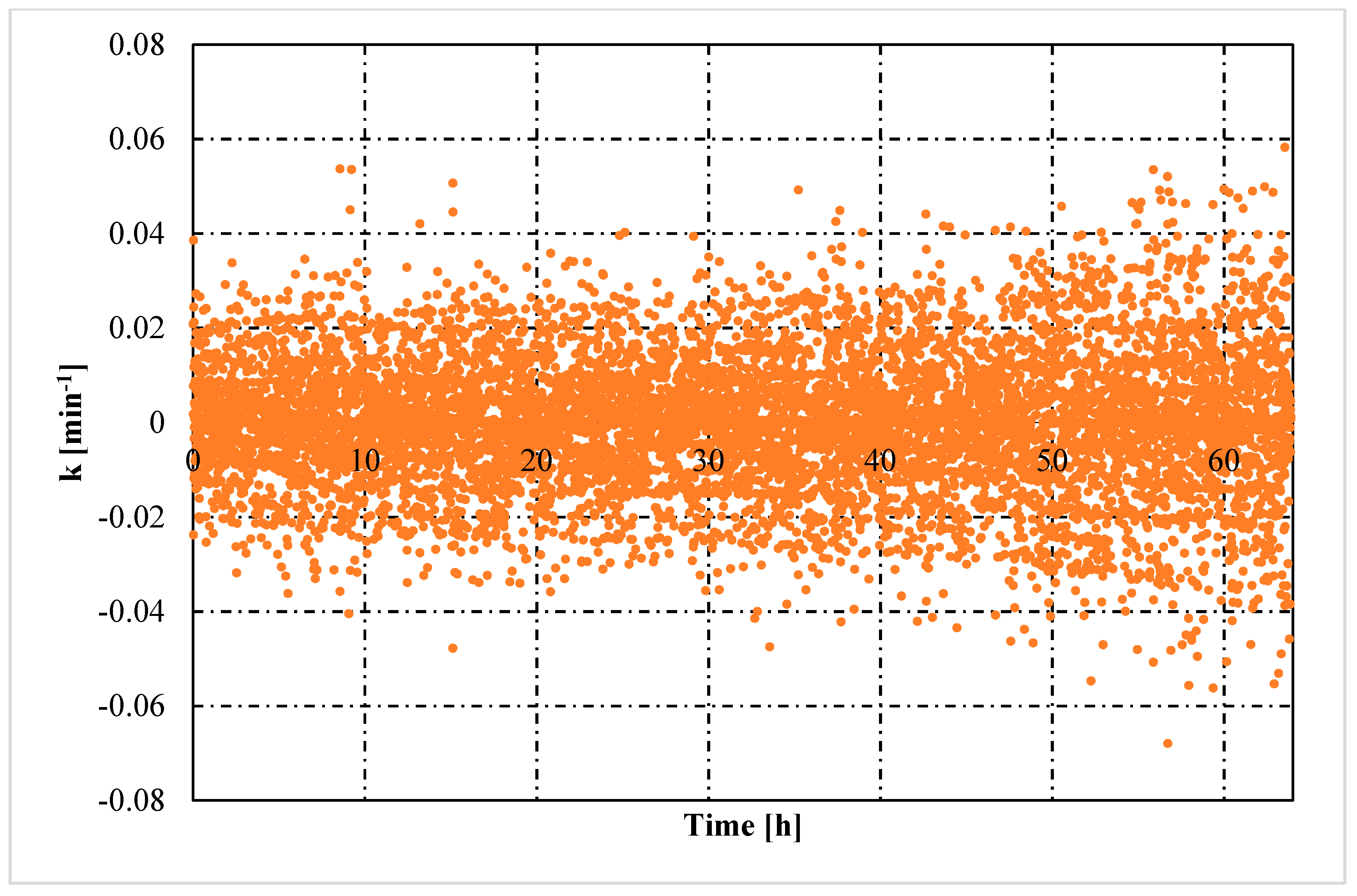
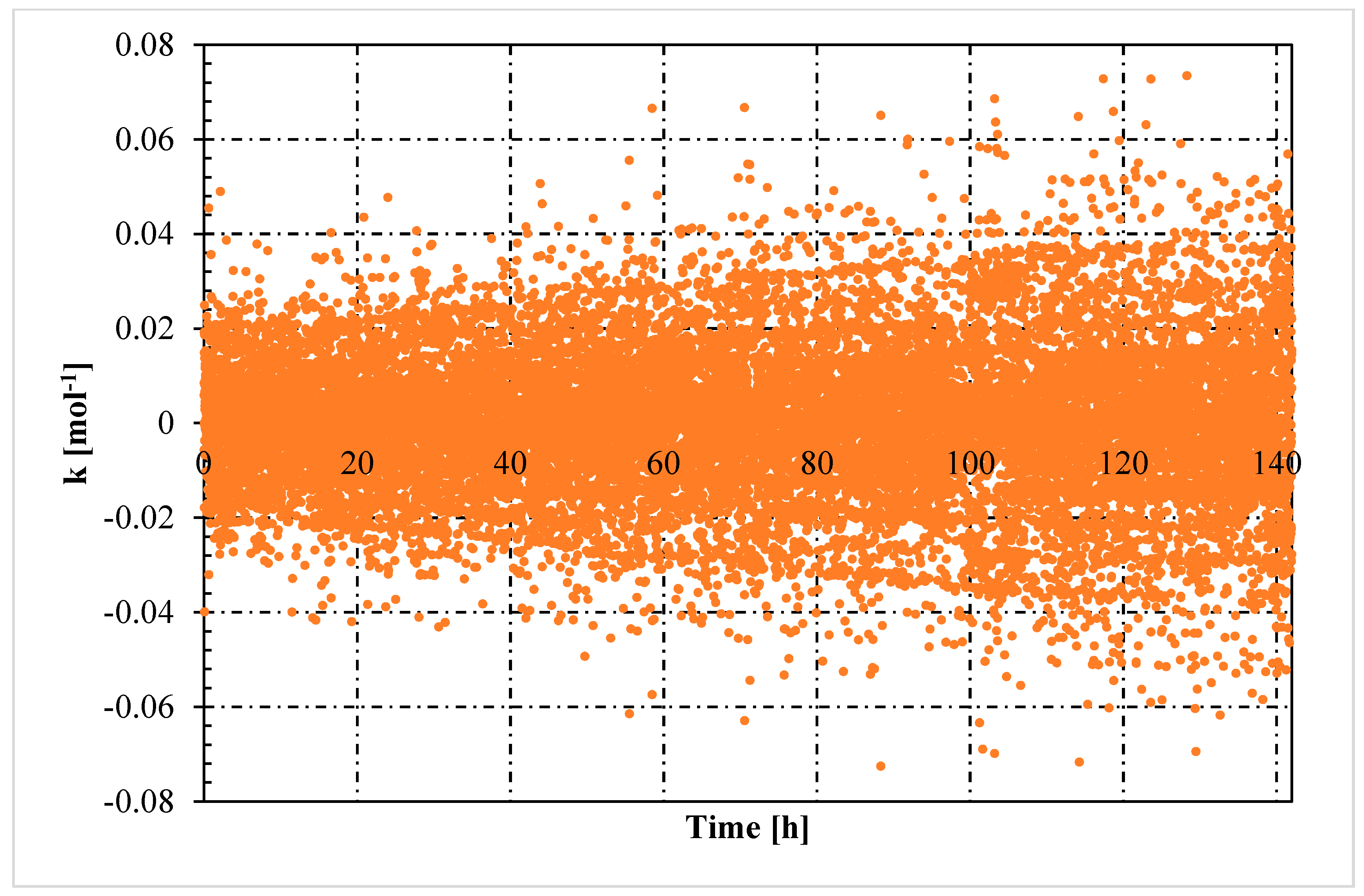
- The rate constant showed an oscillating trend in both situations, proving that the hydrates formed and dissociated continuously, according to the Labile Cluster Theory. However, in Tests with memory, oscillations were less pronounced.
- Moreover, in tests with memory, the rate constant remained constant over time, proving that the process was less dependent from the local thermodynamic conditions and. In particular, from their variation (remaining them within the hydrate stability zone). Differently, in tests without memory, the rate constant increased with time. Such phenomenon was explained with the decrease between experimental and equilibrium thermodynamic condition, which gradually reduced in intensity the driving forces of the process.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gambelli, A.M.; Filipponi, M.; Rossi, F. How methane release may affect carbon dioxide storage during replacement processes in natural gas hydrate reservoirs. J. Pet. Sci. Eng. 2021, 205, 108895. [Google Scholar] [CrossRef]
- Sloan, E.D.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Gambelli, A.M. Analyses on CH4 and CO2 hydrate formation to define the optimal pressure for CO2 injection to maximize the replacement efficiency into natural gas hydrate in presence of silica-based porous medium, via depressurization techniques. Chem. Eng. Process. 2021, 167, 108512. [Google Scholar] [CrossRef]
- Englezos, P. Clathrate hydrates. Ind. Eng. Chem. Res. 1993, 32, 1251–1274. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kawamura, T.; Yamamoto, Y.; Yoon, J.H. Phase Equilibrium Studies of Tetrahydrofuran (THF) + CH4, THF + CO2, CH4 + CO2, and THF + CO2 + CH4 Hydrates. J. Chem. Eng. 2012, 57, 3543–3548. [Google Scholar] [CrossRef]
- Li, X.S.; Xu, C.G.; Zhang, Y.; Ruan, X.K.; Li, G.; Wang, Y. Investigation into gas production from natural gas hydrate: A review. Appl. Energy 2016, 172, 286–322. [Google Scholar] [CrossRef] [Green Version]
- Handa, Y.P.; Stupin, D. Thermodynamic Properties and Dissociation Characteristics of Methane and Propane Hydrates in 70 Å—Radius Silica gel Pores. J. Phys. Chem. 1992, 96, 8599–8603. [Google Scholar] [CrossRef]
- Uchida, T.; Ebinuma, T.; Ishizaki, T. Dissociation condition measurements of methane hydrate in confined small pores of porous glass. J. Phys. Chem. B 1999, 103, 3659–3662. [Google Scholar] [CrossRef]
- Wilder, J.W.; Sheshardi, K.; Smith, D.H. Resolving apparent contradictions in equilibrium measurements for clathrate hydrates in porous media. J. Phys. Chem. B 2001, 105, 9970–9972. [Google Scholar] [CrossRef]
- Anderson, R.; Llamendo, M.; Tohidi, B.; Burgass, R.W. Experimental measurement of methane and carbon dioxide clathrate hydrate equilibria in mesoporous silica. J. Phys. Chem. B 2003, 107, 3507–3514. [Google Scholar] [CrossRef]
- Smith, D.H.; Wilder, J.W.; Sheshardi, K. Methane hydrate equilibria in silica gels with broad pore-size distribution. AlCHE J. 2002, 48, 393–400. [Google Scholar] [CrossRef]
- Liu, H.; Zhan, S.; Guo, P.; Fan, S.; Zhang, S. Understanding the characteristic of methane hydrate equilibrium in materials and its potential application. Chem. Eng. J. 2018, 349, 775–781. [Google Scholar] [CrossRef]
- Kang, S.P.; Lee, J.W.; Ryu, H.J. Phase behavior of methane and carbon dioxide hydrates in meso- and micro-sized porous media. Fluid Phase Equilib. 2008, 274, 68–72. [Google Scholar] [CrossRef]
- Lee, S.; Seo, Y. Experimental measurement and thermodynamic modelling of the mixed CH4 + C3H8 clathrate hydrate equilibria in silica gel pores: Effects of pore size and salinity. Langmuir 2010, 26, 9742–9748. [Google Scholar] [CrossRef] [PubMed]
- Zarifi, M.; Javanmardi, J.; Hashemi, H.; Eslamimanesh, A.; Mohammadi, A.H. Experimental study and thermodynamic modelling of methane mixed C1 + C2 + C3 clathrate hydrates in the presence of mesoporous silica gel. Fluid Phase Equilib. 2016, 423, 17–24. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.S.; Wang, Y.; Chen, Z.Y.; Yan, K.F. Decomposition conditions of methane hydrate in marine sediments from South China Sea. Fluid Phase Equilib. 2016, 413, 110–115. [Google Scholar] [CrossRef]
- Uchida, T.; Ebinuma, T.; Takeya, S.; Nagao, J.; Narita, H. Effects of pore sizes on dissociation temperatures and pressures of methane, carbon dioxide, and propane hydrates in porous media. J. Phys. Chem. B 2002, 106, 820–826. [Google Scholar] [CrossRef]
- Anderson, R.; Llamendo, M.; Tohidi, B.; Burgass, R.W. Characteristics of clathrate hydrate equilibria in mesopores and interpretation of experimental data. J. Phys. Chem. B 2003, 107, 3500–3506. [Google Scholar] [CrossRef]
- Clennell, M.B.; Henry, P.; Hovland, M.; Booth, J.S.; Winters, W.J. Formation of natural gas hydrates in marine sediments: 1. Conceptual model of gas hydrate growth conditioned by host sediments. J. Geophys. Res. B 1999, 104, 22985–23003. [Google Scholar] [CrossRef]
- Henry, P.; Thomas, M.; Clennell, M.B. Formation of natural gas hydrates in marine sediments 2. Thermodynamic calculations of stability conditions in porous sediments. J. Geophys. Res. B 1999, 104, 23005–23022. [Google Scholar] [CrossRef] [Green Version]
- Van der Waals, J.H.; Platteeuw, J.C. Clathrate Solutions. Adv. Chem. Phys. 1959, 2, 1–57. [Google Scholar]
- Holder, G.D.; Zetts, S.P.; Pradhan, N. Phase behavior in systems containing clathrate hydrates. Rev. Chem. Eng. 1988, 5, 1–70. [Google Scholar] [CrossRef]
- Holder, G.D.; Corbin, G.; Papadopoulos, K.D. Thermodynamic and molecular properties of gas hydrates from mixtures containing methane, argon, and krypton. Ind. Eng. Chem. 1980, 19, 282–286. [Google Scholar] [CrossRef]
- Zhou, J.; Liang, W.; Wei, C. Phase Equilibrium Condition for Pore Hydrate: Theoretical Formulation and Experimental Validation. J. Geophys. Res. Solid Earth 2019, 124, 12703–12721. [Google Scholar] [CrossRef]
- Li, S.L.; Ma, Q.L.; Sun, C.Y.; Chen, L.T.; Liu, B.; Feng, X.J.; Wang, X.Q.; Chen, G.J. A fractal approach on modelling gas hydrate phase equilibria in porous media. Fluid Phase Equilib. 2013, 356, 277–283. [Google Scholar] [CrossRef]
- Sun, R.; Duan, Z. An accurate model to predict the thermodynamic stability of methane hydrate and methane solubility in marine environments. Chem. Geol. 2007, 244, 248–262. [Google Scholar] [CrossRef]
- Peddireddy, S.; Lee, S.Y.; Lee, J.W. Variable contact angle model for gas hydrate equilibrium in porous media. AlCHE J. 2006, 52, 1228–1234. [Google Scholar] [CrossRef]
- Ullerich, J.W.; Selim, M.S.; Sloan, E.D. Theory and measurement of hydrate dissociation. AlCHE J. 1987, 33, 747–752. [Google Scholar] [CrossRef]
- Kamath, V.A.; Holder, G.D. Dissociation heat-transfer characteristics of methane hydrates. AlCHE J. 1989, 35, 1049–1052. [Google Scholar] [CrossRef]
- Katsuki, D.; Ohmura, R.; Ebinuma, T.; Narita, H. Visual observation of dissociation of methane hydrate crystals in a glass micro model: Production and transfer of methane. J. Appl. Phys. 2008, 104, 083514. [Google Scholar] [CrossRef]
- Almenningen, S.; Flatlandsmo, J.; Ferno, M.A.; Ersiand, G. Multiscale laboratory verification of depressurization for production of sedimentary methane hydrates. SPE J. 2017, 22, 138–147. [Google Scholar] [CrossRef]
- Beltràn, G.; Servio, P. Morphological investigation of Methane-Hydrate Films Formed on a Glass Surface. Cryst. Growth Des. 2010, 10, 4339–4347. [Google Scholar] [CrossRef]
- Em, I.; Stoporev, A.S.; Semenov, A.P.; Glotov, A.; Smirnova, E.; Villevand, G.; Vinokurov, V.A.; Manakov, A.Y.; Lvov, Y.M. Methane Hydrate Formation in Halloyte Clay Nanotubes. ACS Sustain. Chem. Eng. 2020, 8, 7860–7868. [Google Scholar] [CrossRef]
- Esmail, S.; Beltràn, J.G. Methane hydrate propagation on surfaces of varying wettability. J. Nat. Gas Sci. Eng. 2016, 35, 1535–1543. [Google Scholar] [CrossRef]
- Filarsky, F.; Schmuck, C.; Schultz, H.J. Impact of Modified Silica Beads on Methane Hydrate Formation in a Fixed—Bed Reactor. Ind. Eng. Chem. Res. 2019, 58, 16687–16695. [Google Scholar] [CrossRef]
- Em, I.; Zakharenko, A.M.; Kosyanov, D.Y.; Stoporev, A.S.; Gulkov, A.N. Enhancement of gas hydrates synthesis with CNT surfaces. AIP Conf. Proc. 2018, 2051, 020072. [Google Scholar]
- Ripmeester, J.A.; Alavi, S. Some current challenges in clathrate hydrate science: Nucleation, decomposition and the memory effect. Curr. Opin. Solid State Mater. Sci. 2016, 20, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Gambelli, A.M.; Rossi, F. Thermodynamic and kinetic characterization of methane hydrate nucleation, growth and dissociation processes, according to the Labile Cluster theory. Chem. Eng. J. 2021, 425, 130706. [Google Scholar] [CrossRef]
- Li, Y.; Wu, N.; Ning, F.; Gao, D.; Hao, X.; Chen, Q.; Liu, C.; Sun, J. Hydrate-induced clogging of sand-control screen and its implication on hydrate production operation. Energy 2020, 206, 118030. [Google Scholar] [CrossRef]
- Roozeboom, H.W.B. Sur l’hydrate de l’acide sulfureux. Recl. Trav. Chim. Pays-Bas 1884, 3, 29–58. [Google Scholar] [CrossRef]
- Wilson, P.W.; Haymet, A.D.J. Hydrate formation and re-formation in nucleating THF/water mixtures show no evidence to support a “memory” effect. Chem. Eng. J. 2010, 161, 146–150. [Google Scholar] [CrossRef]
- Li, Y.; Wu, N.; He, C.; Sun, Z.; Zhang, Z.; Hao, X.; Chen, Q.; Bu, Q.; Liu, C.; Sun, J. Nucleation probability and memory effect of methane-propane mixed gas hydrate. Fuel 2021, 291, 120103. [Google Scholar] [CrossRef]
- Takeya, S.; Hori, A.; Hondoh, T.; Uchida, T. Freezing-Memory Effect of Water on Nucleation of CO2 Hydrate Crystals. J. Phys. Chem. B 2000, 104, 4164–4168. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, B. Memory effect on the Pressure-Temperature Condition and Induction Time of Gas Hydrate nucleation. J. Nat. Gas Chem. 2010, 19, 446–451. [Google Scholar] [CrossRef]
- Duchateau, C.; Glénat, P.; Pou, T.-E.; Hidalgo, M.; Dicharry, C. Hydrate Precursor Test Method for the Laboratory Evaluation of Kinetic Hydrate Inhibitors. Energy Fuels 2010, 24, 616–623. [Google Scholar] [CrossRef]
- Merkel, S.F.; Schultz, H.J. Methane Extraction from Natural Gas Hydrate Reservoirs with Simultaneous Storage of Carbon Dioxide. Chem. Ing. Tech. 2015, 87, 475–483. [Google Scholar] [CrossRef]
- Filarsky, F.; Schmuck, C.; Schultz, H.J. Development of a biogas production and purification process using promoted gas hydrate formation—A feasibility study. Chem. Eng. Res. Des. 2018, 134, 257–267. [Google Scholar] [CrossRef]
- Zeng, H.; Wilson, L.D.; Walker, V.K.; Ripmeester, J.A. Effect of Antifreeze Proteins on the Nucleation, Growth, and the Memory Effect during Tetrahydrofuran Clathrate Hydrate Formation. J. Am. Chem. Soc. 2006, 128, 2844–2850. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Rossi, F. Formation rate as parameter to distinguish nucleation from hydrate massive growth phase: Experimental investigation in presence of two different porous media. Exp. Therm. Fluid Sci. 2022, 131, 110525. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Presciutti, A.; Rossi, F. Kinetic considerations and formation rate for carbon dioxide hydrate, formed in presence of a natural silica-based porous medium: How initial thermodynamic conditions may modify the process kinetic. Thermochim. Acta 2021, 705, 179039. [Google Scholar] [CrossRef]
- Sloan, E.D.; Fleyfel, F. A molecular mechanism for gas hydrate nucleation from ice. AIChE J. 1991, 37, 1281–1292. [Google Scholar] [CrossRef]
- Muller-Bongartz, B.; Wildeman, T.R.; Sloan, E.D. A hypothesis for hydrate nucleation phenomena. In Proceedings of the Second international Offshore and Polar Engineering Conference, San Francisco, CA, USA, 14–19 June 1992; Available online: https://onepetro.org/ISOPEIOPEC/proceedings-abstract/ISOPE92/All-ISOPE92/ISOPE-I-92-089/22828 (accessed on 10 November 2021).
- Gambelli, A.M.; Rossi, F. Experimental investigation on the possibility of defining the feasibility of CO2/CH4 exchange into a natural gas hydrate marine reservoir via fast analysis of sediment properties. Chem. Eng. Res. Des. 2021, 171, 327–339. [Google Scholar] [CrossRef]
- Gambelli, A.M. An experimental description of the double positive effect of CO2 injection in methane hydrate deposits in terms of climate change mitigation. Chem. Eng. Sci. 2021, 233, 116430. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, J.C.; Li, X.S.; Zhang, Y. Experimental investigation of optimization of well spacing for gas recovery from methane hydrate reservoir in sandy sediment by heat stimulation. Appl. Energy 2017, 207, 562–572. [Google Scholar] [CrossRef]
- Yuan, Q.; Sun, C.Y.; Yang, X.; Ma, P.C.; Ma, Z.W.; Liu, B.; Ma, Q.L.; Yang, L.Y.; Chen, G.J. Recovery of methane from hydrate reservoir with gaseous carbon dioxide using a three-dimensional middle-size reactor. Energy 2012, 40, 47–58. [Google Scholar] [CrossRef]
- Yin, Z.; Huang, L.; Linga, P. Effect of wellbore design on the production behaviour of methane hydrate-bearing sediments induced by depressurization. Appl. Energy 2019, 254, 113365. [Google Scholar] [CrossRef]
- Rossi, F.; Gambelli, A.M.; Sharma, D.K.; Castellani, B.; Nicolini, A.; Castaldi, M.J. Experiments on methane hydrates formation in seabed deposits and gas recovery adopting carbon dioxide replacement strategies. Appl. Therm. Eng. 2019, 148, 371–381. [Google Scholar] [CrossRef]
- James, C.; Armstrong, M.; Biaglow, A. Comparison of pure component thermodynamic properties from CHEMCAD with direct calculation using the Peng-Robinson equation of state. Chem. Data Collect. 2020, 30, 100571. [Google Scholar] [CrossRef]
- Takeya, S.; Kida, M.; Minami, H.; Sakagami, H.; Hachikubo, A.; Takahashi, N.; Shoji, H.; Soloviev, V.; Wallmann, K.; Biebow, N.; et al. Structure and thermal expansion of natural gas clathrate hydrates. Chem. Eng. Sci. 2006, 61, 2670–2674. [Google Scholar] [CrossRef]
- Aregba, A.G. Gas hydrate-Properties, Formation and Benefits. Open J. Yangtze Oil Gas 2017, 2, 27–44. [Google Scholar] [CrossRef] [Green Version]
- Stoporev, A.S.; Svarovskaya, L.I.; Strelets, L.A.; Altunina, L.K.; Manakov, A.Y. Effect of reactor wall material on the nucleation of methane hydrate in water-in-oil emulsions. Mendelev Commun. 2018, 28, 343–344. [Google Scholar] [CrossRef]
- Nema, Y.; Ohmura, R.; Senaha, I.; Yasuda, K. Quadruple point determination in carbon dioxide hydrate forming system. Fluid Phase Equilib. 2017, 441, 49–53. [Google Scholar] [CrossRef]
- Nagashima, H.D.; Fukushima, N.; Ohmura, R. Phase equilibrium condition measurements in carbon dioxide clathrate hydrate forming system from 199.1 K to 247.1 K. Fluid Phase Equilib. 2016, 413, 53–56. [Google Scholar] [CrossRef]
- Khan, M.S.; Partoon, B.; Bavoh, C.B.; Lal, B.; Mellon, B.M. Influence of tetramethylammonium hydroxide on methane and carbon dioxide gas hydrate phase equilibrium conditions. Fluid Phase Equilib. 2017, 440, 1–8. [Google Scholar] [CrossRef]
- Khan, M.S.; Bavoh, C.; Partoon, B.; Lal, B.; Bustam, M.A.; Shariff, A.M. Thermodynamic effect of ammonium based ionic liquids on CO2 hydrates phase boundary. J. Mol. Liq. 2017, 238, 533–539. [Google Scholar] [CrossRef]
- Sadeq, D.; Iglauer, S.; Lebedev, M.; Smith, C.; Barifcani, A. Experimental determination of hydrate phase equilibrium for different gas mixtures containing methane, carbon dioxide and nitrogen with motor current measurements. J. Nat. Gas Sci. Eng. 2017, 38, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Sun, C.; Chen, G.; Nie, Y.; Sun, Z.; Liu, Y. Measurements of Hydrate Equilibrium Conditions for CH4, CO2, and CH4 + C2H6 + C3H8 in Various Systems by Step-heating Method. Chin. J. Chem. Eng. 2009, 17, 635–641. [Google Scholar] [CrossRef]
- Jarrahian, A.; Nakhaee, A. Hydrate-liquid-vapor equilibrium condition for N2 + CO2 + H2O system: Measurement and modelling. Fuel 2019, 237, 769–774. [Google Scholar] [CrossRef]
- Kyung, D.; Lee, K.; Kim, H.; Lee, W. Effect of marine environmental factors on the phase equilibrium of CO2 hydrate. Int. J. Greenh. Gas Control 2014, 20, 285–292. [Google Scholar] [CrossRef]
- Seo, Y.T.; Lee, H. Multiple-phase hydrate equilibria of the ternary carbon dioxide, methane, and water mixtures. J. Phys. Chem. B 2001, 105, 10084–10090. [Google Scholar] [CrossRef]
- Rossi, F.; Gambelli, A.M. Thermodynamic phase equilibrium of single-guest hydrate and formation data of hydrate in presence of chemical additives: A review. Fluid Phase Equilib. 2021, 536, 112958. [Google Scholar] [CrossRef]
- Shicai, S.; Yong, Z.; Changling, L.; Yufeng, L. Preliminary study on measurement technology for hydrate phase equilibrium. Fluid Phase Equilib. 2015, 403, 60–69. [Google Scholar] [CrossRef]
- Yu, Y.S.; Zhou, S.D.; Li, X.S.; Wang, S.L. Effect of graphite nanoparticles on CO2 hydrate phase equilibrium. Fluid Phase Equilib. 2016, 414, 23–28. [Google Scholar] [CrossRef]
- Pahlavanzadeh, H.; Farhoudi, A.; Manteghian, M. Experimental measurement of carbon dioxide clathrate hydrate in the presence of adamantane and other water soluble and insoluble additives. J. Chem. Thermodyn. 2019, 135, 352–358. [Google Scholar] [CrossRef]
- Herri, J.M.; Bouchemoua, A.; Kwatersky, M.; Fezoua, A.; Ouabbas, Y.; Cameirao, A. Gas hydrate equilibria for CO2-N2 and CO2-CH4 gas mixtures—Experimental studies and thermodynamic modelling. Fluid Phase Equilib. 2011, 301, 171–190. [Google Scholar] [CrossRef] [Green Version]
- Kvamme, B. Kinetics of hydrate formation, dissociation and reformation. Chem. Thermodyn. Therm. Anal. 2021, 1–2, 100004. [Google Scholar] [CrossRef]
- Kvamme, B.; Zhao, J.Z.; Wei, N.; Sun, W.T.; Saeidi, N.; Pei, J.; Kuznetsova, T. Hydrate production philosophy and thermodynamic calculations. Energies 2020, 13, 672. [Google Scholar] [CrossRef] [Green Version]
- Kvamme, B. Kinetics of hydrate formation from nucleation theory. Int. J. Offshore Polar. 2002, 12, 256–263. [Google Scholar]
- Kvamme, B. Droplets of dry ice and cold liquid CO2 for self-transport of CO2 to large depths. Int. J. Offshore Polar. 2003, 13, 139–146. [Google Scholar]
- Chaturvedi, E.; Maiti, M.; Laik, S.; Mandal, A. Mineralogical and structural characterization of the sediments of Krishna Godavari and Mahanadi Basin and their influences on hydrate formation kinetics. Adv. Powder Technol. 2021, 32, 1247–1263. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, F.; Li, Y.; Gambelli, A.M. Thermodynamic and Kinetic Description of the Main Effects Related to the Memory Effect during Carbon Dioxide Hydrates Formation in a Confined Environment. Sustainability 2021, 13, 13797. https://doi.org/10.3390/su132413797
Rossi F, Li Y, Gambelli AM. Thermodynamic and Kinetic Description of the Main Effects Related to the Memory Effect during Carbon Dioxide Hydrates Formation in a Confined Environment. Sustainability. 2021; 13(24):13797. https://doi.org/10.3390/su132413797
Chicago/Turabian StyleRossi, Federico, Yan Li, and Alberto Maria Gambelli. 2021. "Thermodynamic and Kinetic Description of the Main Effects Related to the Memory Effect during Carbon Dioxide Hydrates Formation in a Confined Environment" Sustainability 13, no. 24: 13797. https://doi.org/10.3390/su132413797
APA StyleRossi, F., Li, Y., & Gambelli, A. M. (2021). Thermodynamic and Kinetic Description of the Main Effects Related to the Memory Effect during Carbon Dioxide Hydrates Formation in a Confined Environment. Sustainability, 13(24), 13797. https://doi.org/10.3390/su132413797