
Representativeness of Fecal Microbiota Is Limited to Cecum and Colon in Domestic Yak


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. DNA Extraction, Amplification, and Sequencing
2.3. Bioinformatic Pipeline
2.4. Statistical Analysis
2.5. Source-Tracking Analysis
2.6. The Ecological Assembly Process of Microbiota of Eight Intestinal Parts
3. Results
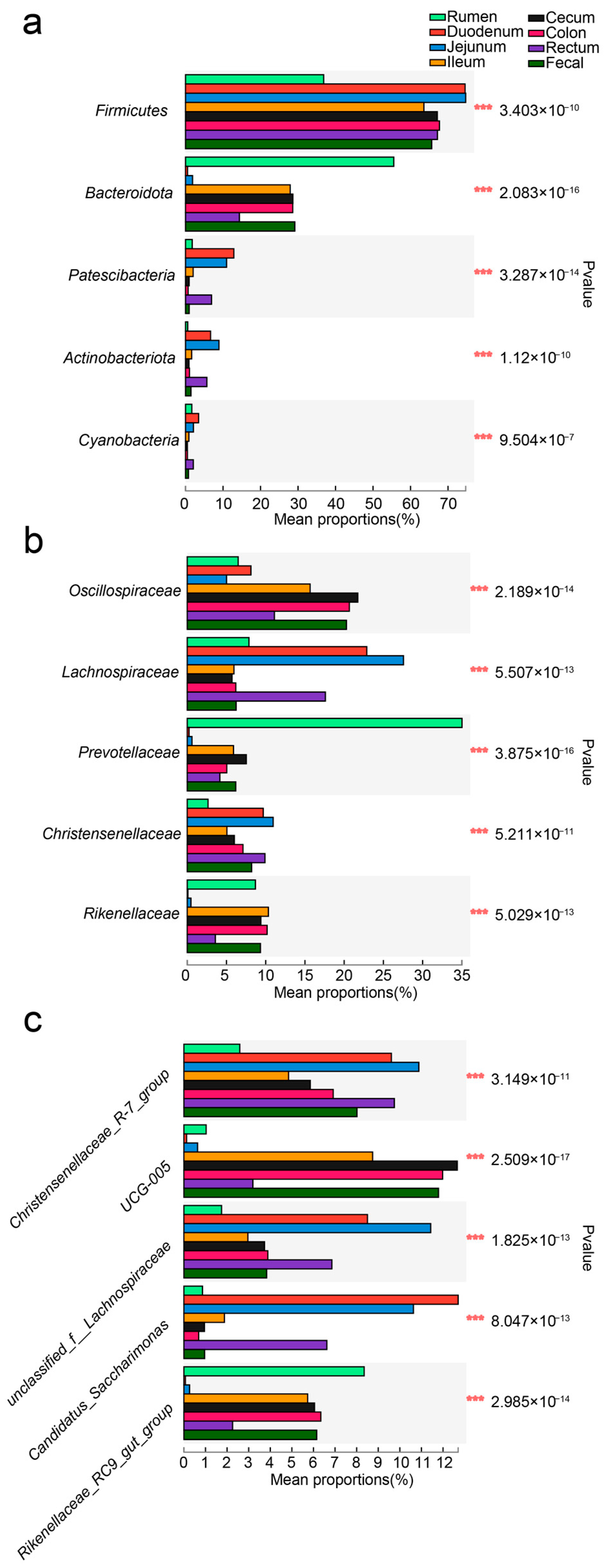
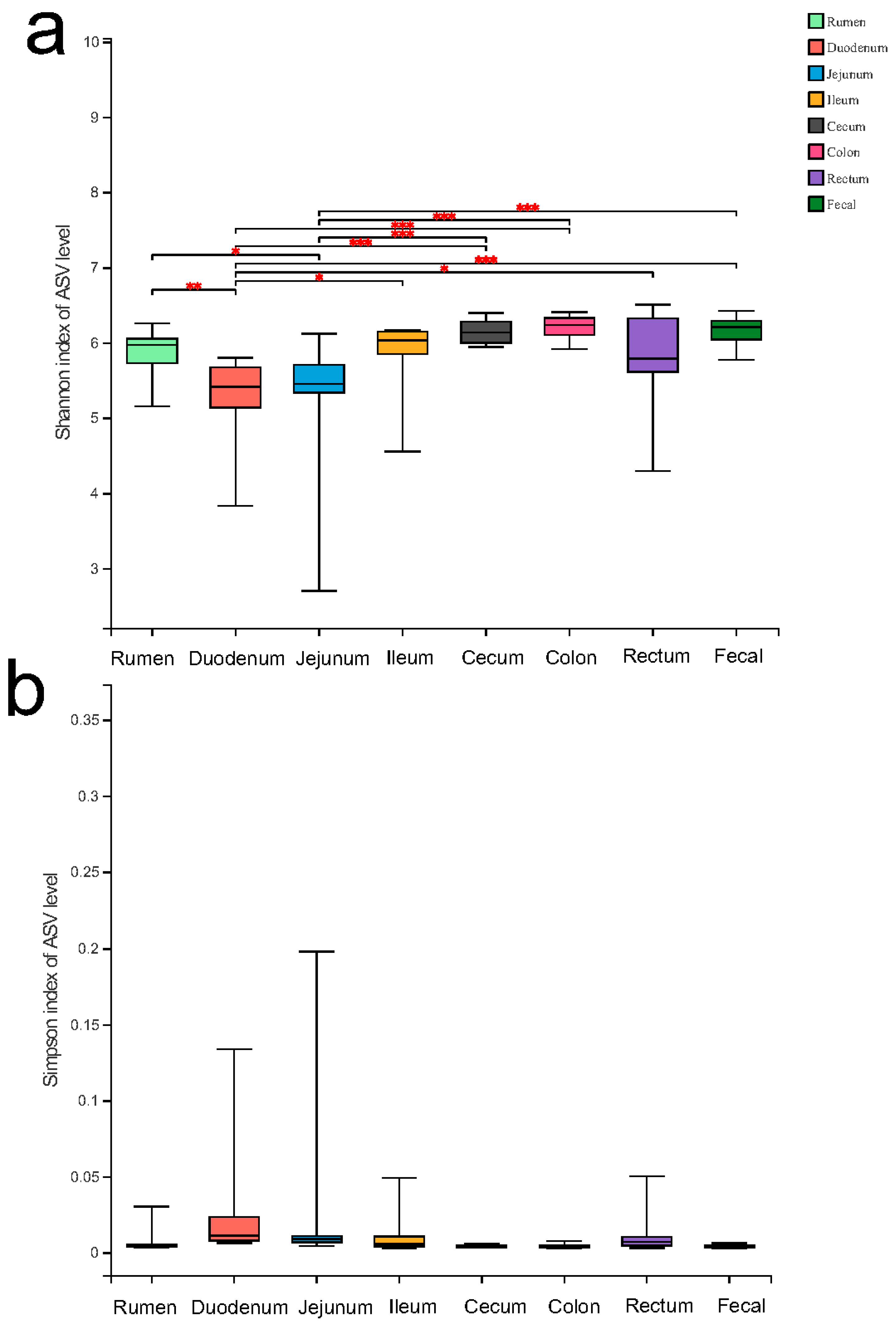
3.1. Comparison of the Gut Microbial Diversity among Eight Intestinal Parts
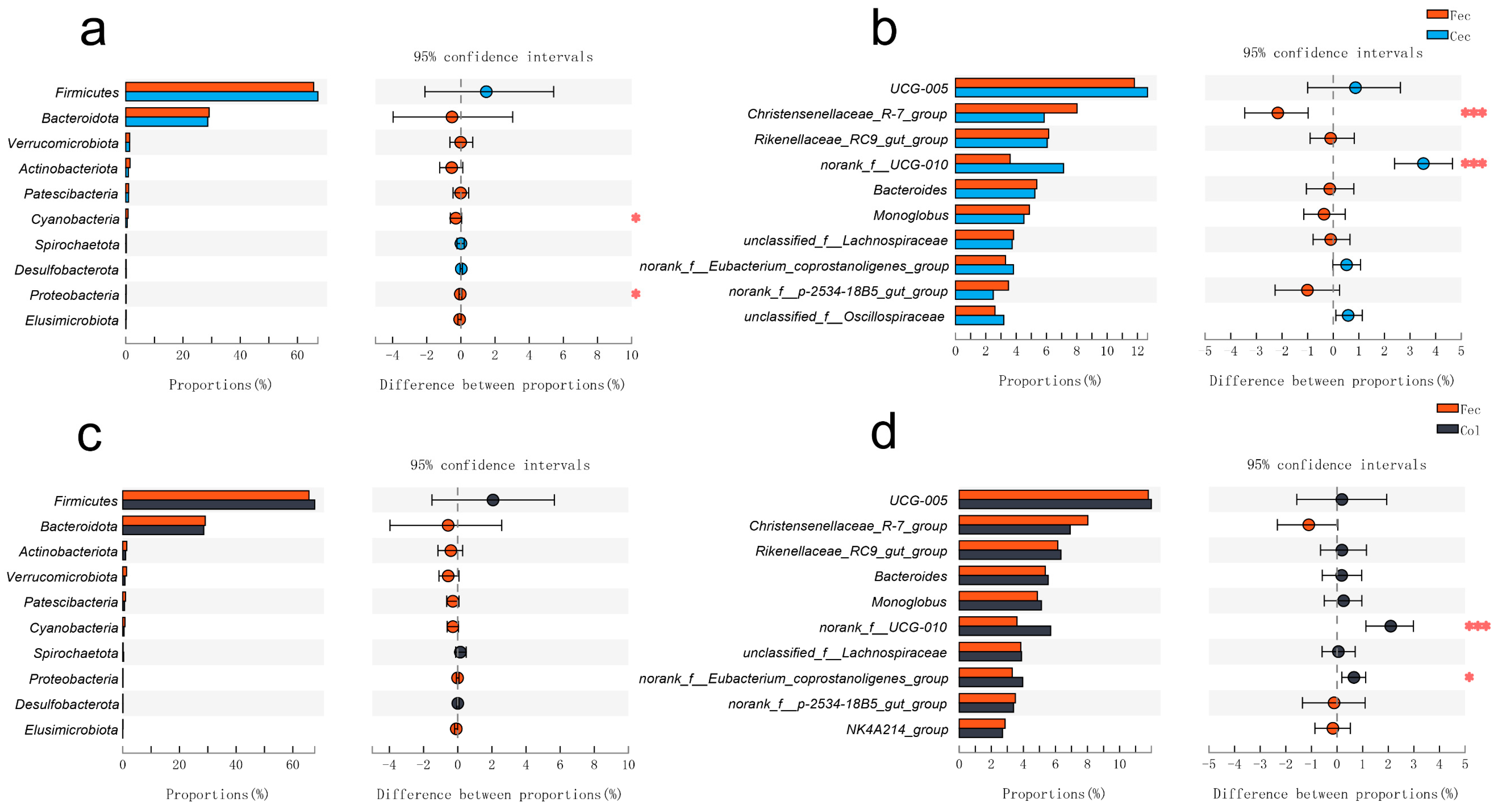
3.2. The Differences in Gut Microbial Microbiota between the Feces and Cecum, and the Feces and Colon
3.3. Comparison of the Functions in the Gut Microbiota among the Eight Intestinal Parts
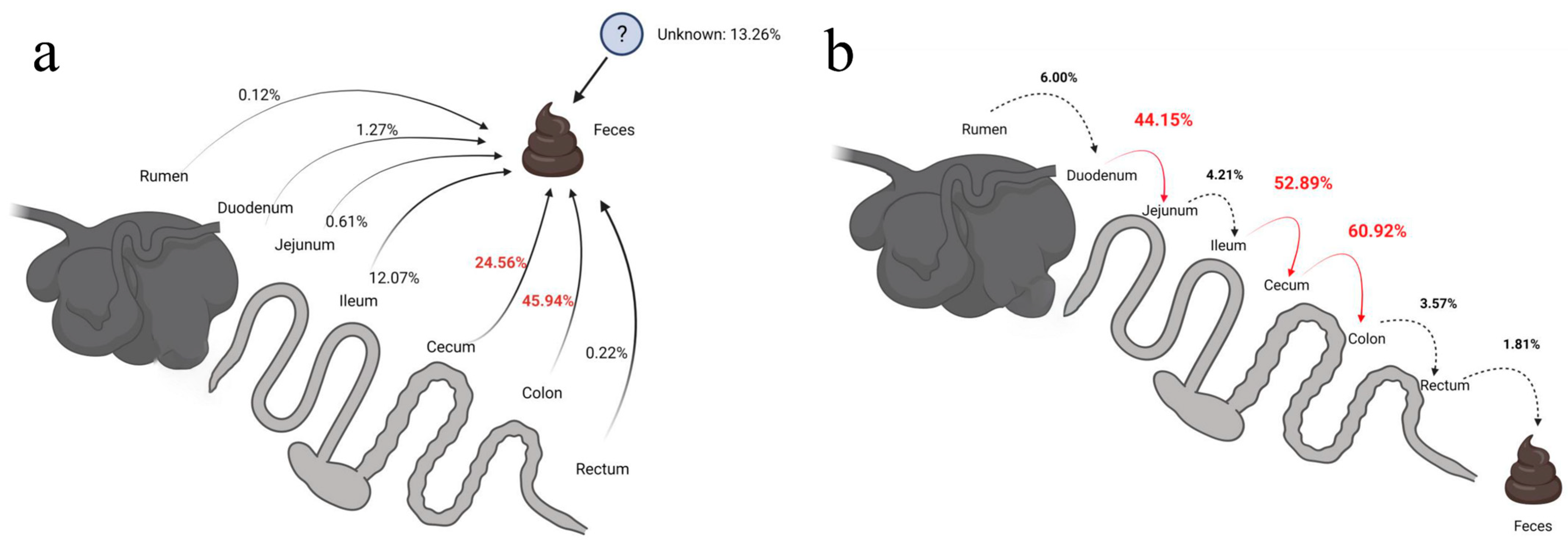
3.4. Source-Tracking Analysis
3.5. The Ecological Assembly Processes of Gut Microbiota in Eight Intestinal Parts
4. Discussion
4.1. The Digestive Process May Contribute to Fecal Microbiota Being More Similar to the Cecum and Colon
4.2. Main Factors That Influence the Contribution of Fecal Microbiota That Originated from the Rectum
4.3. Main Factors That Would Affect the Similarities between Fecal and Other Intestinal Parts in Microbiota
4.4. The Ecological Assembly Processes on the Gut Microbiota Communities among Eight Intestinal Parts
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Df | SumsOfSqs | MeanSqs | F.Models | R2 | Pr (>F) |
|---|---|---|---|---|---|---|
| Fec_Cec | 1 | 0.208128 | 0.208128 | 1.244843 | 0.037445 | 0.109 |
| Residuals | 32 | 5.350159 | 0.167192 | - | 0.962555 | - |
| Total | 33 | 5.558287 | - | - | 1 | - |
| Fec_Col | 1 | 0.09718 | 0.09718 | 0.59051 | 0.018693 | 0.977 |
| Residuals | 31 | 5.101666 | 0.16457 | - | 0.981307 | - |
| Total | 32 | 5.198846 | - | - | 1 | - |
References
- Ding, B.A.; Ma, S.Q.; Li, Z.R.; Li, X.L.; Madigosky, S.R. Seasonal Changes of Rumen and Intestine Morphology of the Qinghai Yak (Bos Grunniens). Vet. World 2018, 11, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Cui, Z.; Chen, X.; Wang, P.; Yao, J. Changed Caecal Microbiota and Fermentation Contribute to the Beneficial Effects of Early Weaning with Alfalfa Hay, Starter Feed, and Milk Replacer on the Growth and Organ Development of Yak Calves. Animals 2019, 9, 921. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Wu, S.; Li, J.; Yang, Q.-E.; Chai, S.; Wang, L.; Wang, X.; Zhang, X.; Liu, S.; Yao, J. Effect of Alfalfa Hay and Starter Feeding Intervention on Gastrointestinal Microbial Community, Growth and Immune Performance of Yak Calves. Front. Microbiol. 2020, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Rudi, K.; Moen, B.; Sekelja, M.; Frisli, T.; Lee, M.R. An Eight-Year Investigation of Bovine Livestock Fecal Microbiota. Vet. Microbiol. 2012, 160, 369–377. [Google Scholar] [CrossRef]
- Guo, W.; Zhou, M.; Ma, T.; Bi, S.; Wang, W.; Zhang, Y.; Huang, X.; Guan, L.L.; Long, R. Survey of Rumen Microbiota of Domestic Grazing Yak during Different Growth Stages Revealed Novel Maturation Patterns of Four Key Microbial Groups and Their Dynamic Interactions. Anim. Microbiome 2020, 2, 23. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Zhang, X.X.; Li, F.D.; Li, C.; Li, G.Z.; Zhang, D.Y.; Song, Q.Z.; Li, X.L.; Zhao, Y.; Wang, W.M. Characterization of the Rumen Microbiota and Its Relationship with Residual Feed Intake in Sheep. Animal 2021, 15, 100161. [Google Scholar] [CrossRef]
- Guan, Y.; Yang, H.; Han, S.; Feng, L.; Wang, T.; Ge, J. Comparison of the Gut Microbiota Composition between Wild and Captive Sika Deer (Cervus Nippon Hortulorum) from Feces by High-Throughput Sequencing. AMB Express 2017, 7, 212. [Google Scholar] [CrossRef]
- Rodriguez, C.; Taminiau, B.; Brevers, B.; Avesani, V.; Broeck, J.V.; Leroux, A.; Gallot, M.; Bruwier, A.; Amory, H.; Delmee, M.; et al. Faecal Microbiota Characterisation of Horses Using 16 Rdna Barcoded Pyrosequencing, and Carriage Rate of Clostridium Difficile at Hospital Admission. BMC Microbiol. 2015, 15, 1–14. [Google Scholar] [CrossRef]
- Fan, Q.; Wanapat, M.; Hou, F. Rumen Bacteria Influence Milk Protein Yield of Yak Grazing on the Qinghai-Tibet Plateau. Asian-Australas. J. Anim. Sci. 2021, 34, 1466–1478. [Google Scholar] [CrossRef]
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; et al. The Yak Genome and Adaptation to Life at High Altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef]
- Guo, N.; Wu, Q.; Shi, F.; Niu, J.; Zhang, T.; Degen, A.A.; Fang, Q.; Ding, L.; Shang, Z.; Zhang, Z.; et al. Seasonal Dynamics of Diet-Gut Microbiota Interaction in Adaptation of Yaks to Life at High Altitude. Npj Biofilms Microbiomes 2021, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.; Zhang, C.; Ji, Q.; Li, C.; Hu, Y.; Wang, J. Study on Meat Production and Quality Properties of Leiwuqi Yak. Hubei Agric. Sci. 2017, 56, 501–505. [Google Scholar]
- Zhong, M.; Wang, J.; Chai, Z.; Wang, H.; Wang, J.; Wu, Z.; Xin, J.; Zhang, C.; Ji, Q.; Zhong, J. Cloning and Tissue Expression of C1QA, C1QB and C1QC Genes in Yak. Acta Agric. Boreali-Sin. 2020, 35, 211–221. [Google Scholar]
- Liu, C.; Wu, H.; Liu, S.; Chai, S.; Meng, Q.; Zhou, Z. Dynamic Alterations in Yak Rumen Bacteria Community and Metabolome Characteristics in Response to Feed Type. Front. Microbiol. 2019, 10, 1116. [Google Scholar] [CrossRef]
- Cui, Z.; Wu, S.; Liu, S.; Sun, L.; Feng, Y.; Cao, Y.; Chai, S.; Zhang, G.; Yao, J. From Maternal Grazing to Barn Feeding During Pre-Weaning Period: Altered Gastrointestinal Microbiota Contributes to Change the Development and Function of the Rumen and Intestine of Yak Calves. Front. Microbiol. 2020, 11, 485. [Google Scholar] [CrossRef]
- Fan, Q.; Wanapat, M.; Yan, T.; Hou, F. Altitude Influences Microbial Diversity and Herbage Fermentation in the Rumen of Yaks. BMC Microbiol. 2020, 20, 370. [Google Scholar] [CrossRef]
- Hume, I. Digestive Strategies of Mammals. Acta Zool. Sin. 2002, 48, 1–19. [Google Scholar]
- Romero-Pérez, G.A.; Ominski, K.H.; McAllister, T.A.; Krause, D.O. Effect of Environmental Factors and Influence of Rumen and Hindgut Biogeography on Bacterial Communities in Steers. Appl. Environ. Microbiol. 2011, 77, 258–268. [Google Scholar] [CrossRef]
- Guo, W.; Li, Y.; Wang, L.; Wang, J.; Xu, Q.; Yan, T.; Xue, B. Evaluation of Composition and Individual Variability of Rumen Microbiota in Yaks by 16S RRNA High-Throughput Sequencing Technology. Anaerobe 2015, 34, 74–79. [Google Scholar] [CrossRef]
- Xue, D.; Chen, H.; Luo, X.; Guan, J.; He, Y.; Zhao, X. Microbial Diversity in the Rumen, Reticulum, Omasum, and Abomasum of Yak on a Rapid Fattening Regime in an Agro-Pastoral Transition Zone. J. Microbiol. 2018, 56, 734–743. [Google Scholar] [CrossRef]
- Xin, J.; Ji, Q. Research Progress of Yak Rumen and Intestinal Microbial Diversity and Influcing Factors. Acta Ecol. Anim. Domastici 2021, 42, 1–7. [Google Scholar]
- Yang, C.; Ding, X.; Qian, J.; Wu, X.; Liang, C.; Bao, P.; Long, R.; Yan, P. Research Progress on Adaptation on the Histology and Anatomy in Yak (Bos Grunniens ) in Qinghai-Tibetan Plateau. Chin. J. Anim. Sci. 2017, 53, 18–24. [Google Scholar]
- Yao, F. Determination of Intestinal Anatomical Indices in Plateau Yak. J. Anhui Agric. Sci. 2014, 42, 8623–8624. [Google Scholar]
- Li, D. Study on the Histological Structure and Distributive Changes of Mucosal Immunity-Associated Cells in Large Intestine of Yak. Master’s Thesis, Gansu Agriculture University, Lanzhou, China, 2013. [Google Scholar]
- Tian, B.; Li, D.; Fang, M.; Jia, N. Obervation on the Distribution of Mucosal Immunity-Associated Cells and Microstructure of Yak Large Intestine. Chin. J. Vet. Sci. 2014, 34, 1989–1994. [Google Scholar]
- Ma, J.; Zhu, Y.; Wang, Z.; Yu, X.; Hu, R.; Wang, X.; Cao, G.; Zou, H.; Shah, A.M.; Peng, Q.; et al. Comparing the Bacterial Community in the Gastrointestinal Tracts Between Growth-Retarded and Normal Yaks on the Qinghai–Tibetan Plateau. Front. Microbiol. 2020, 11, 600516. [Google Scholar] [CrossRef]
- Meale, S.J.; Li, S.; Azevedo, P.; Derakhshani, H.; Plaizier, J.C.; Khafipour, E.; Steele, M.A. Development of Ruminal and Fecal Microbiomes Are Affected by Weaning but not Weaning Strategy in Dairy Calves. Front. Microbiol. 2016, 7, 582. [Google Scholar] [CrossRef]
- Quan, J.; Cai, G.; Yang, M.; Zeng, Z.; Ding, R.; Wang, X.; Zhuang, Z.; Zhou, S.; Li, S.; Yang, H.; et al. Exploring the Fecal Microbial Composition and Metagenomic Functional Capacities Associated with Feed Efficiency in Commercial DLY Pigs. Front. Microbiol. 2019, 10, 52. [Google Scholar] [CrossRef]
- Gong, G.; Zhou, S.; Luo, R.; Gesang, Z.; Suolang, S. Metagenomic Insights into the Diversity of Carbohydrate-Degrading Enzymes in the Yak Fecal Microbial Community. BMC Microbiol. 2020, 20, 302. [Google Scholar] [CrossRef]
- Fu, H.; Zhang, L.; Fan, C.; Liu, C.; Li, W.; Li, J.; Zhao, X.; Jia, S.; Zhang, Y. Domestication Shapes the Community Structure and Functional Metagenomic Content of the Yak Fecal Microbiota. Front. Microbiol. 2021, 12, 594075. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Q.; Song, J.; Xin, J.; Zhang, S.; Lei, Y.; Yang, Y.; Xie, P.; Suo, H. Comparison of Gut Microbiota of Yaks from Different Geographical Regions. Front. Microbiol. 2021, 12, 666940. [Google Scholar] [CrossRef]
- Menke, S.; Meier, M.; Mfune, J.K.E.; Melzheimer, J.; Wachter, B.; Sommer, S. Effects of Host Traits and Land-Use Changes on the Gut Microbiota of the Namibian Black-Backed Jackal (Canis Mesomelas). FEMS Microbiol. Ecol. 2017, 93, fix123. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Piazuelo, D.; Migura-Garcia, L.; Estelle, J.; Criado-Mesas, L.; Revilla, M.; Castello, A.; Munoz, M.; Garcia-Casco, J.M.; Fernandez, A.I.; Ballester, M.; et al. Association between the Pig Genome and Its Gut Microbiota Composition. Sci. Rep. 2019, 9, 8791. [Google Scholar] [CrossRef] [PubMed]
- Daniel, H.; Gholami, A.M.; Berry, D.; Desmarchelier, C.; Hahne, H.; Loh, G.; Mondot, S.; Lepage, P.; Rothballer, M.; Walker, A.; et al. High-Fat Diet Alters Gut Microbiota Physiology in Mice. ISME J. 2014, 8, 295–308. [Google Scholar] [CrossRef]
- Han, G.; Lee, H.J.; Jeong, S.E.; Jeon, C.O.; Hyun, S. Comparative Analysis of Drosophila Melanogaster Gut Microbiota with Respect to Host Strain, Sex, and Age. Microb. Ecol. 2017, 74, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.; Kim, N. Roles of Sex Hormones and Gender in the Gut Microbiota. J. Neurogastroenterol. Motil. 2021, 27, 314–325. [Google Scholar] [CrossRef]
- Ren, T.; Boutin, S.; Humphries, M.M.; Dantzer, B.; Gorrell, J.C.; Coltman, D.W.; McAdam, A.G.; Wu, M. Seasonal, Spatial, and Maternal Effects on Gut Microbiome in Wild Red Squirrels. Microbiome 2017, 5, 163. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Gaskins, H.R.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat Degradation Impacts Black Howler Monkey (Alouatta Pigra) Gastrointestinal Microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef]
- Linnenbrink, M.; Wang, J.; Hardouin, E.A.; Kunzel, S.; Metzler, D.; Baines, J.F. The Role of Biogeography in Shaping Diversity of the Intestinal Microbiota in House Mice. Mol. Ecol. 2013, 22, 1904–1916. [Google Scholar] [CrossRef]
- Bergmann, G.T.; Craine, J.M.; Robeson, M.S.; Fierer, N. Seasonal Shifts in Diet and Gut Microbiota of the American Bison (Bison Bison). PLoS ONE 2015, 10, e0142409. [Google Scholar] [CrossRef]
- Moeller, A.H.; Peeters, M.; Ndjango, J.-B.; Li, Y.; Hahn, B.H.; Ochman, H. Sympatric Chimpanzees and Gorillas Harbor Convergent Gut Microbial Communities. Genome Res. 2013, 23, 1715–1720. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, X.; Liang, C.; Guo, X.; Ding, X.; Chu, M.; Wang, H.; Pei, J.; Bao, P.; Yan, P. Bibliometric Analysis of Yak Based on the China National Knowledge Infrastructure Database. Pratacultural Sci. 2019, 36, 2151–2158. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Ii, M.S.R.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible Sequence Taxonomy Reference Database Management. PLoS Comput. Biol. 2021, 17, e1009581. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Community Ecology Package. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 22 December 2021).
- Wickham, H.; Chang, W.; Henry, L.; Pedersen, T.L.; Takahashi; Wilke, C.; Woo, K.; Yutani, H. Create Elegant Data Visualisations Using the Grammar of Graphics. 2019. Available online: https://ggplot2.tidyverse.org/reference/ggplot2-package.html (accessed on 22 December 2021).
- Ning, D.; Deng, Y.; Tiedje, J.M.; Zhou, J. A General Framework for Quantitatively Assessing Ecological Stochasticity. Proc. Natl. Acad. Sci. USA 2019, 116, 16892–16898. [Google Scholar] [CrossRef]
- Mo, Y.; Peng, F.; Gao, X.; Xiao, P.; Logares, R.; Jeppesen, E.; Ren, K.; Xue, Y.; Yang, J. Low Shifts in Salinity Determined Assembly Processes and Network Stability of Microeukaryotic Plankton Communities in a Subtropical Urban Reservoir. Microbiome 2021, 9, 128. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Zhang, P.; Xue, K.; Liang, Y.; Van Nostrand, J.D.; Yang, Y.; He, Z.; Wu, L.; Stahl, D.A.; et al. Stochasticity, Succession, and Environmental Perturbations in a Fluidic Ecosystem. Proc. Natl. Acad. Sci. USA 2014, 111, E836–E845. [Google Scholar] [CrossRef]
- Stone, L.; Roberts, A. The Checkerboard Score and Species Distributions. Oecologia 1990, 85, 74–79. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Yang, S.; Zhou, J.; Zhang, T.; Qi, L.; Sun, X.; Fan, M.; Xu, S.; Cha, M.; et al. Comparative Analysis of the Gut Microbiota Composition between Captive and Wild Forest Musk Deer. Front. Microbiol. 2017, 8, 1705. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, M.; Xue, J.; Huang, J.; Zhuang, R.; Zhou, X.; Zhang, H.; Fu, Q.; Hao, Y. Body Mass Index Differences in the Gut Microbiota Are Gender Specific. Front. Microbiol. 2018, 9, 1250. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Y.; Dong, L.; Hu, H.; Meng, L.; Liu, H.; Zheng, N.; Wang, J. Microbial Characteristics and Safety of Dairy Manure ComPosting for Reuse as Dairy Bedding. Biology 2021, 10, 13. [Google Scholar] [CrossRef]
- Jiao, J.-Z.; Wang, P.-P.; Tang, S.X.; Zhou, C.-S.; Tan, Z.-L. Quantity and Distribution Characteristics of Functional Microorganisms in Gastrointestinal Tract of Liuyang Black Goats. Acta Vet. Zootech. Sin. 2013, 44, 1590–1599. [Google Scholar]
- Li, Y.; Song, H.; Su, S.; Qirigeri; Tao, J.; Zhang, J.; Dugarjaviin, M.; Zhao, Y. Analysis of Screening, Identification and Enzymatic Characteristics for Cellulolytic Bacteria in Mongolian Horse Cecum. Feed Ind. 2022, 43, 52–58. [Google Scholar]
- Birchenough, G.M.H.; Johansson, M.E.V. Forming a Mucus Barrier along the Colon. Science 2020, 370, 402–403. [Google Scholar] [CrossRef]
- Wang, J.; Fan, H.; Han, Y.; Zhao, J.; Zhou, Z. Characterization of the Microbial Communities along the Gastrointestinal Tract of Sheep by 454 Pyrosequencing Analysis. Asian-Australas. J. Anim. Sci. 2017, 30, 100–110. [Google Scholar] [CrossRef]
- Zhang, H.; Shao, M.; Huang, H.; Wang, S.; Ma, L.; Wang, H.; Hu, L.; Wei, K.; Zhu, R. The Dynamic Distribution of Small-Tail Han Sheep Microbiota across Different Intestinal Segments. Front. Microbiol. 2018, 9, 32. [Google Scholar] [CrossRef]
- Mao, S.; Zhang, M.; Liu, J.; Zhu, W. Characterising the Bacterial Microbiota across the Gastrointestinal Tracts of Dairy Cattle: Membership and Potential Function. Sci. Rep. 2015, 5, 16116. [Google Scholar] [CrossRef]
- Vrieze, A.; Holleman, F.; Zoetendal, E.G.; de Vos, W.M.; Hoekstra, J.B.; Nieuwdorp, M. The Environment within: How Gut Microbiota May Influence Metabolism and Body Composition. Diabetologia 2010, 53, 606–613. [Google Scholar] [CrossRef]
- Li, H.; Zhou, R.; Zhu, J.; Huang, X.; Qu, J. Environmental Filtering Increases with Elevation for the Assembly of Gut Microbiota in Wild Pikas. Microb. Biotechnol. 2019, 12, 976–992. [Google Scholar] [CrossRef]
- Grieneisen, L.; Charpentier, M.; Alberts, S.; Blekhman, R.; Bradburd, G.; Tung, J.; Archie, E. Genes, Geology and Germs: Gut Microbiota across a Primate Hybrid Zone Are Explained by Site Soil Properties, not Host Species. Proc. R. Soc. B Biol. Sci. 2019, 286, 20190431. [Google Scholar] [CrossRef]
- Thomas, N.A.; Olvera-Ramirez, A.M.; Abecia, L.; Adam, C.L.; Edwards, J.E.; Cox, G.F.; Findlay, P.A.; Destables, E.; Wood, T.A.; McEwan, N.R. Characterisation of the Effect of Day Length, and Associated Differences in Dietary Intake, on the Gut Microbiota of Soay Sheep. Arch. Microbiol. 2019, 201, 889–896. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of Diet in Shaping Gut Microbiota Revealed by a Comparative Study in Children from Europe and Rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef]
| Intestinal Parts | Cellular Processes | Environmental Information Processing | Genetic Information Processing | Human Diseases | Metabolism | Organismal Systems |
|---|---|---|---|---|---|---|
| Rum | 3.7001% * | 3.9605% * | 8.7812% | 3.0962% * | 78.6070% * | 1.8550% * |
| Duo | 4.4970% * | 5.3893% | 9.1073% * | 3.0120% * | 76.3568% * | 1.6376% * |
| Jej | 4.5849% | 5.5159% * | 8.9574% | 3.0152% * | 76.2777% * | 1.6489% * |
| Ile | 4.2582% * | 4.9648% | 8.6554% * | 3.2195% | 77.0959% | 1.8063% |
| Cec | 4.3132% | 4.8865% | 8.7487% | 3.1709% | 77.0875% | 1.7933% |
| Col | 4.3304% | 4.9005% | 8.7659% | 3.1921% | 77.0174% | 1.7938% |
| Rec | 4.4378% | 5.2128% * | 8.8489% | 3.0839% * | 76.7052% * | 1.7114% * |
| Fec | 4.2829% | 4.8426% | 8.7704% | 3.1813% | 77.1327% | 1.7901% |
| Intestinal Parts | Metabolic Pathways | Biosynthesis of Secondary Metabolites | Biosynthesis of Amino Acids | Microbial Metabolism in Diverse Environments | Carbon Metabolism |
|---|---|---|---|---|---|
| Rum | 47.7223% * | 23.7620% * | 10.8200% * | 10.6050% * | 7.0907% * |
| Duo | 45.9547% * | 23.6977% * | 11.9351% | 11.2317% * | 7.1808% * |
| Jej | 46.0799% * | 23.6476% | 11.9187% | 11.2531% * | 7.1007% * |
| Ile | 46.7765% | 23.4297% | 11.0974% * | 11.3556% | 7.3408% * |
| Cec | 46.6225% | 23.4675% | 11.2990% | 11.2321% | 7.3790% |
| Col | 46.5940% | 23.4863% | 11.3051% | 11.2381% | 7.3765% |
| Rec | 46.4532% * | 23.5800% | 11.5579% | 11.2289% * | 7.1800% * |
| Fec | 46.6905% | 23.4640% | 11.2265% | 11.2476% | 7.3714% |
| Phenotypes | Rumen | Duodenum | Jejunum | Ileum | Cecum | Colon | Rectum | Fecal | p-Value |
|---|---|---|---|---|---|---|---|---|---|
| Stress_Tolerant | 22.81% | 21.94% | 21.90% | 22.77% | 23.35% | 23.57% | 22.03% * | 23.24% | 4.452 × 10−8 |
| Contains_Mobile_Elements | 13.93% | 24.18% | 23.44% | 19.50% | 19.94% | 19.90% | 21.33% * | 19.65% | 5.738 × 10−16 |
| Anaerobic | 20.21% | 17.47% | 18.43% | 18.58% | 18.73% | 18.72% | 17.92% | 18.62% | 3.318 × 10−5 |
| Gram_Positive | 10.45% | 21.29% | 20.98% | 17.47% | 18.79% | 19.06% | 18.60% | 18.31% | 1.259 × 10−13 |
| Potentially_Pathogenic | 15.84% | 6.68% | 7.05% | 11.86% | 11.74% | 11.44% | 9.65% | 11.46% | 1.507 × 10−15 |
| Gram_Negative | 14.75% | 3.20% | 2.79% | 6.71% | 5.48% | 5.38% | 5.66% | 6.33% | 2.81 × 10−13 |
| Forms_Biofilms | 1.31% | 3.76% | 4.04% | 1.80% | 1.07% | 1.05% | 3.27% * | 1.39% | 3.857 × 10−7 |
| Aerobic | 0.23% | 1.03% | 0.88% | 0.56% | 0.50% | 0.35% | 0.72% | 0.52% | 0.0010 |
| Facultatively_Anaerobic | 0.47% | 0.46% | 0.49% | 0.75% | 0.40% | 0.53% | 0.82% | 0.49% | 0.2504 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, W.; Song, P.; Li, J.; Xie, J.; Zhang, S. Representativeness of Fecal Microbiota Is Limited to Cecum and Colon in Domestic Yak. Sustainability 2022, 14, 10263. https://doi.org/10.3390/su141610263
Qin W, Song P, Li J, Xie J, Zhang S. Representativeness of Fecal Microbiota Is Limited to Cecum and Colon in Domestic Yak. Sustainability. 2022; 14(16):10263. https://doi.org/10.3390/su141610263
Chicago/Turabian StyleQin, Wen, Pengfei Song, Jirong Li, Jiuxiang Xie, and Shoudong Zhang. 2022. "Representativeness of Fecal Microbiota Is Limited to Cecum and Colon in Domestic Yak" Sustainability 14, no. 16: 10263. https://doi.org/10.3390/su141610263