
A Comprehensive Metagenomic Analysis Framework Revealing Microbiome Profile and Potential for Hydrocarbon Degradation and Carbohydrate Metabolism in a Himalayan Artificial Lake


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Extraction of Metagenomic DNA
2.2. Metagenome Sequencing, Assembly, and Elementary Analysis
2.3. Taxonomic and Functional Profiling
2.4. Exploration of Hydrocarbon Degradation and Carbohydrate Metabolism Pathways
2.5. Statistical Comparison with Other Lake Metagenomes
2.6. ICP-MS Analysis
3. Results and Discussion
3.1. Site Description and Physicochemical Analysis
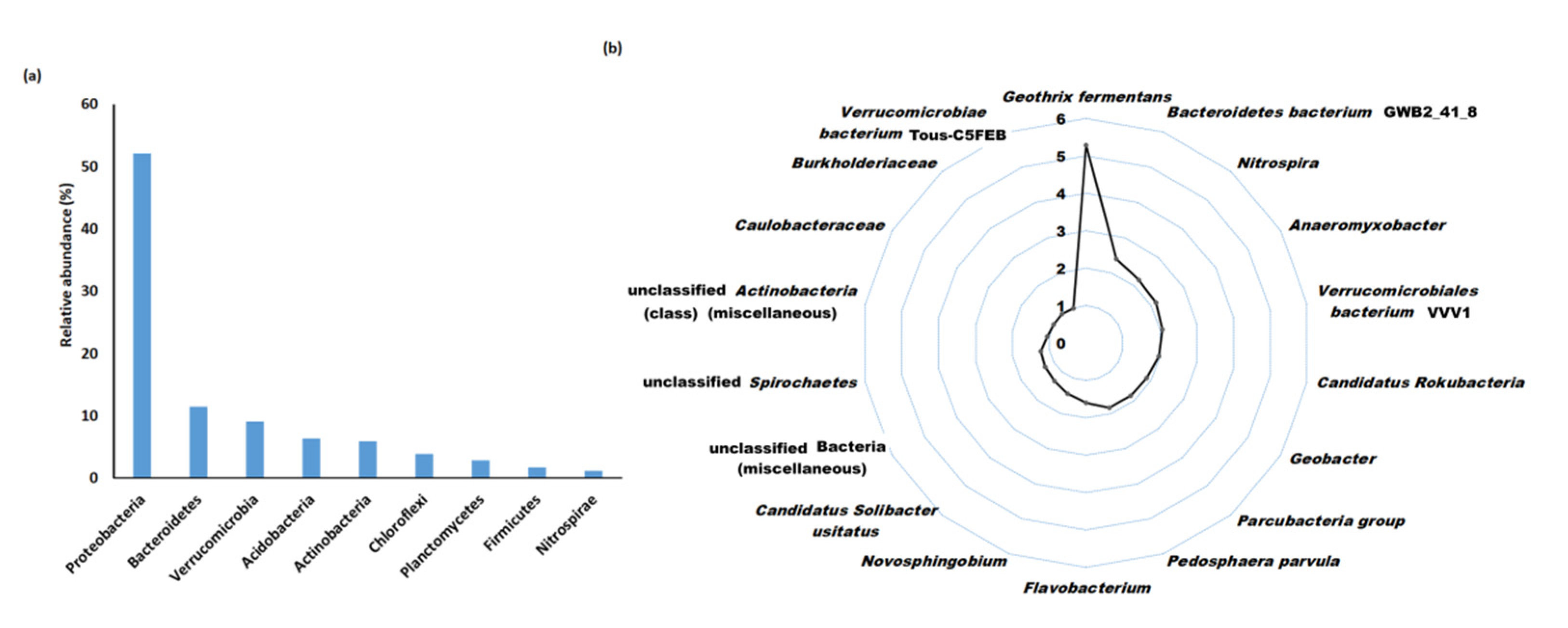
3.2. Assembly Statistics and Taxonomic Distribution
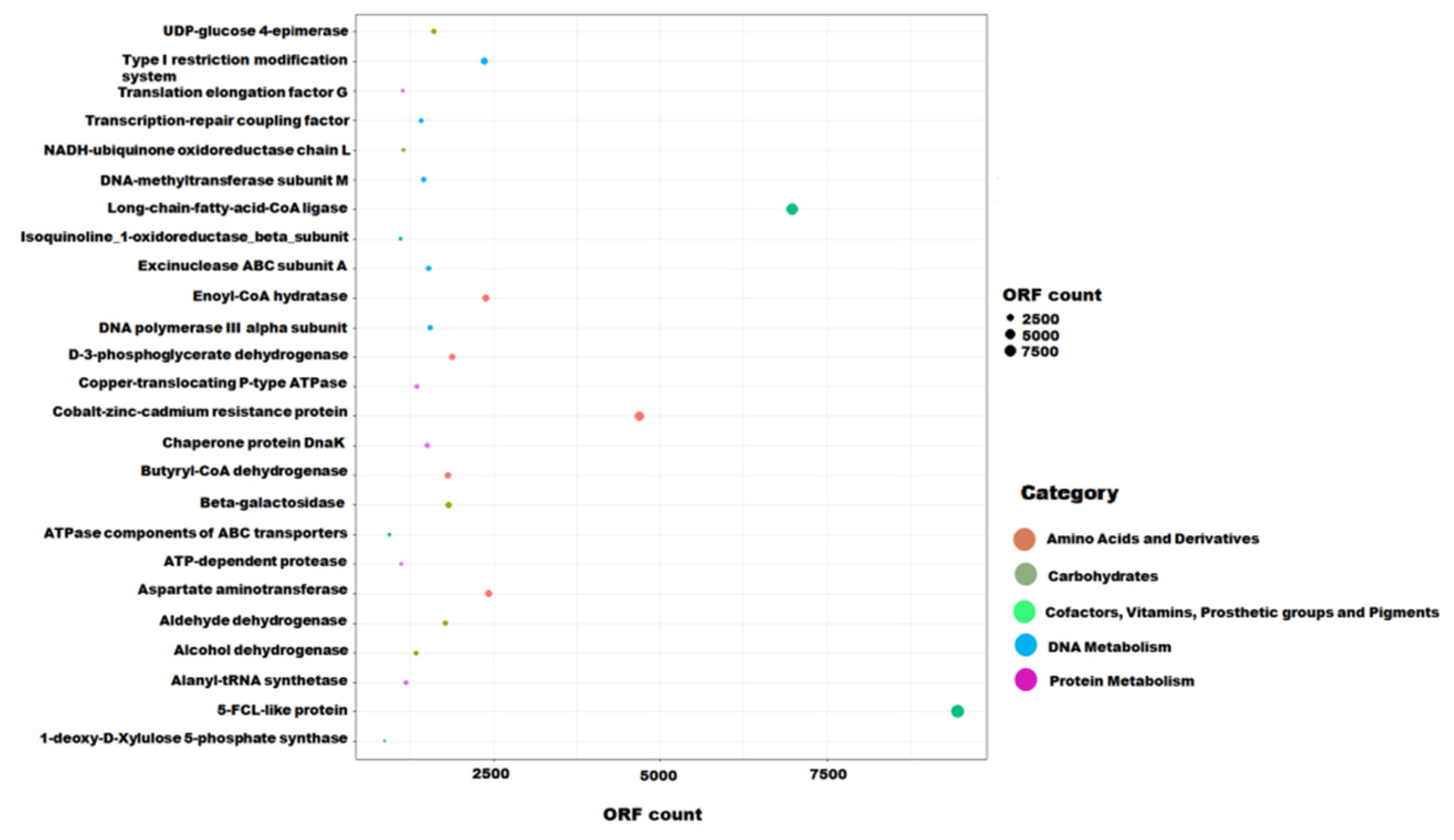
3.3. Functional Potential
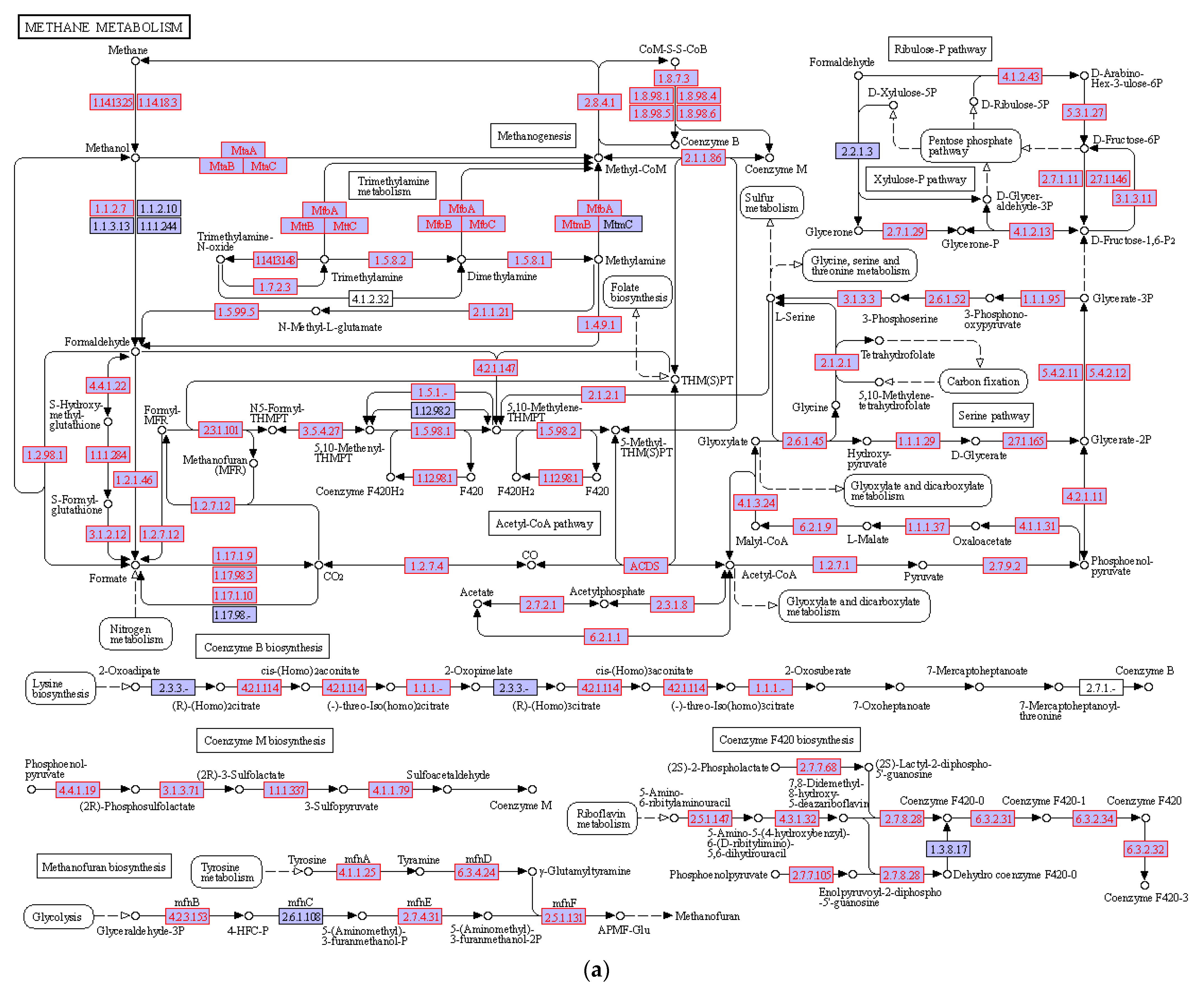
3.4. Hydrocarbon Degradation Pathway
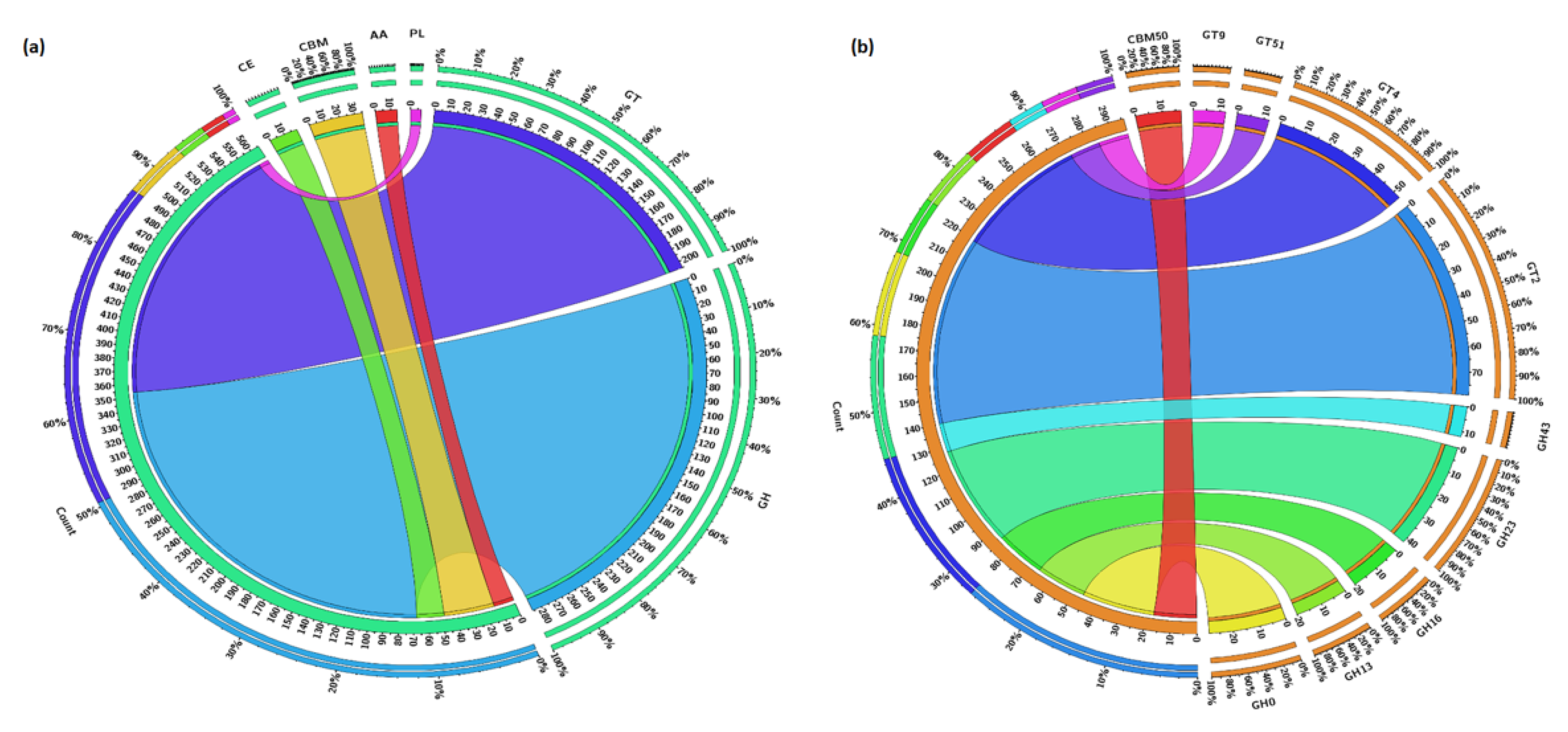
3.5. Carbohydrate Metabolism
3.6. Antibiotic Resistance Genes
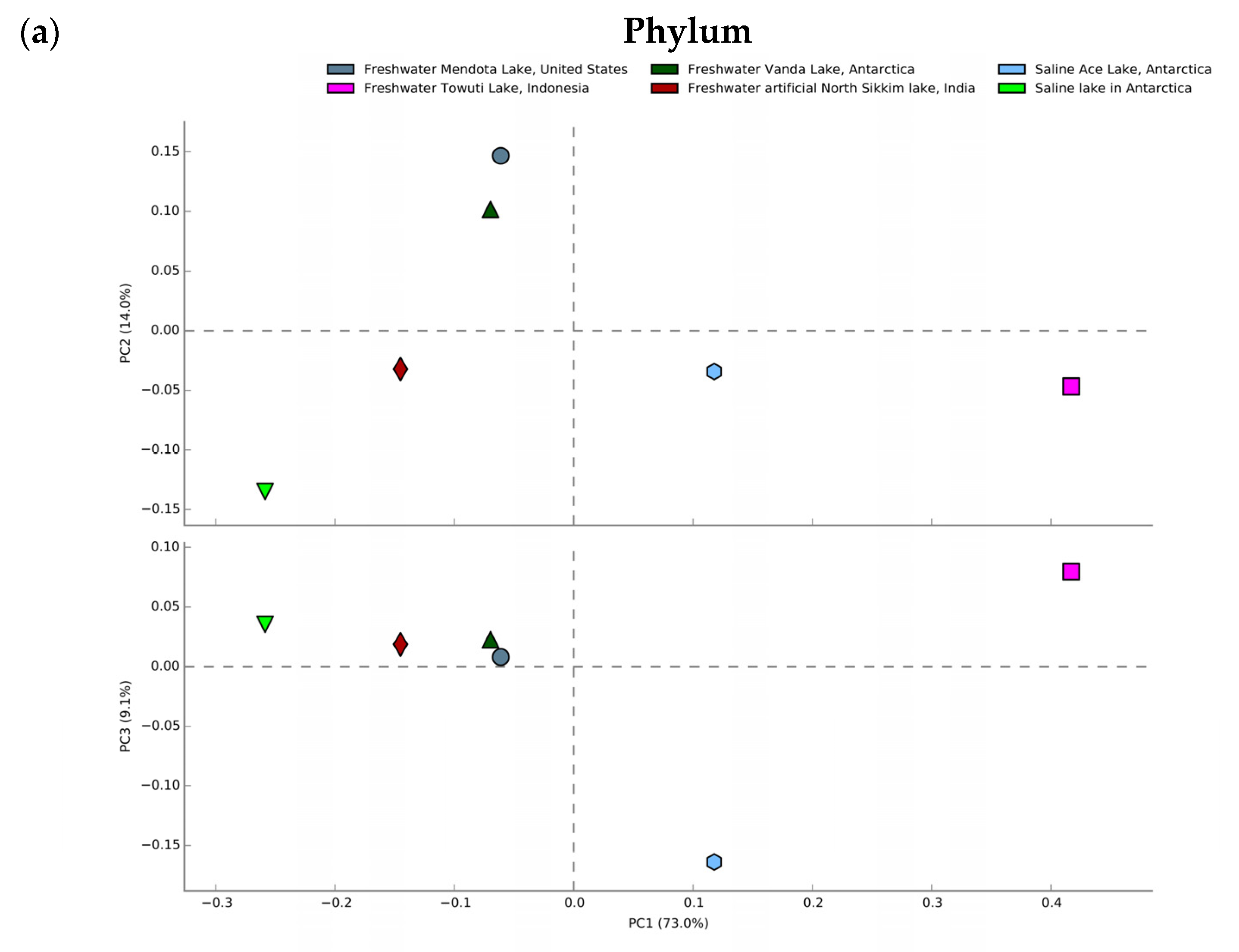
3.7. Comparative Metagenomics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Badar, B.; Romshoo, S.A.; Khan, M.A. Integrating biophysical and socioeconomic information for prioritizing watersheds in a Kashmir Himalayan lake: A remote sensing and GIS approach. Environ. Monit. Assess. 2013, 185, 6419–6445. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Gupta, G.; Sharma, A.; Kaur, B.; El-Sheikh, M.A.; Alyemeni, M.N. Metagenomic analysis exploring taxonomic and functional diversity of bacterial communities of a Himalayan urban fresh water lake. PLoS ONE 2021, 16, e0248116. [Google Scholar] [CrossRef] [PubMed]
- Skvortsov, T.; De Leeuwe, C.; Quinn, J.P.; McGrath, J.W.; Allen, C.C.R.; McElarney, Y.; Watson, C.; Arkhipova, K.; Lavigne, R.; Kulakov, L.A. Metagenomic characterisation of the viral community of Lough Neagh, the largest freshwater lake in Ireland. PLoS ONE 2016, 11, e0150361. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.; Jeon, J.H.; Kang, I.; Park, K.S.; Lee, K.; Cha, C.-J.; Lee, S.H.; Cho, J.-C. Freshwater viral metagenome reveals novel and functional phage-borne antibiotic resistance genes. Microbiome 2020, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rathour, R.; Gupta, J.; Mishra, A.; Rajeev, A.C.; Dupont, C.L.; Thakur, I.S. A comparative metagenomic study reveals microbial diversity and their role in the biogeochemical cycling of Pangong lake. Sci. Total Environ. 2020, 731, 139074. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Bhattacharjee, A. Molecular profiling of microbial community structure and their CAZymes via metagenomics, from Tsomgo lake in the Eastern Himalayas. Arch. Microbiol. 2021, 203, 3135–3146. [Google Scholar] [CrossRef]
- Vavourakis, C.D.; Ghai, R.; Rodriguez-Valera, F.; Sorokin, D.Y.; Tringe, S.G.; Hugenholtz, P.; Muyzer, G. Metagenomic insights into the uncultured diversity and physiology of microbes in four hypersaline soda lake brines. Front. Microbiol. 2016, 7, 211. [Google Scholar] [CrossRef]
- Chakraborty, J.; Sapkale, V.; Shah, M.; Rajput, V.; Mehetre, G.; Agawane, S.; Kamble, S.; Dharne, M. Metagenome sequencing to unveil microbial community composition and prevalence of antibiotic and metal resistance genes in hypersaline and hyperalkaline Lonar Lake, India. Ecol. Indic. 2020, 110, 105827. [Google Scholar] [CrossRef]
- Panwar, P.; Allen, M.A.; Williams, T.J.; Hancock, A.M.; Brazendale, S.; Bevington, J.; Roux, S.; Páez-Espino, D.; Nayfach, S.; Berg, M. Influence of the polar light cycle on seasonal dynamics of an Antarctic lake microbial community. Microbiome 2020, 8, 1–24. [Google Scholar] [CrossRef]
- Liang, H.; Wang, F.; Mu, R.; Huang, J.; Zhao, R.; Li, X.; Yu, K.; Li, B. Metagenomics analysis revealing the occurrence of antibiotic resistome in salt lakes. Sci. Total Environ. 2021, 790, 148262. [Google Scholar] [CrossRef]
- Phukon, L.C.; Chourasia, R.; Kumari, M.; Godan, T.K.; Sahoo, D.; Parameswaran, B.; Rai, A.K. Production and characterisation of lipase for application in detergent industry from a novel Pseudomonas helmanticensis HS6. Bioresour. Technol. 2020, 309, 123352. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Sharma, N.; Rai, A.K.; Singh, S.P. A novel cold-active type I pullulanase from a hot-spring metagenome for effective debranching and production of resistant starch. Bioresour. Technol. 2021, 320, 124288. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Sahoo, D.; Rai, A.K.; Singh, S.P. A highly alkaline pectate lyase from the Himalayan hot spring metagenome and its bioscouring applications. Process Biochem. 2022, 115, 100–109. [Google Scholar] [CrossRef]
- Singh, A.K.; Kumari, M.; Sharma, N.; Rai, A.K.; Singh, S.P. Metagenomic views on taxonomic and functional profiles of the Himalayan Tsomgo cold lake and unveiling its deterzome potential. Curr. Genet. 2022, 1–15. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Noguchi, H.; Taniguchi, T.; Itoh, T. MetaGeneAnnotator: Detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 2008, 15, 387–396. [Google Scholar] [CrossRef]
- Mount, D.W. Using the basic local alignment search tool (BLAST). Cold Spring Harb. Protoc. 2007, 2007, 17. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2016, 45, 566–573. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active enZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.M.A.; Chu, K.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Huntemann, M.; Varghese, N.; White, J.R.; Seshadri, R. IMG/M v. 5.0: An integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 2019, 47, D666–D677. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Coutinho, F.H.; Cabello-Yeves, P.J.; Gonzalez-Serrano, R.; Rosselli, R.; López-Pérez, M.; Zemskaya, T.I.; Zakharenko, A.S.; Ivanov, V.G.; Rodriguez-Valera, F. New viral biogeochemical roles revealed through metagenomic analysis of Lake Baikal. Microbiome 2020, 8, 163. [Google Scholar] [CrossRef]
- Debroas, D.; Humbert, J.-F.; Enault, F.; Bronner, G.; Faubladier, M.; Cornillot, E. Metagenomic approach studying the taxonomic and functional diversity of the bacterial community in a mesotrophic lake (Lac du Bourget–France). Environ. Microbiol. 2009, 11, 2412–2424. [Google Scholar] [CrossRef]
- Sharma, N.; Kumar, J.; Abedin, M.M.; Sahoo, D.; Pandey, A.; Rai, A.K.; Singh, S.P. Metagenomics revealing molecular profiling of community structure and metabolic pathways in natural hot springs of the Sikkim Himalaya. BMC Microbiol. 2020, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tran, P.Q.; Kieft, K.; Anantharaman, K. Genome diversification in globally distributed novel marine Proteobacteria is linked to environmental adaptation. ISME J. 2020, 14, 2060–2077. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.J.; Jones, S.E.; Eiler, A.; McMahon, K.D.; Bertilsson, S. A guide to the natural history of freshwater lake bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 14–49. [Google Scholar] [CrossRef]
- Nevin, K.P.; Lovley, D.R. Mechanisms for accessing insoluble Fe(III) oxide during dissimilatory Fe(III) reduction by Geothrix fermentans. Appl. Environ. Microbiol. 2002, 68, 2294–2299. [Google Scholar] [CrossRef] [PubMed]
- Coates, J.D.; Ellis, D.J.; Gaw, C.V.; Lovley, D.R. Geothrix fermentans gen. nov., sp. nov., a novel Fe(III)-reducing bacterium from a hydrocarbon-contaminated aquifer. Int. J. Syst. Bacteriol. 1999, 49 Pt 4, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Onley, J.R.; Ahsan, S.; Sanford, R.A.; Löffler, F.E. Denitrification by Anaeromyxobacter dehalogenans, a Common Soil Bacterium Lacking the Nitrite Reductase Genes nirS and nirK. Appl. Environ. Microbiol. 2018, 84, e01985-17. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.K.; Jindal, R.; Singh, U.B.; Ahluwalia, A.S. Plankton diversity and water quality assessment of three freshwater lakes of Mandi (Himachal Pradesh, India) with special reference to planktonic indicators. Environ. Monit. Assess. 2013, 185, 8355–8373. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, H.; Wang, L.; Liu, R.; Fu, L.; Lin, K. Unique bacterial communities and potential function along the vertical gradient in the deepest marine blue hole. Environ. Microbiol. Rep. 2021, 13, 911–927. [Google Scholar] [CrossRef]
- Kumar, S.; Zhou, J.; Li, M.; Xiang, H.; Zhao, D. Insights into the metabolism pathway and functional genes of long-chain aliphatic alkane degradation in haloarchaea. Extremophiles 2020, 24, 475–483. [Google Scholar] [CrossRef]
- Das, N.; Chandran, P. Microbial Degradation of Petroleum Hydrocarbon Contaminants: An Overview. Biotechnol. Res. Int. 2011, 2011, 941810. [Google Scholar] [CrossRef]
- Bastviken, D.; Cole, J.; Pace, M.; Tranvik, L. Methane emissions from lakes: Dependence of lake characteristics, two regional assessments, and a global estimate. Global Biogeochem. Cycles 2004, 18, 4. [Google Scholar] [CrossRef]
- Masekameni, M.D.; Moolla, R.; Gulumian, M.; Brouwer, D. Risk Assessment of Benzene, Toluene, Ethyl Benzene, and Xylene Concentrations from the Combustion of Coal in a Controlled Laboratory Environment. Int. J. Environ. Res. Public Health 2018, 16, 95. [Google Scholar] [CrossRef]
- Yan, Z.; Song, N.; Wang, C.; Jiang, H. Functional potential and assembly of microbes from sediments in a lake bay and adjoining river ecosystem for polycyclic aromatic hydrocarbon biodegradation. Environ. Microbiol. 2021, 23, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Derendorp, L.; Holzinger, R.; Röckmann, T. UV-induced emissions of C2–C5 hydrocarbons from leaf litter. Environ. Chem. 2011, 8, 602–611. [Google Scholar] [CrossRef]
- Guo, J.; Fang, J.; Cao, J. Characteristics of petroleum contaminants and their distribution in Lake Taihu, China. Chem. Cent. J. 2012, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wu, R.; Zhang, H.; Wu, H. Structural and biochemical analysis of a bacterial glycosyltransferase. Glycosyltransferases 2013, 1022, 29–39. [Google Scholar]
- Schmid, J.; Heider, D.; Wendel, N.J.; Sperl, N.; Sieber, V. Bacterial Glycosyltransferases: Challenges and Opportunities of a Highly Diverse Enzyme Class Toward Tailoring Natural Products. Front. Microbiol. 2016, 7, 182. [Google Scholar] [CrossRef]
- Yakovlieva, L.; Walvoort, M.T.C. Processivity in Bacterial Glycosyltransferases. ACS Chem. Biol. 2020, 15, 3–16. [Google Scholar] [CrossRef]
- Oehme, D.P.; Shafee, T.; Downton, M.T.; Bacic, A.; Doblin, M.S. Differences in protein structural regions that impact functional specificity in GT2 family β-glucan synthases. PLoS ONE 2019, 14, e0224442. [Google Scholar] [CrossRef]
- Palcic, M.M. Glycosyltransferases as biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 226–233. [Google Scholar] [CrossRef]
- Narita, S.I.; Tokuda, H. Bacterial lipoproteins; biogenesis, sorting and quality control. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2017, 1862, 1414–1423. [Google Scholar] [CrossRef]
- Takahashi-Íñiguez, T.; Aburto-Rodríguez, N.; Vilchis-González, A.L.; Flores, M.E. Function, kinetic properties, crystallization, and regulation of microbial malate dehydrogenase. J. Zhejiang Univ. Sci. B 2016, 17, 247–261. [Google Scholar] [CrossRef]
- Jenkins, C.H.; Wallis, R.; Allcock, N.; Barnes, K.B.; Richards, M.I.; Auty, J.M.; Galyov, E.E.; Harding, S.V.; Mukamolova, G.V. The lytic transglycosylase, LtgG, controls cell morphology and virulence in Burkholderia pseudomallei. Sci. Rep. 2019, 9, 11060. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Kumar Rai, A.; Singh, S.P. An acid-tolerant and cold-active β-galactosidase potentially suitable to process milk and whey samples. Appl. Microbiol. Biotechnol. 2022, 106, 3599–3610. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, W.; Lin, H.; Wang, W.; Du, L.; Xing, W. Antibiotics and antibiotic resistance genes in global lakes: A review and meta-analysis. Environ. Int. 2018, 116, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Ohore, O.E.; Addo, F.G.; Han, N.; Li, X.; Zhang, S. Profiles of ARGs and their relationships with antibiotics, metals and environmental parameters in vertical sediment layers of three lakes in China. J. Environ. Manag. 2020, 255, 109583. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Boulund, F.; Fick, J.; Kristiansson, E.; Larsson, D.G.J. Shotgun metagenomics reveals a wide array of antibiotic resistance genes and mobile elements in a polluted lake in India. Front. Microbiol. 2014, 5, 648. [Google Scholar] [CrossRef]
- Sharma, N.; Kumari, R.; Thakur, M.; Rai, A.K.; Singh, S.P. Molecular dissemination of emerging antibiotic, biocide, and metal co-resistomes in the Himalayan hot springs. J. Environ. Manag. 2022, 307, 114569. [Google Scholar] [CrossRef]
- Demanèche, S.; Kay, E.; Gourbière, F.; Simonet, P. Natural transformation of Pseudomonas fluorescens and Agrobacterium tumefaciens in soil. Appl. Environ. Microbiol. 2001, 67, 2617–2621. [Google Scholar] [CrossRef]
- Mesleh, M.F.; Rajaratnam, P.; Conrad, M.; Chandrasekaran, V.; Liu, C.M.; Pandya, B.A.; Hwang, Y.S.; Rye, P.T.; Muldoon, C.; Becker, B.; et al. Targeting Bacterial Cell Wall Peptidoglycan Synthesis by Inhibition of Glycosyltransferase Activity. Chem. Biol. Drug Des. 2016, 87, 190–199. [Google Scholar] [CrossRef]
- Sarkar, M.K.; Paul, K.; Blair, D. Chemotaxis signaling protein CheY binds to the rotor protein FliN to control the direction of flagellar rotation in Escherichia coli. Proc. Natl. Acad. Sci. USA 2010, 107, 9370. [Google Scholar] [CrossRef]
- Paget, M.S.; Helmann, J.D. The sigma70 family of sigma factors. Genome Biol. 2003, 4, 203. [Google Scholar] [CrossRef] [PubMed]
- Mosbahi, K.; Wojnowska, M.; Albalat, A.; Walker, D. Bacterial iron acquisition mediated by outer membrane translocation and cleavage of a host protein. Proc. Natl. Acad. Sci. USA 2018, 115, 6840. [Google Scholar] [CrossRef]
- Lau, C.K.Y.; Krewulak, K.D.; Vogel, H.J. Bacterial ferrous iron transport: The Feo system. FEMS Microbiol. Rev. 2016, 40, 273–298. [Google Scholar] [CrossRef] [PubMed]
- Eiler, A.; Zaremba-Niedzwiedzka, K.; Martínez-García, M.; McMahon, K.D.; Stepanauskas, R.; Andersson, S.G.E.; Bertilsson, S. Productivity and salinity structuring of the microplankton revealed by comparative freshwater metagenomics. Environ. Microbiol. 2014, 16, 2682–2698. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Nobu, M.K.; Kusada, H.; Meng, X.-Y.; Hosogi, N.; Uematsu, K.; Yoshioka, H.; Kamagata, Y.; Tamaki, H. Isolation of a member of the candidate phylum ‘Atribacteria’ reveals a unique cell membrane structure. Nat. Commun. 2020, 11, 6381. [Google Scholar] [CrossRef] [PubMed]
- Genderjahn, S.; Alawi, M.; Mangelsdorf, K.; Horn, F.; Wagner, D. Desiccation- and saline-tolerant bacteria and archaea in Kalahari pan sediments. Front. Microbiol. 2018, 9, 2082. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Sample ID in Given Analysis | Temperature | Reference |
|---|---|---|---|
| Saline Ace Lake, Antarctica | S1_3300005936 | 1 °C to 6 °C | https://link.springer.com/article/10.1007/s00300-014-1553-3 (accessed date 12 July 2022) |
| Freshwater Vanda Lake, Antarctica | S2_3300015360 | 4 °C to 10 °C | https://www.mdpi.com/2076-2607/3/3/391 (accessed date 12 July 2022) |
| Saline organic lake in Antarctica | S3_3300031218 | −10 °C to −14 °C | https://eprints.utas.edu.au/14434/ (accessed date 15 July 2022) |
| Freshwater Towuti Lake, Indonesia | S4_3300031587 | 26 °C to 29 °C | https://www.frontiersin.org/articles/10.3389/fmicb.2016.01007/full (accessed date 15 July 2022) |
| Freshwater Mendota Lake, United States | S5_3300035663 | 21 °C to 23 °C | https://onlinelibrary.wiley.com/doi/abs/10.1046/j.1365-2427.2002.01011.x (accessed date 15 July 2022) |
| Freshwater artificial North Sikkim lake, India | S6_3300047516 | 12 °C to 14 °C | (This study) |
| Element | Concentration (ppm) |
|---|---|
| Fe | 126.69 |
| K | 33.83 |
| Mg | 30.83 |
| Al | 22.89 |
| Ca | 2.19 |
| Mn | 1.86 |
| Na | 1.0 |
| Rb | 0.64 |
| Ba | 0.48 |
| Zn | 0.29 |
| V | 0.23 |
| Li | 0.19 |
| Cr | 0.16 |
| Cu | 0.097 |
| As | 0.096 |
| Ni | 0.091 |
| Cs | 0.073 |
| U | 0.061 |
| Co | 0.047 |
| Pb | 0.046 |
| Ga | 0.044 |
| Sr | 0.038 |
| Se | 0.008 |
| Be | 0.004 |
| Tl | 0.0005 |
| Cd | 0.0003 |
| Ag | 0.000006 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaushal, G.; Thakur, M.; Rai, A.K.; Singh, S.P. A Comprehensive Metagenomic Analysis Framework Revealing Microbiome Profile and Potential for Hydrocarbon Degradation and Carbohydrate Metabolism in a Himalayan Artificial Lake. Sustainability 2022, 14, 11455. https://doi.org/10.3390/su141811455
Kaushal G, Thakur M, Rai AK, Singh SP. A Comprehensive Metagenomic Analysis Framework Revealing Microbiome Profile and Potential for Hydrocarbon Degradation and Carbohydrate Metabolism in a Himalayan Artificial Lake. Sustainability. 2022; 14(18):11455. https://doi.org/10.3390/su141811455
Chicago/Turabian StyleKaushal, Girija, Monika Thakur, Amit Kumar Rai, and Sudhir P. Singh. 2022. "A Comprehensive Metagenomic Analysis Framework Revealing Microbiome Profile and Potential for Hydrocarbon Degradation and Carbohydrate Metabolism in a Himalayan Artificial Lake" Sustainability 14, no. 18: 11455. https://doi.org/10.3390/su141811455
APA StyleKaushal, G., Thakur, M., Rai, A. K., & Singh, S. P. (2022). A Comprehensive Metagenomic Analysis Framework Revealing Microbiome Profile and Potential for Hydrocarbon Degradation and Carbohydrate Metabolism in a Himalayan Artificial Lake. Sustainability, 14(18), 11455. https://doi.org/10.3390/su141811455