


Stimuli-Responsive Macromolecular Self-Assembly

Abstract
:1. Introduction
2. Mechanism of Stimuli-Responsive Macromolecular Self-Assembly
3. Research Progress of Stimuli-Responsive Macromolecular Self-Assembly
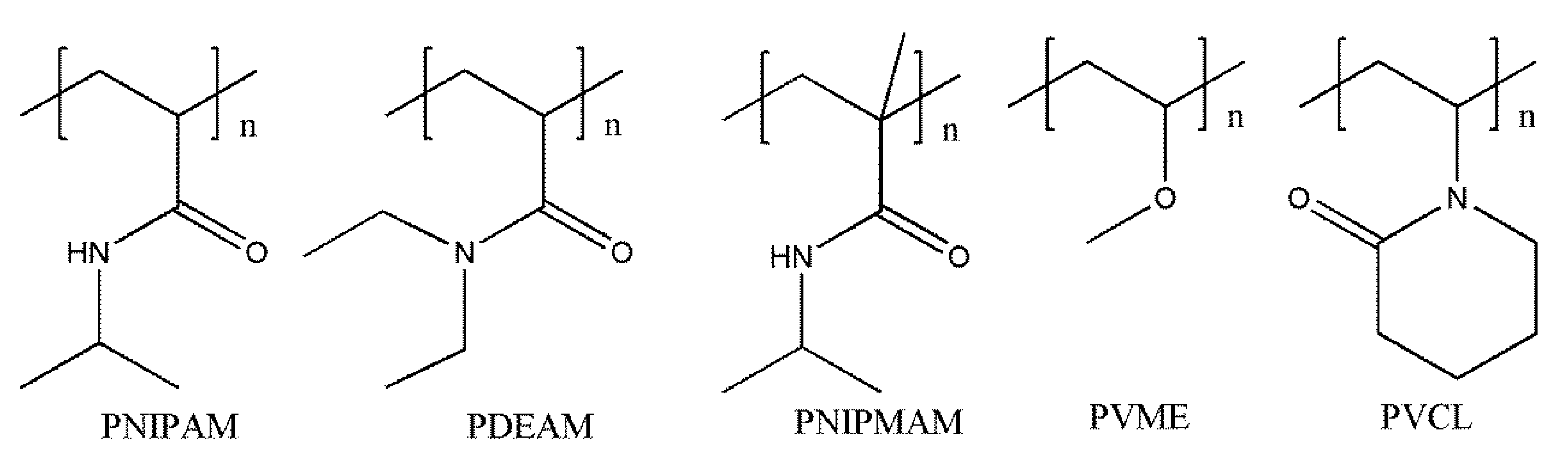
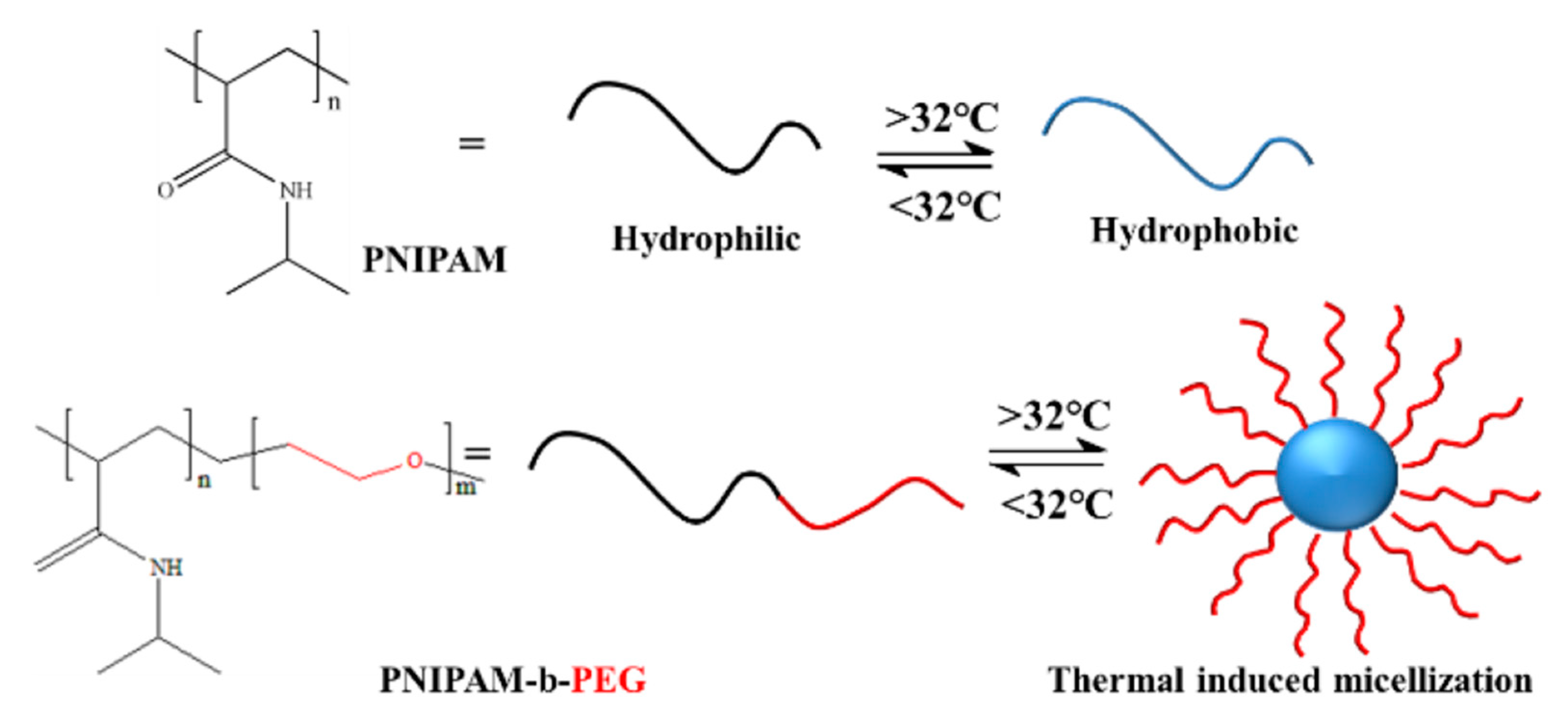
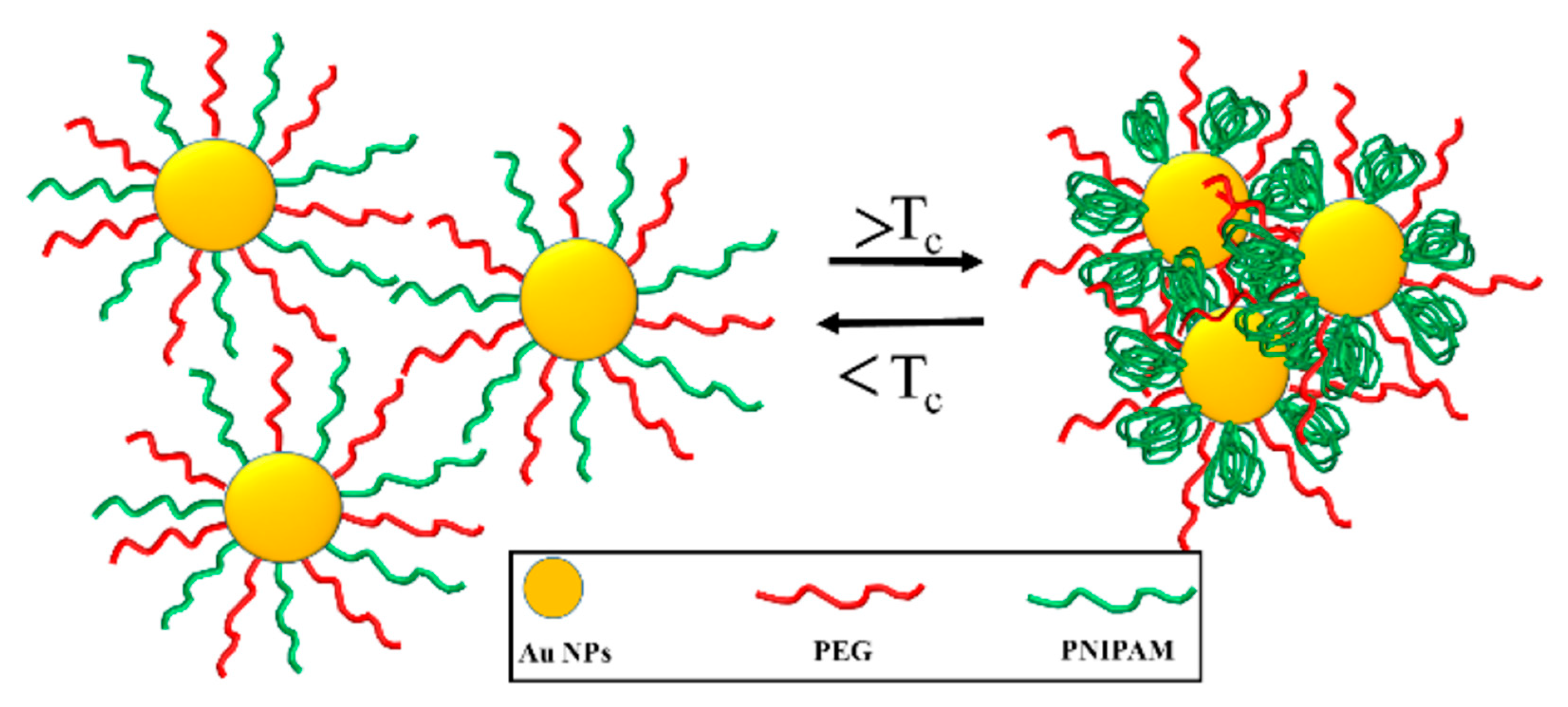
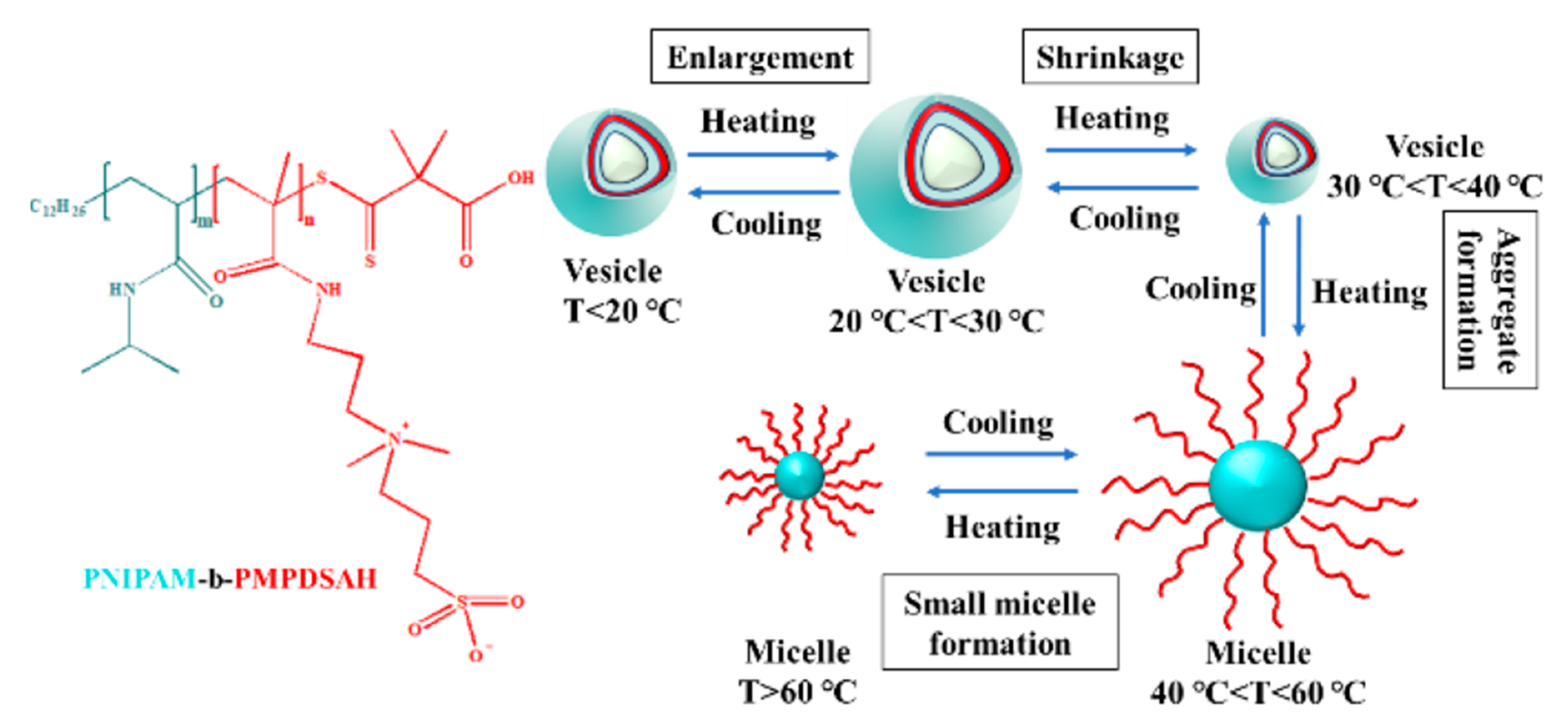
3.1. Temperature-Responsive Macromolecular Self-Assembly
3.2. Light-Responsive Macromolecule Self-Assembly
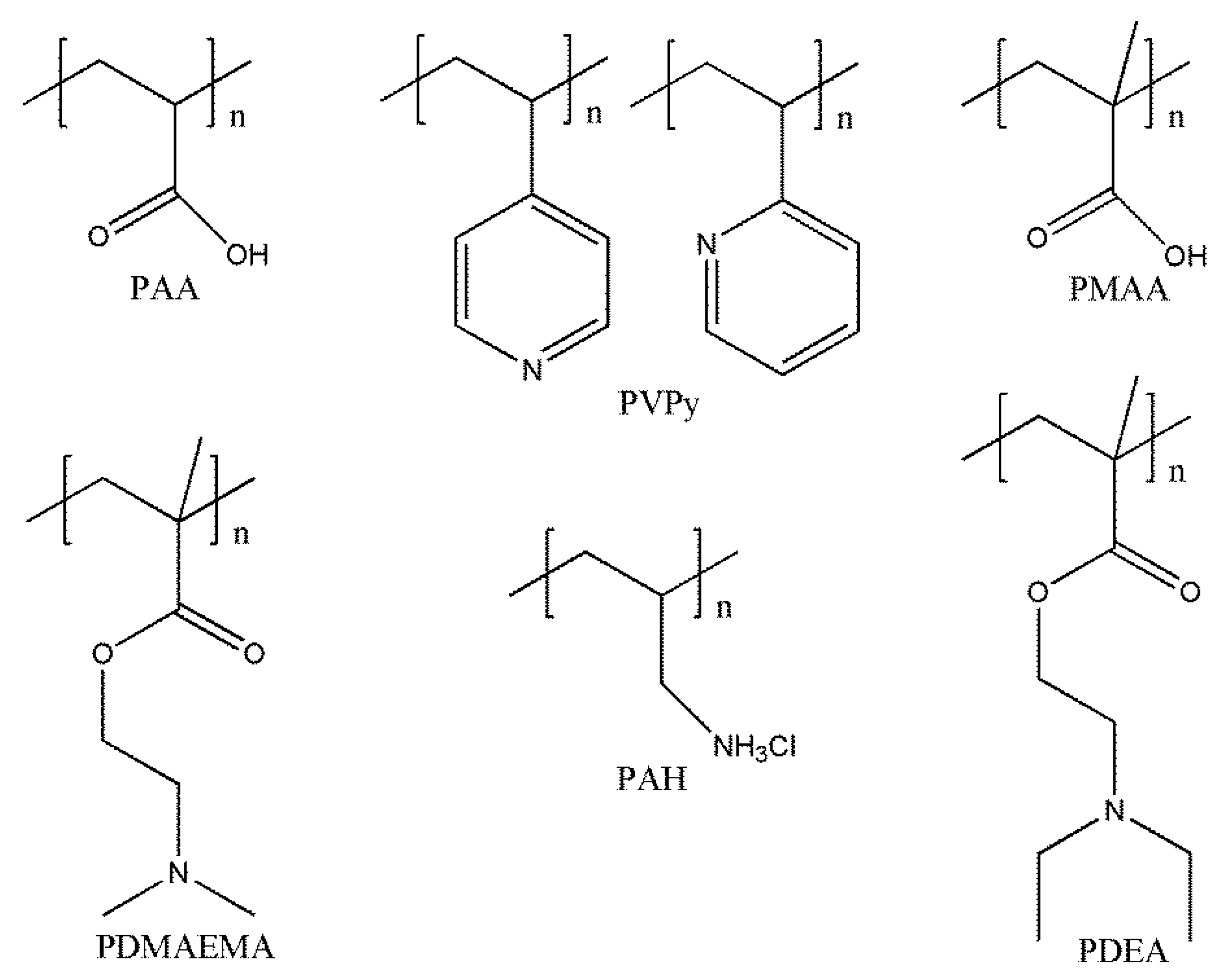
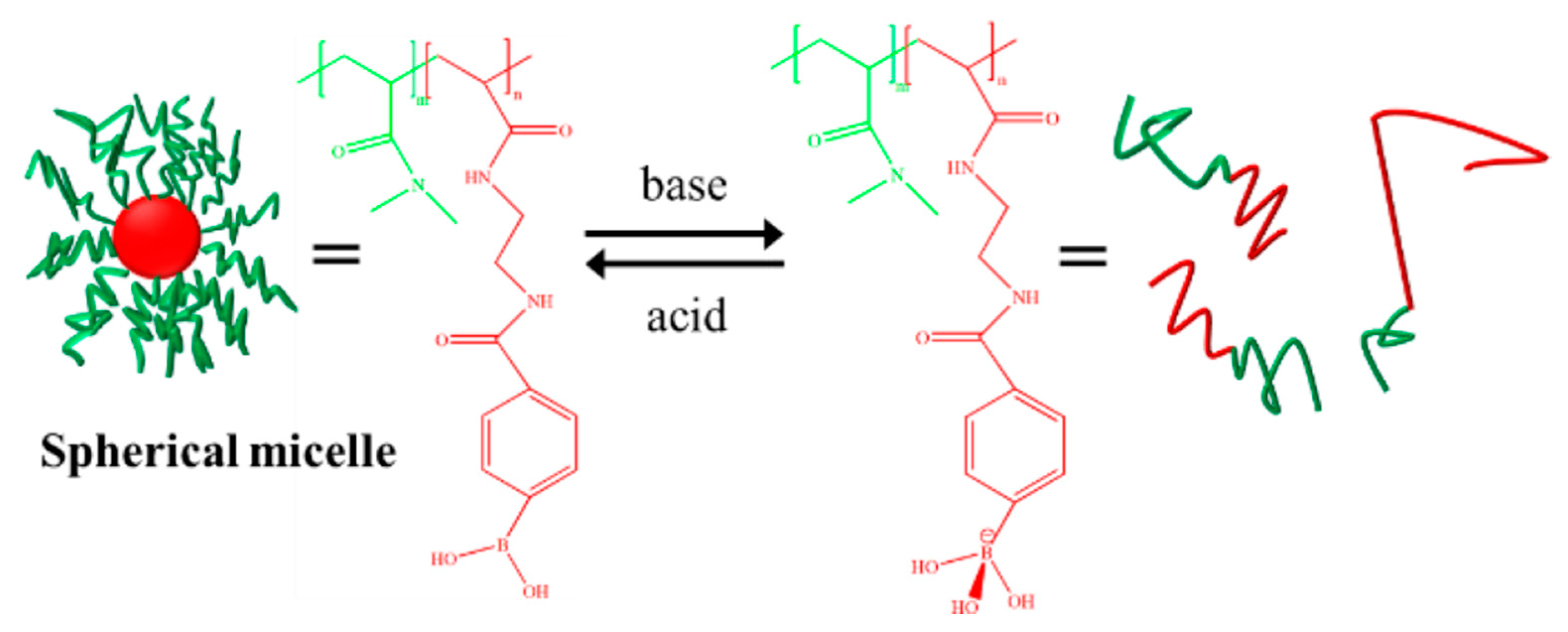
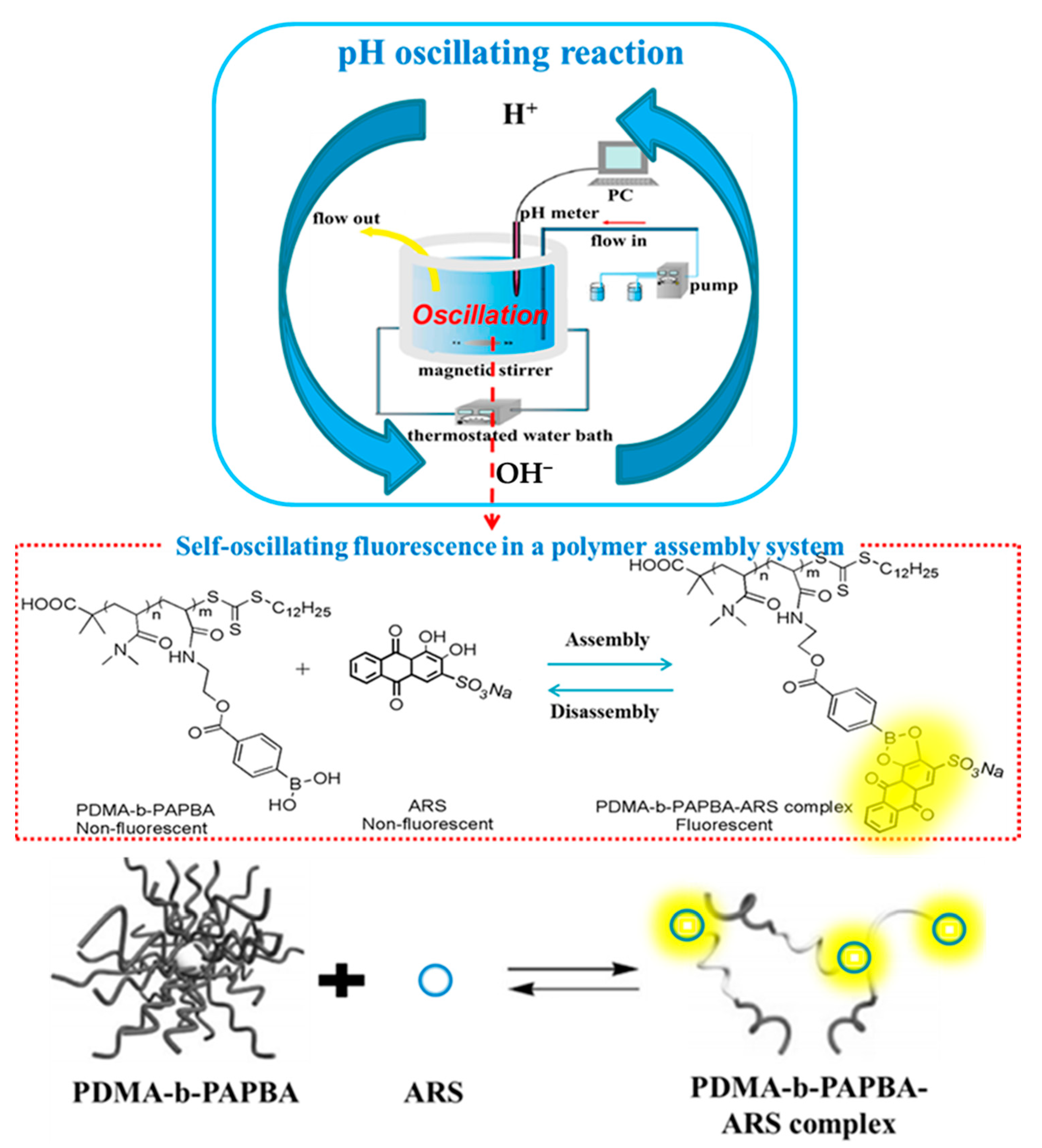
3.3. pH-Responsive Macromolecule Self-Assembly
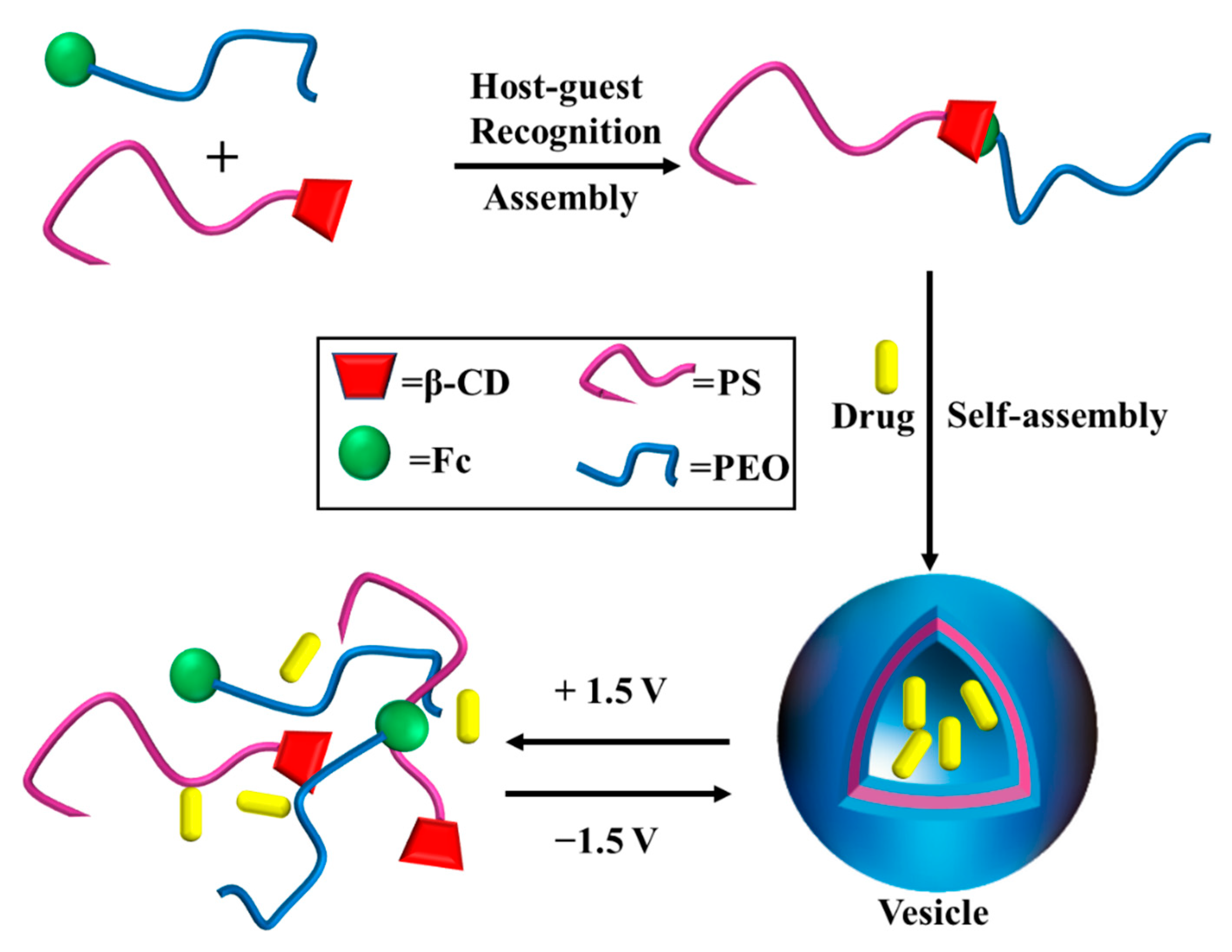
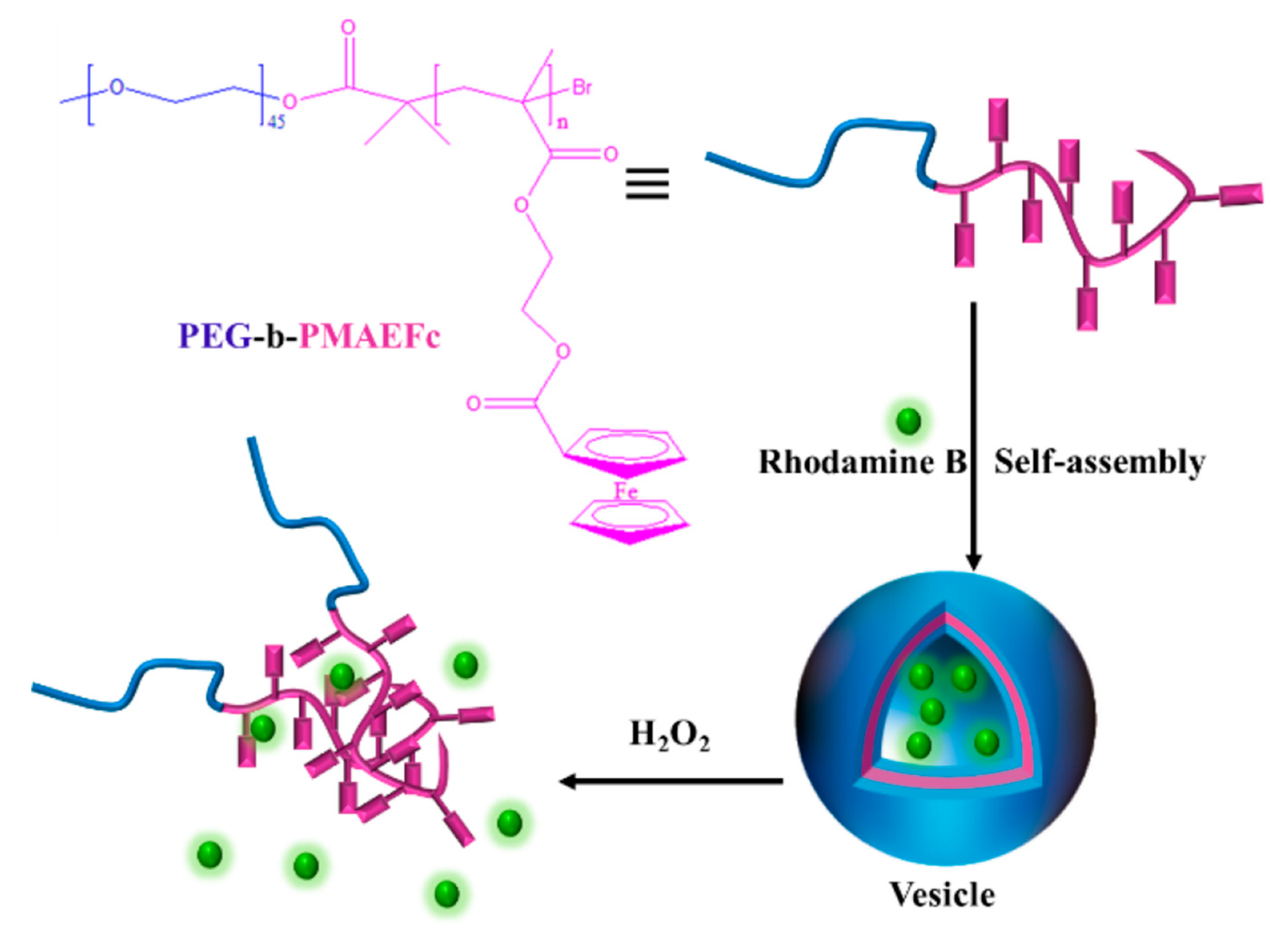
3.4. Redox-Responsive Macromolecule Self-Assembly
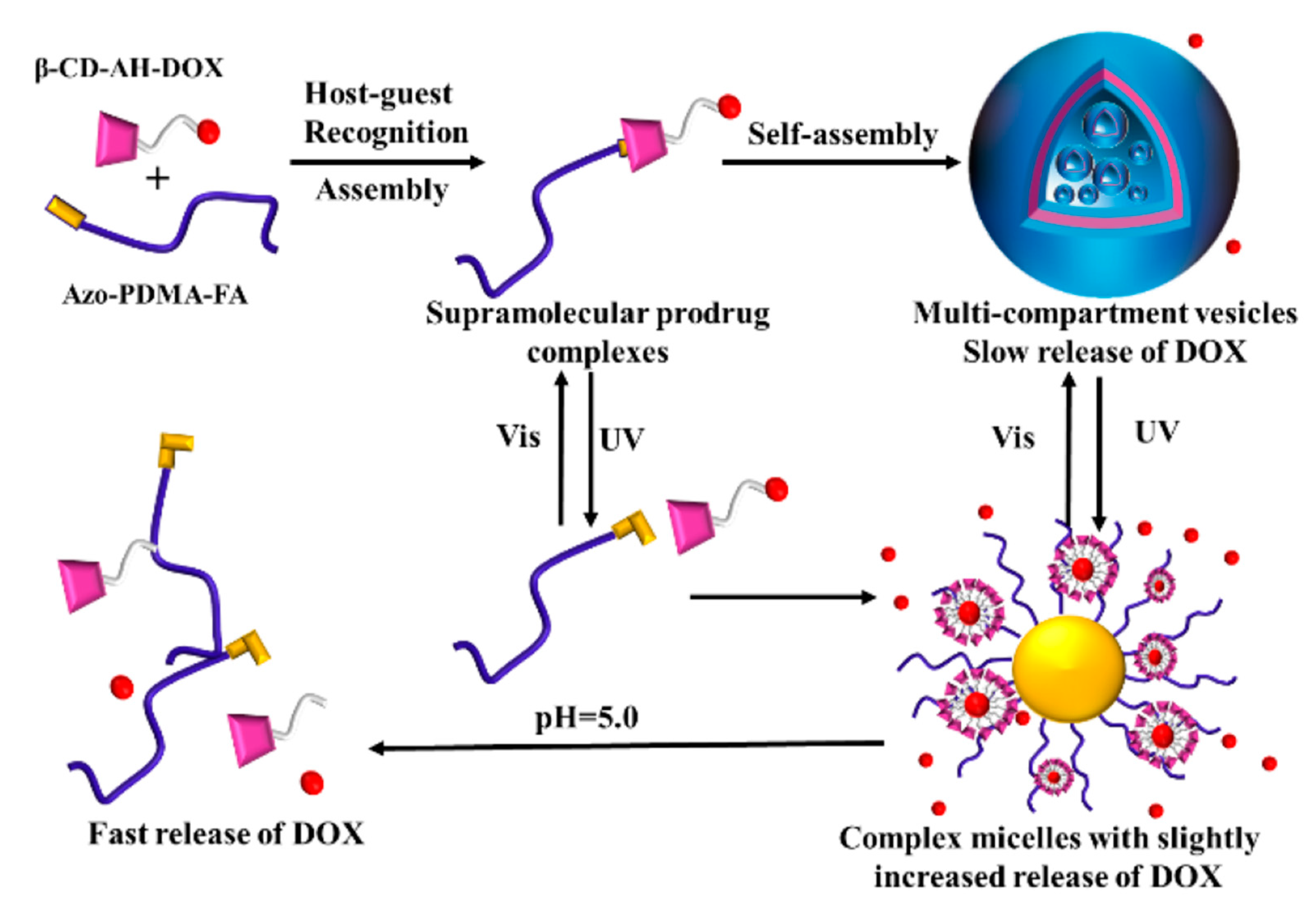
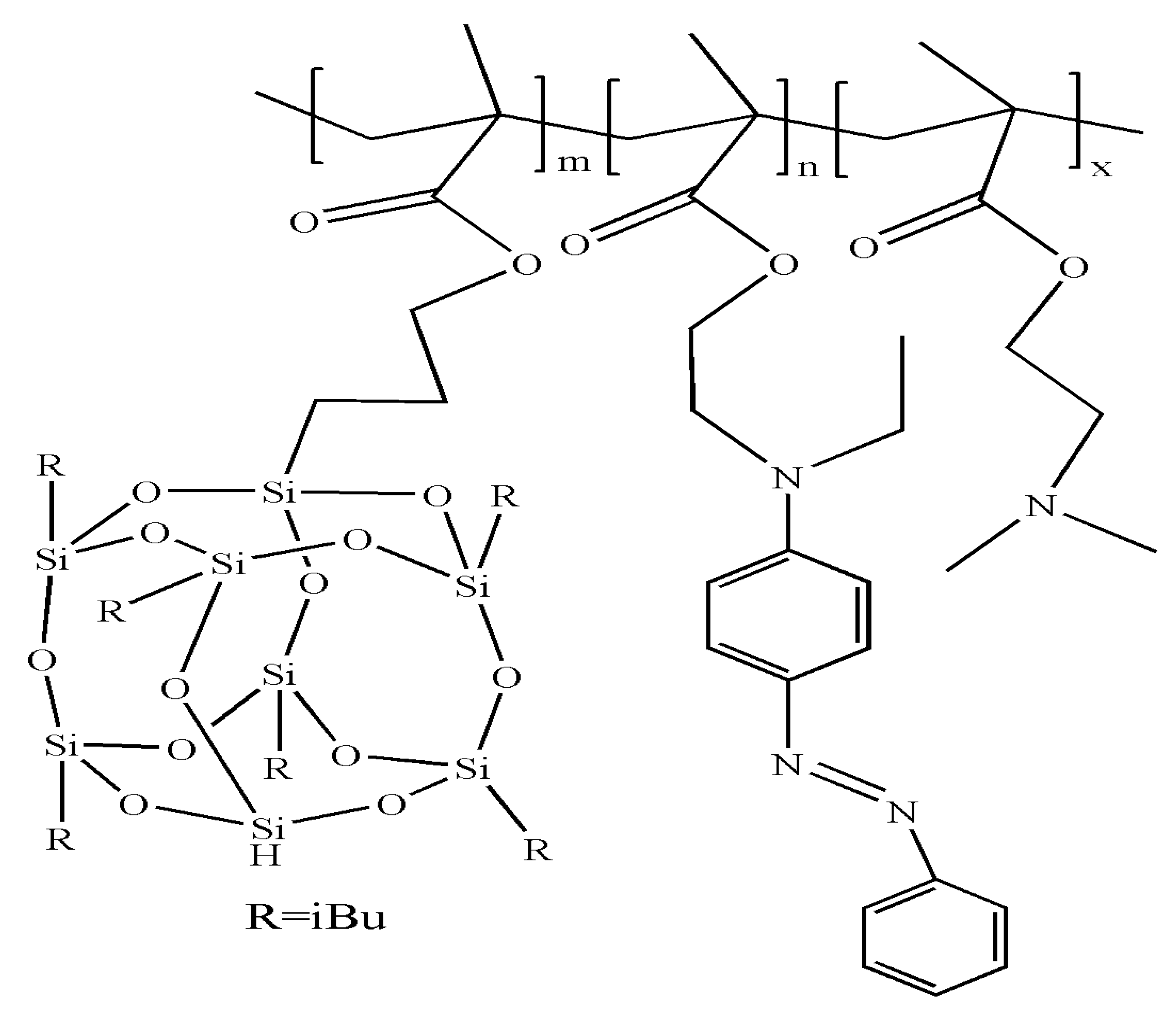
3.5. Multiple-Responsive Self-Assembly
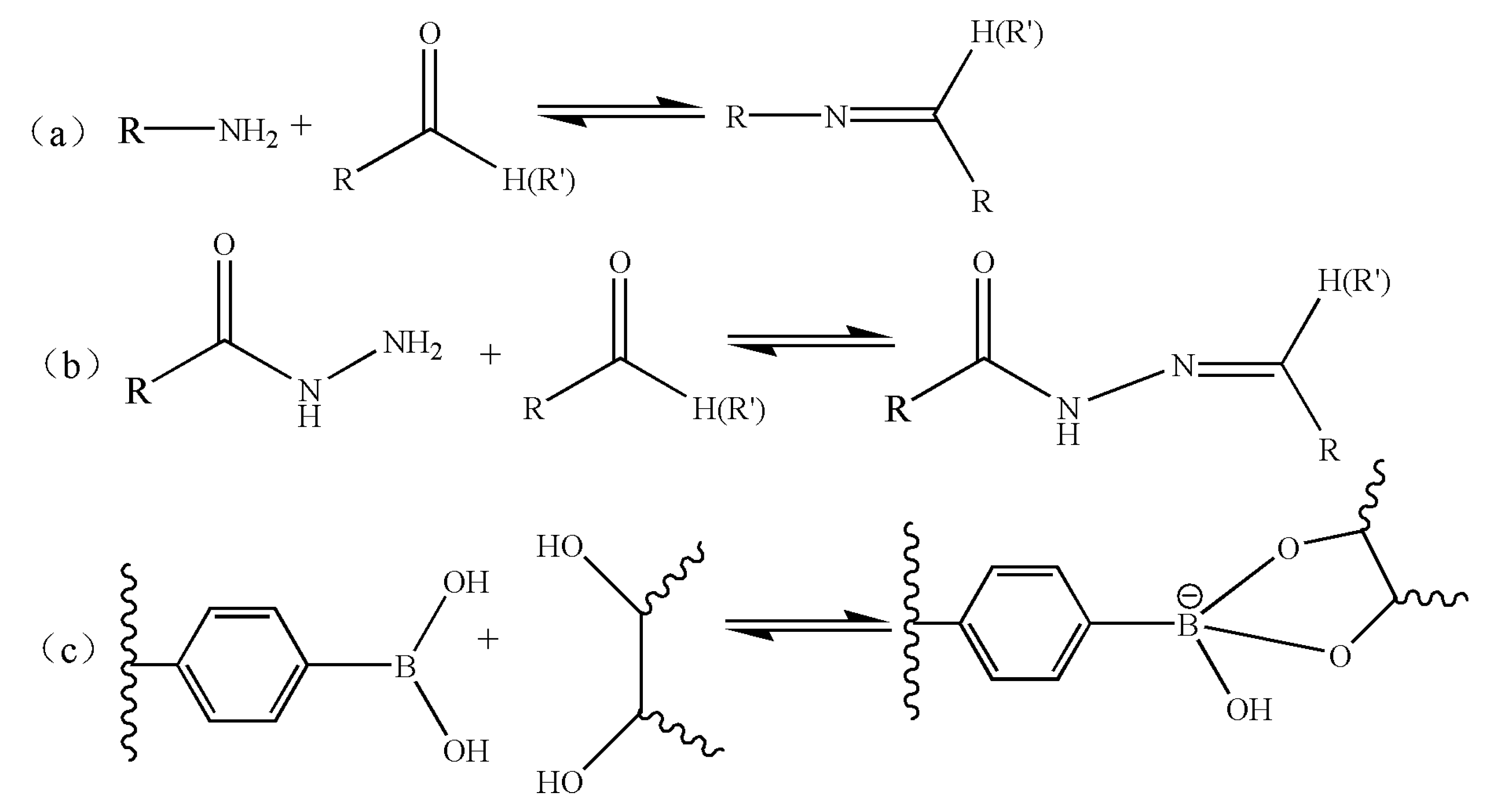
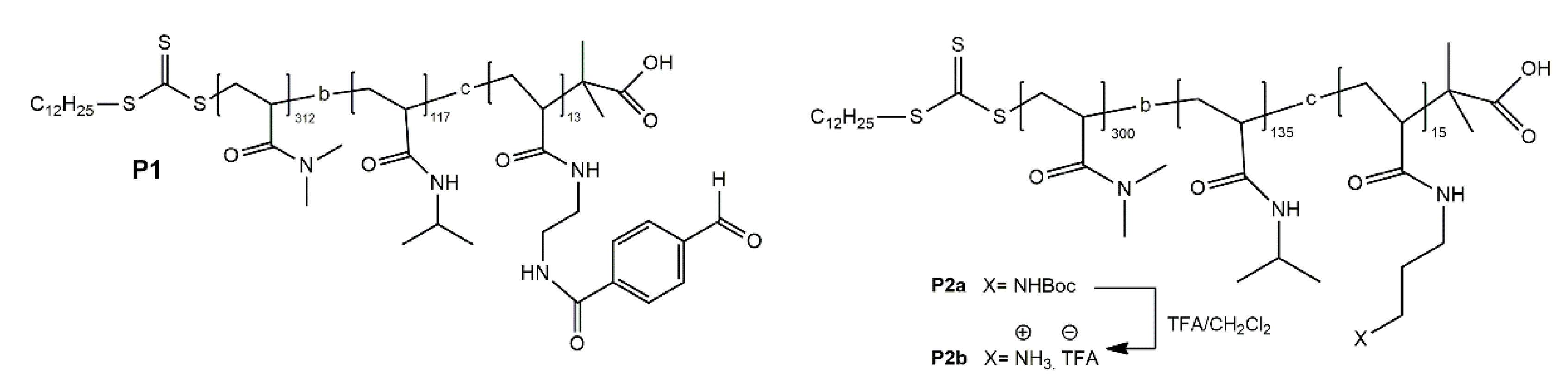
4. Dynamic Self-Assembly of Macromolecules
5. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, J.M.; Jiang, J.H.; Lin, S.; Cornel, E.J.; Li, C.; Du, J.Z. Polymersomes: From Macromolecular Self-Assembly to Particle Assembly. Chin. J. Chem. 2022, 40, 1842–1855. [Google Scholar] [CrossRef]
- Drexler, K.E. Molecular engineering: An approach to the development of general capabilities for molecular manipulation. Proc. Natl. Acad. Sci. USA 1981, 78, 5275–5278. [Google Scholar] [CrossRef] [PubMed]
- Li, D.H.; Wu, M.J.; Chen, X.F.; Liu, J.H.; Sun, Y.; Huang, J.; Zou, Y.L.; Wang, X.P.; Chen, D.Y.; Zhang, K.K. Boosting Organic Afterglow Performance via a Two-Component Design Strategy Extracted from Macromolecular Self-Assembly. J. Phys. Chem. Lett. 2022, 13, 5030–5039. [Google Scholar] [CrossRef]
- Theato, P.; Sumerlin, B.S.; O’Reilly, R.K. Stimuli responsive materials. Chem. Sov. Rev. 2013, 42, 7055–7056. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Shao, Y.G.; Yan, F.Z.; Zhang, Z.B.; Li, S.J. Construction of Supramolecular Polymers with Different Topologies by Orthogonal Self-Assembly of Cryptand-Paraquat Recognition and Metal Coordination. Molecules 2021, 26, 952. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.J.; Pashuck, E.T. (Macro)molecular self-assembly for hydrogel drug delivery. Adv. Drug Deliv. Rev. 2021, 172, 275–295. [Google Scholar] [CrossRef]
- Xing, P.; Chen, H.; Bai, L.; Hao, A.; Zhao, Y. Superstructure Formation and Topological Evolution Achieved by Self-Organization of a Highly Adaptive Dynamer. ACS Nano 2016, 10, 2716–2727. [Google Scholar] [CrossRef]
- Ke, D.; Zhan, C.; Xu, S.; Ding, X.; Peng, A.; Sun, J.; He, S.; Li, A.D.; Yao, J. Self-assembled hollow nanospheres strongly enhance photoluminescence. J. Am. Chem. Soc. 2011, 133, 11022–11025. [Google Scholar] [CrossRef]
- Li, L.Y.; Raghupathi, K.; Song, C.; Prasad, P.; Thayumanavan, S. Self-assembly of random copolymers. Chem. Commun. 2014, 50, 13417–13432. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.J.; Lou, Z.C. Recent Advancements in MOF/Biomass and Bio-MOF Multifunctional Materials: A Review. Sustainability 2022, 14, 5768. [Google Scholar] [CrossRef]
- Liu, D.D.; Yang, S.Q.; Peng, S.J.; Chen, Y.; Zhang, L.; Tan, J.B. Simultaneous Synthesis and Self-Assembly of Bottlebrush Block Copolymers at Room Temperature via Photoinitiated RAFT Dispersion Polymerization. Macromol. Rapid Commun. 2022, 43, 2100921. [Google Scholar] [CrossRef] [PubMed]
- Dergham, M.; Lin, S.M.; Geng, J. Supramolecular Self-Assembly in Living Cells. Angew. Chem. Int. 2022, 61, e202114267. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Zhen, F.H.; Chao, W.; Lei, Z.; Qian, Y.X.; Su, Y.W.; Yan, H.W.; Duo, P.; Bin, B.D.; Ren, B.W.; et al. The recent progress of synergistic supramolecular polymers: Preparation, properties and applications. Chem. Commun. 2021, 57, 1413–1429. [Google Scholar] [CrossRef]
- Zhang, Z.K.; Ma, R.J.; Shi, L.Q. Cooperative macromolecular self-assembly toward polymeric assemblies with multiple and bioactive functions. Acc. Chem. Res. 2014, 47, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.; Steed, J.W. Supramolecular Gel Phase Crystallization: Orthogonal Self-assembly under Non-equilibrium Conditions. Chem. Soc. Rev. 2014, 43, 2080–2088. [Google Scholar] [CrossRef]
- Mishra, R.K.; Das, S.; Vedhanarayanan, B.; Das, G.; Ajayaghosh, A. Chapter 7: Stimuli-responsive Supramolecular Gels. In Molecular Gels: Structure and Dynamics; Royal Society of Chemistry: London, UK, 2018; pp. 190–226. [Google Scholar] [CrossRef]
- Zhang, D.W.; Zhao, X.; Hou, J.L.; Li, Z.T. Aromatic Amide Foldamers: Structures, Properties, and Functions. Chem. Rev. 2012, 112, 5271–5316. [Google Scholar] [CrossRef]
- Cheng, X.X.; Miao, T.F.; Qian, Y.L.; Zhang, Z.B.; Zhang, W.; Zhu, X.L. Supramolecular Chirality in Azobenzene-Containing Polymer System: Traditional Postpolymerization Self-Assembly Versus In Situ Supramolecular Self-Assembly Strategy. Int. J. Mol. Sci. 2020, 21, 6186. [Google Scholar] [CrossRef]
- Tao, H.; Galati, E.; Kumacheva, E. Temperature-Responsive Self-Assembly of Nanoparticles Grafted with UCST Polymer Ligands. Macromolecules 2018, 51, 6021–6027. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, Y.; Wu, G.; Liu, J.R.; Cao, N.; Wang, L.; Wang, Y.; Li, X.; Hong, X.; Yang, C.; et al. Temperature-dependent Self-Assembly of a Purely Organic Cage in Water. Chem. Commun. 2018, 54, 3138–3141. [Google Scholar] [CrossRef]
- Pan, Q.; Zong, Z.; Shen, J.; Xue, H.; Pu, Y. Synthesis, self-assembly, and pH-responsive Drug Release of PHMEMA-PEG-PHMEMA ABA triblock Copolymers. J. Macromol. Sci. A 2018, 55, 691–697. [Google Scholar] [CrossRef]
- Wang, T.P.; Shen, L.Y.; Lin, F.Y.; Forrester, M.; Torres, S.W.; Robison, T.W.; Lee, T.H.; Goyal, S.; Cochran, E.W. Standalone Block Copolymer Nanoballoons: Decoupling Self-Assembly from Implementation in Nanomanufacturing. ACS Appl. Polym. Mater. 2022, 4, 5134–5143. [Google Scholar] [CrossRef]
- Yi, M.H.; Guo, J.Q.; He, H.J.; Tan, W.Y.; Harmon, N.; Ghebreyessus, K.; Xu, B. Phosphobisaromatic motifs enable rapid enzymatic self-assembly and hydrogelation of short peptides. Soft Matter. 2021, 17, 8590–8594. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, K.; Kelderman, J.; Coleman, H.; Aguilar, M.I.; Parkington, H.; Del Borgo, M. Self-assembly of trifunctional tripeptides to form neural scaffolds. J. Mater. Chem. B 2021, 9, 4475–4479. [Google Scholar] [CrossRef] [PubMed]
- Percec, V.; Peterca, M.; Tadjiev, T.; Zeng, X.; Ungar, G.; Leowanawat, P.; Aqad, E.; Imam, M.R.; Rosen, B.M.; Akbey, U.; et al. Self-Assembly of Dendronized Perylene Bisimides into Complex Helical Columns. J. Am. Chem. Soc. 2011, 133, 12197–12219. [Google Scholar] [CrossRef] [PubMed]
- Avestro, A.J.; Belowich, M.E.; Stoddart, J.F. Cooperative Self-assembly: Producing Synthetic Polymers with Precise and Concise Primary Structures. Chem. Soc. Rev. 2012, 41, 5881–5895. [Google Scholar] [CrossRef]
- Forgan, R.S.; Gassensmith, J.J.; Cordes, D.B.; Boyle, M.M.; Hartlieb, K.J.; Friedman, D.C.; Slawin, A.M.Z.; Stoddart, J.F. Self-Assembly of a 2 Pseudorota 3 Catenane in Water. J. Am. Chem. Soc. 2012, 134, 17007–17010. [Google Scholar] [CrossRef]
- Gassensmith, J.J.; Erne, P.M.; Paxton, W.F.; Frasconi, M.; Donakowski, M.D.; Stoddart, J.F. Patterned Assembly of Quantum Dots onto Surfaces Modified with Click Microcontact Printing. Adv. Mater. 2013, 25, 223–226. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, Y.; Jiang, M.; Chen, D. Cascade Molecule-Particle-Molecule Self-Assemblies for Fabricating Narrowly Size-Distributed Polymeric Superparticles with a Bicontinuous Nanostructure. ACS Macro Lett. 2012, 1, 1312–1316. [Google Scholar] [CrossRef]
- Wei, K.; Li, J.; Chen, G.; Jiang, M. Dual Molecular Recognition Leading to a Protein-Polymer Conjugate and Further Self-Assembly. ACS Macro Lett. 2013, 2, 278–283. [Google Scholar] [CrossRef]
- Zhang, K.; Jiang, M.; Chen, D. DNA/Polymeric Micelle Self-Assembly Mimicking Chromatin Compaction. Angew. Chem. Int. Ed. 2012, 51, 8744–8747. [Google Scholar] [CrossRef]
- Zhang, K.; Jiang, M.; Chen, D. Self-assembly of Particles: The regulatory role of Particle Flexibility. Prog. Polym. Sci. 2012, 37, 445–486. [Google Scholar] [CrossRef]
- Guo, W.; Wang, X.P.; Wang, X.P.; Zhang, K.K. Achieving Purely-Organic Room-Temperature Aqueous Phosphorescence via a Two-Component Macromolecular Self-Assembly Strategy. Chem. Asian J. 2020, 15, 3469–3474. [Google Scholar] [CrossRef]
- Tang, Y.; Zhou, L.P.; Li, J.; Luo, Q.A.; Huang, X.; Wu, P.; Wang, Y.G.; Xu, J.Y.; Shen, J.C.; Liu, J.Q. Giant Nanotubes Loaded with Artificial Peroxidase Centers: Self-Assembly of Supramolecular Amphiphiles as a Tool To Functionalize Nanotubes. Angew. Chem. Int. Ed. 2010, 49, 3920–3924. [Google Scholar] [CrossRef]
- Wang, G.; Wang, C.; Wang, Z.; Zhang, X. Bolaform Superamphiphile Based on A Dynamic Covalent Bond and Its Self-Assembly in Water. Langmuir 2011, 27, 12375–12380. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, G.T.; Wang, Z.Q.; Zhang, X. A pH-Responsive Superamphiphile Based on Dynamic Covalent Bonds. Chem. Eur. J. 2011, 17, 3322–3325. [Google Scholar] [CrossRef] [PubMed]
- Huan, X.Y.; Wang, D.L.; Dong, R.J.; Tu, C.L.; Zhu, B.S.; Yan, D.Y.; Zhu, X.Y. Supramolecular ABC Miktoarm Star Terpolymer Based on Host-Guest Inclusion Complexation. Macromolecules 2012, 45, 5941–5947. [Google Scholar] [CrossRef]
- Sun, W.Q.; He, X.H.; Gao, C.Y.; Liao, X.J.; Xie, M.R.; Lin, S.L.; Yan, D.Y. Novel Amphiphilic and Photo-responsive ABC 3-miktoarm Star Terpolymers: Synthesis, Self-assembly and Photo-responsive behavior. Polym. Chem. 2013, 4, 1939–1949. [Google Scholar] [CrossRef]
- Tao, W.; Liu, Y.; Jiang, B.; Yu, S.; Huang, W.; Zhou, Y.; Yan, D. A Linear-Hyperbranched Supramolecular Amphiphilic and Its Self-Assembly into Vesicles with Great Ductility. J. Am. Chem. Soc. 2012, 134, 762–764. [Google Scholar] [CrossRef]
- Kathyayani, D.; Mahesh, B.; Chamaraja, N.A.; Madhukar, B.S.; Channe Gowda, D. Synthesis and structural characterization of elastin-based polypentapeptide/hydroxypropylmethylcellulose blend films: Assessment of miscibility, thermal stability and surfacecharacteristics. Colloids Surf. A 2022, 649, 129503–129517. [Google Scholar] [CrossRef]
- Zhou, J.; He, R.; Ma, J. RAFT-Mediated Polymerization-Induced Self-Assembly of Poly(Acrylic Acid)-b-Poly(Hexafluorobutyl Acrylate): Effect of the pH on the Synthesis of Self-Stabilized Particles. Polymers 2016, 8, 207. [Google Scholar] [CrossRef]
- Xiao, D.; Jia, H.Z.; Ma, N.; Zhuo, R.X.; Zhang, X.Z. A Redox-Responsive Mesoporous Silica Nanoparticle Capped with Amphiphilic Peptieds by Self-assembly for Cancer Targeting Drug Delivery. Nanoscale 2015, 7, 10071–10077. [Google Scholar] [CrossRef] [PubMed]
- Chong, D.; Tan, J.; Zhang, J.; Zhou, Y.; Wan, X.; Zhang, J. Dual electrical switching permeability of vesicles via redox-responsive self-assembly of amphiphilic block copolymers and polyoxometalates. Chem. Commun. 2018, 54, 7838–7841. [Google Scholar] [CrossRef]
- Picco, A.S.; Yameen, B.; Knoll, W.; Ceolín, M.R.; Azzaroni, O. Temperature-driven self-assembly of self-limiting uniform supraparticles from non-uniform unimolecular micelles. J. Colloid Interface Sci. 2016, 471, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yuan, D.; Yuan, T.; Dong, J.; Feng, N.; Han, G. A Visible Light Responsive Azobenzene-Functionalized Polymer: Synthesis, Self-Assembly, and Photoresponsive Properties. J. Polym. Sci. A 2015, 53, 2768–2775. [Google Scholar] [CrossRef]
- Wu, A.; Lu, F.; Sun, P.; Gao, X.; Shi, L.; Zheng, L.Q. Photo-Responsive Self-Assembly of Surface Active Ionic Liquid. Langmuir 2016, 32, 8163–8170. [Google Scholar] [CrossRef]
- Shao, L.; Hua, B.; Sun, J.; Li, Q.; Yang, J.; Yu, G. A cucurbit[7]uril-based supra-amphiphile: Photo-responsive self-assembly and application in controlled release. Tetrahedron Lett. 2017, 58, 1863–1867. [Google Scholar] [CrossRef]
- Jiang, Y.; Geng, T.; Li, Q.; Li, G.; Ju, H. Influences of temperature, pH and salinity on the surface property and self-assembly of 1:1 salt-free catanionic surfactant. J. Mol. Liq. 2014, 199, 1–6. [Google Scholar] [CrossRef]
- Chung, C.Y.S.; Yam, V.W.W. Dual pH- and Temperature-Responsive Metallosupramolecular Block Copolymers with Tunable Critical Micelle Temperature by Modulation of the Self-Assembly of NIR-Emissive Alkynylplatinum(II) Complexes Induced by Changes in Hydrophilicity and Electrostatic Effects. Chem. Eur. J. 2013, 19, 13182–13192. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, W.G.; Li, J.W.; Hong, C.Y.; Zhang, W.J.; You, Y.Z. Preparation of pH- and reductive-responsive prodrug nanoparticles via polymerization-induced self-assembly. Sci. China Chem. 2018, 61, 1159–1166. [Google Scholar] [CrossRef]
- Guo, J.; Lu, Y.; Bi, C.; Fan, J.; Xu, G.H.; Ma, J. Stimuli-Responsive Peptides Self-Assembly and Its Application. Prog. Chem. 2019, 31, 83–93. [Google Scholar] [CrossRef]
- Dong, S.; Luo, Y.; Yan, X.; Zheng, B.; Ding, X.; Yu, Y.; Ma, Z.; Zhao, Q.; Huang, F. A dual-responsive supramolecular polymer gel formed by crown ether based molecular recognition. Angew. Chem. Int. Ed. 2011, 50, 1905–1909. [Google Scholar] [CrossRef] [PubMed]
- Beves, J.E.; Campbell, C.J.D.; Leigh, A.; Pritchard, R.G. Tetrameric Cyclic Double Helicates as a Scaffold for a Molecular Solomon Link. Angew. Chem. Int. Ed. 2013, 52, 6464–6467. [Google Scholar] [CrossRef] [PubMed]
- Rodler, F.; Linders, J.; Fenske, T.; Rehm, T.; Mayer, C.; Schmuck, C. pH-Switchable Vesicles from a Serine-Derived Guanidiniocabonyl Pyrrole Carboxylate Zwitterion in DMSO. Angew. Chem. Int. Ed. 2010, 49, 8747–8750. [Google Scholar] [CrossRef] [PubMed]
- Gröger, G.; Meyer-Zaika, W.; Böttcher, C.; Gröhn, F.; Ruthard, C.; Schmuck, C. Switchable Supramolecular Polymers from the Self-assembly of a Small Monomer with Two Orthogonal Binding Interactions. J. Am. Chem. Soc. 2011, 133, 8961–8971. [Google Scholar] [CrossRef] [PubMed]
- Yuen, F.; Tam, K.C. Cyclodextrin-Assisted Assembly of Stimuli-Responsive Polymers in Aqueous Media. Soft Matter 2010, 6, 4613–4630. [Google Scholar] [CrossRef]
- Xu, J.F.; Chenm, Y.Z.; Wu, L.Z.; Tung, C.H.; Yang, Q.Z. Dynamic Covalent Bond Based on Reversible Photo [4+4]cycloaddition of Anthracene for Construction of Double-Dynamic Polymers. Org. Lett. 2013, 15, 6148–6151. [Google Scholar] [CrossRef]
- Qi, Z.; Schalley, C.A. Exploring Macrocycles in Functional Supramolecular Gels: From Stimuli Responsiveness to Systems Chemistry. Acc. Chem. Res. 2014, 47, 2222–2233. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, D.; Yan, X.; Chen, J.; Dong, S.; Zheng, B.; Huang, F. Self-Healing Supramolecular Gels Formed by Crown Ether Based Host-Guest Interactions. Angew. Chem. Int. Ed. 2012, 51, 7011–7015. [Google Scholar] [CrossRef]
- Harada, A.; Kobayashi, R.; Takashima, Y.; Hashidzume, A.; Yamaguchi, H. Macroscopic Self-Assembly through Molecular Recognition. Nat. Chem. 2011, 3, 34–37. [Google Scholar] [CrossRef]
- Liu, G.; Ma, R.J.; Ren, J.; Li, Z.; Zhang, H.X.; Zhang, Z.K.; An, Y.L.; Shi, L.Q. A glucose-responsive complex polymeric micelle enabling repeated on–off release and insulin protection. Soft Matter 2013, 9, 1636–1644. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, C.M.; Dong, H.Y.; Chen, D.Y. Solution-based fabrication of narrow-disperse ABC three-segment and Θ-shaped nanoparticles. Angew. Chem. Int. Ed. 2016, 128, 6290–6294. [Google Scholar] [CrossRef]
- Lee, H.J.; Koo, A.N.; Lee, S.W.; Lee, M.H.; Lee, S.C. Catechol-functionalized adhesive polymer nanoparticles for controlled local release of bone morphogenetic protein-2 from titanium surface. J. Control. Release 2013, 170, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Authimoolam, S.P.; Vasilakes, A.L.; Shah, N.M.; Puleo, D.A.; Dziubla, T.D. Synthetic oral mucin mimic from polymer micelle networks. Biomacromolecules 2014, 15, 3099–3111. [Google Scholar] [CrossRef] [PubMed]
- Authimoolam, S.P.; Lakes, A.L.; Puleo, D.A.; Dziubla, T.D. Layer-by-layers of polymeric micelles as a biomimetic drug-releasing network. Macromol. Biosci. 2016, 16, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Kong, J.H.; Sun, J.T.; Li, H.; He, C.B. Stable superhydrophobic porous coatings from hybrid ABC triblock copolymers and their anticorrosive performance. ACS Appl. Mater. Interfaces 2017, 9, 30056–30063. [Google Scholar] [CrossRef] [PubMed]
- Tambunlertchai, S.; Srisang, S.; Nasongkla, N. Development of antimicrobial coating by later-by-layer dip coating of chlorhexidine-loaded micelles. J. Mater. Sci. Mater. Med. 2017, 28, 90. [Google Scholar] [CrossRef]
- Yang, Y.Q.; Yi, C.L.; Luo, J.; Liu, R.; Liu, J.K.; Jiang, J.Q.; Liu, X.Y. Glucose sensors based on electrodeposition of molecularly imprinted polymeric micelles: A novel strategy for MIP sensors. Biosens. Bioelectron. 2011, 26, 2607–2612. [Google Scholar] [CrossRef]
- Zhu, X.W.; Liu, M.H. Self-assembly and morphology control of new L-glutamic acid-based amphiphilic random copolymers: Giant vesicles, vesicles, spheres, and honeycomb film. Langmuir 2011, 27, 12844–12850. [Google Scholar] [CrossRef]
- Falentin-Daudré, C.; Faure, E.; Svaldo-Lanero, T.; Farina, F.; Jerome, C.; Van, de.; Weerdt, C.; Martial, J.; Duwez, A.S.; Detrembleur, C. Antibacterial polyelectrolyte micelles for coating stainless steel. Langmuir 2012, 28, 7233–7241. [Google Scholar] [CrossRef]
- Sun, Y.X.; Ren, K.F.; Zhao, Y.X.; Liu, X.S.; Chang, G.X.; Ji, J. Construction of redox-active multilayer film for electrochemically controlled release. Langmuir 2013, 29, 11163–11168. [Google Scholar] [CrossRef]
- Wang, F.; Yang, X.W.; Zhang, L.L.; Xu, W.S.; Liu, H. Novel macromolecular emulsifiers as coatings with water-tolerant antifogging properties based on coumarin-containing copolymeric micelles. Macromol. Mater. Eng. 2017, 302, 1700173. [Google Scholar] [CrossRef]
- Sun, J.D.; Zhu, Y.; Meng, L.; Chen, P.; Shi, T.T.; Liu, X.Y.; Zheng, Y.F. Electrophoretic deposition of colloidal particles on Mg with cytocompatibility, antibacterial performance, and corrosion resistance. Acta Biomater. 2016, 45, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Wang, D.Y.; Lin, X.; Politakos, N.; Tuninetti, J.S.; Moya, S.E.; Gao, C.Y. Fabrication of UV responsive micelles-containing multilayers and their influence on cell adhesion. Sci. China Chem. 2018, 61, 54–63. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Y.H.; Yuan, X.Y. Facile preparation of superhydrophobic coating by spraying a fluorinated acrylic random copolymer micelle solution. Soft Matter 2013, 9, 1005–1009. [Google Scholar] [CrossRef]
- Rabnawaz, M.; Liu, G.J. Graft-copolymer-based approach to clear, durable, and anti-smudge polyurethane coatings. Angew. Chem. Int. Ed. 2015, 54, 6516–6520. [Google Scholar] [CrossRef]
- Rabnawaz, M.; Liu, G.J.; Hu, H. Fluorine-free anti-smudge polyurethane coatings. Angew. Chem. Int. Ed. 2015, 54, 12722–12727. [Google Scholar] [CrossRef]
- Iqbal, S.; Zhang, Y.J.; Liu, Y.T.; Akram, R.; Lv, S.S. Polymer micelles as building blocks for layer-by-layer assembly of multilayers under a high-gravity field. Chem. Eng. J. 2016, 293, 302–310. [Google Scholar] [CrossRef]
- Klok, H.A.; Lecommandoux, S. Supramolecular Materials via Block Copolymer Self-assembly. Adv. Mater. 2001, 13, 1217–1229. [Google Scholar] [CrossRef]
- Vriezema, D.M.; Aragonès, M.C.; Elemans, J.A.A.W.; Cornelissen, J.J.L.M.; Rowan, A.E.; Nolte, R.J.M. Self-Assembled Nanoreactors. Chem. Rev. 2005, 705, 1445–1490. [Google Scholar] [CrossRef] [Green Version]
- Blanazs, S.; Armes, S.P.; Ryan, A.J. Self-assembled block copolymer aggregates: From micelles to vesicles and their biological applications. Macromol. Rapid Commun. 2009, 30, 267–277. [Google Scholar] [CrossRef]
- Warren, N.J.; Armes, S.P. Polymerization-induced self-assembly of block copolymer nano-objects via RAFT aqueous dispersion polymerization. J. Am. Chem. Soc. 2014, 136, 10174–10185. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Xu, X.; Kirkland-York, S.E.; Savin, D.A.; McCormick, C.L. “Schizophrenic” Self-assembly of Block Copolymers Synthesized via Aqueous RAFT Polymerization: From Micelles to Vesicles in a Series on Water-soluble Polymers. Macromolecules 2010, 43, 1210–1217. [Google Scholar] [CrossRef]
- Latridi, Z.; Tsitsilianis, C. Water-soluble Stimuli Responsive Star-shaped Segmented Macromolecules. Polymers 2011, 3, 1911–1933. [Google Scholar] [CrossRef]
- Li, G.; Shi, L.; Ma, R.; An, Y.; Huang, N. Formation of Complex Micelles with Double-responsive Channels from Self-assembly of Two Diblock Copolymers. Angew. Chem. Int. Ed. 2006, 45, 4959–4962. [Google Scholar] [CrossRef]
- Kujawa, P.; Segui, F.; Shaban, S.; Diab, C.; Okada, Y.; Tanaka, F.; Winnik, F.M. Impact of End-group Association and Main-chain Hydration on the Thermosensitive Properties of Hydrophobically Modified Telechelic Poly (N-isopropylacrylamides) in Water. Macromolecules 2005, 39, 341–348. [Google Scholar] [CrossRef]
- Xia, Y.; Yin, X.; Burke, N.A.D.; Stover, H.D.H. Thermal Response of Narrow-disperse Poly (N-isopropylacrylamide) Prepared by Atom Transfer Radical Polymerization. Macromolecules 2005, 38, 5937–5943. [Google Scholar] [CrossRef]
- Topp, M.D.C.; Dijkstra, P.J.; Talsma, H.; Feijen, J. Thermosensitive Micelle-Forming Block Copolymers of Poly (ethylene glycol) and Poly (N-isopropylacrylamide). Macromolecules 1997, 30, 8518–8520. [Google Scholar] [CrossRef]
- Dai, L.; Liu, Z.; Zhang, L.; Rong, Y.; Huang, Y.; Chen, T. Temperature-Dependent Self-Assembly/Disassembly of Gold Nanoparticles Oligomers. J. Nanosci. Nanotechnol. 2016, 16, 5826–5832. [Google Scholar] [CrossRef]
- Jiménez, Z.A.; Yoshida, R. Temperature Driven Self-Assembly of a Zwitterionic Block Copolymer That Exhibits Triple Thermoresponsivity and pH Sensitivity. Macromolecules 2015, 48, 4599–4606. [Google Scholar] [CrossRef]
- Martinelli, E.; Guazzelli, E.; Galli, G.; Telling, M.T.F.; Poggetto, G.D.; Immirzi, B.; Domenici, F.; Paradossi, G. Prolate and Temperature-Responsive Self-Assemblies of Amphiphilic Random Copolymers with Perfluoroalkyl and Polyoxyethylene Side Chains in Solution. Macromol. Chem. Phys. 2018, 219, 1800210. [Google Scholar] [CrossRef]
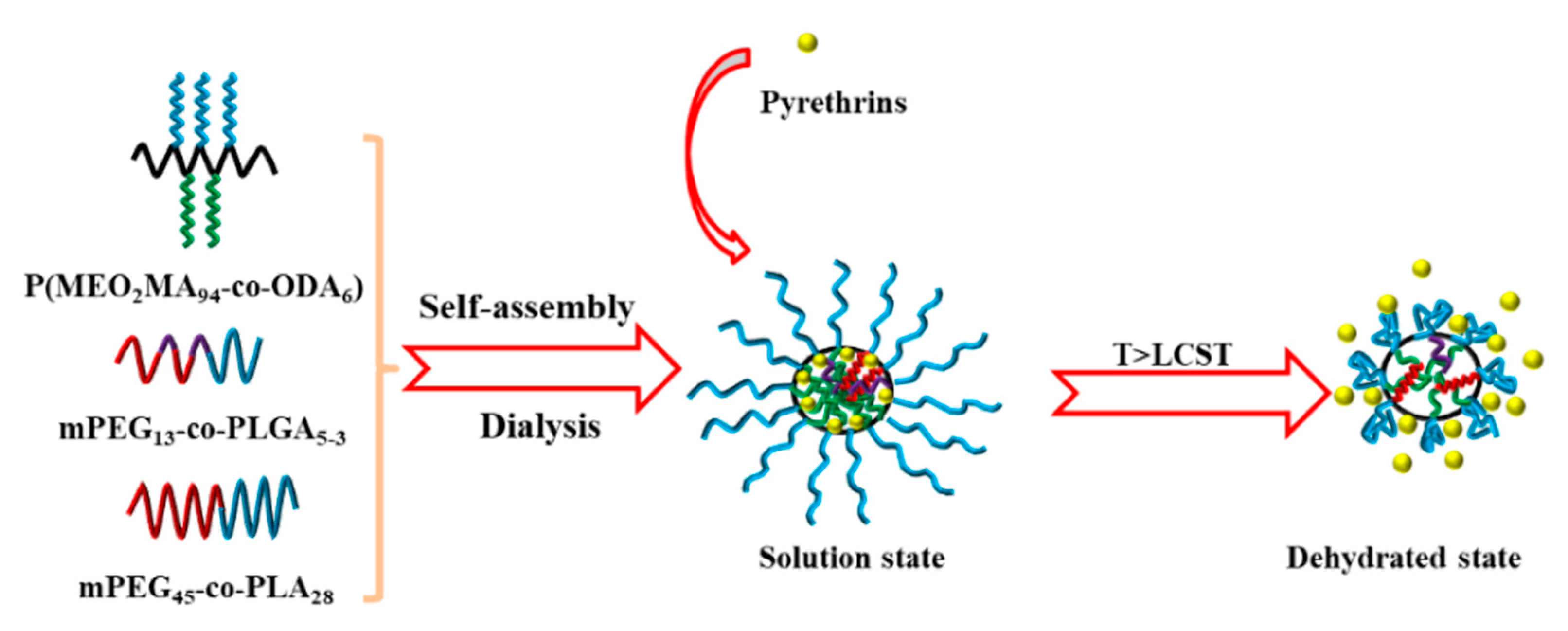
- Zhang, Y.; Chen, W.; Jing, M.; Liu, S.; Feng, J.; Wu, H.; Zhou, Y.; Zhang, X.; Ma, Z. Self-assembled mixed micelle loaded with natural pyrethrins as an intelligent nano-insecticide with a novel temperature-responsive release mode. Chem. Eng. J. 2019, 361, 1381–1391. [Google Scholar] [CrossRef]
- Trenor, S.R.; Shultz, A.R.; Love, B.J.; Long, T.E. Coumarous in Polymers: From Light Harvesting to Photo-cross-linkable Tissue Scaffolds. Chem. Rev. 2004, 104, 3059–3078. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.; Liu, G. Hollow Nanospheres from Polyisoprene-b-poly (2-cinnamoylethyl methacrylate)-b-poly (tert-butyl acrylate). Chem. Mater. 1999, 11, 1048–1054. [Google Scholar] [CrossRef]
- Yan, X.; Liu, G.; Li, Z. Preparation and Phase Segregation of Block Copolymer Nanotube MuItiblocks. J. Am. Chem. Soc. 2004, 126, 10059–10066. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Kreiling, S.; Greiner, A.; Hampp, N. Two-photon-induced Cycloreversion Reaction of Coumarin Photodimers. Chem. Phys. Lett. 2003, 372, 899–903. [Google Scholar] [CrossRef]
- Li, Y.; Deng, Y.; Tong, X.; Wang, X. Formation of Photoresponsive Uniform Colloidal Spheres from an Amphiphilic Azobenzene-containing Random Copolymer. Macromolecules 2006, 39, 1108–1115. [Google Scholar] [CrossRef]
- Wang, G.; Tong, X.; Zhao, Y. Preparation of Azobenzene-containing Amphiphilic Diblock Copolymers for Light-responsive Micellar Aggregates. Macromolecules 2004, 37, 8911–8917. [Google Scholar] [CrossRef]
- Junge, D.M.; Mcgrath, D.V. Photoresponsive Azobenzene-containing Dendrimers with Multiple Discrete States. J. Am. Chem. Soc. 1999, 121, 4912–4913. [Google Scholar] [CrossRef]
- Goodwin, A.P.; Mynar, J.L.; Ma, Y.; Fleming, G.R.; Fréchet, J.M.J. Synthetic Micelle Sensitive to IR Light via a Two-photon Process. J. Am. Chem. Soc. 2005, 127, 9952–9953. [Google Scholar] [CrossRef]
- Jiang, J.; Tong, X.; Morris, D.; Zhao, Y. Toward Photoconlrolled Release Using Light-dissociable Block Copolymer Micelles. Macromolecules 2006, 39, 4633–4640. [Google Scholar] [CrossRef]
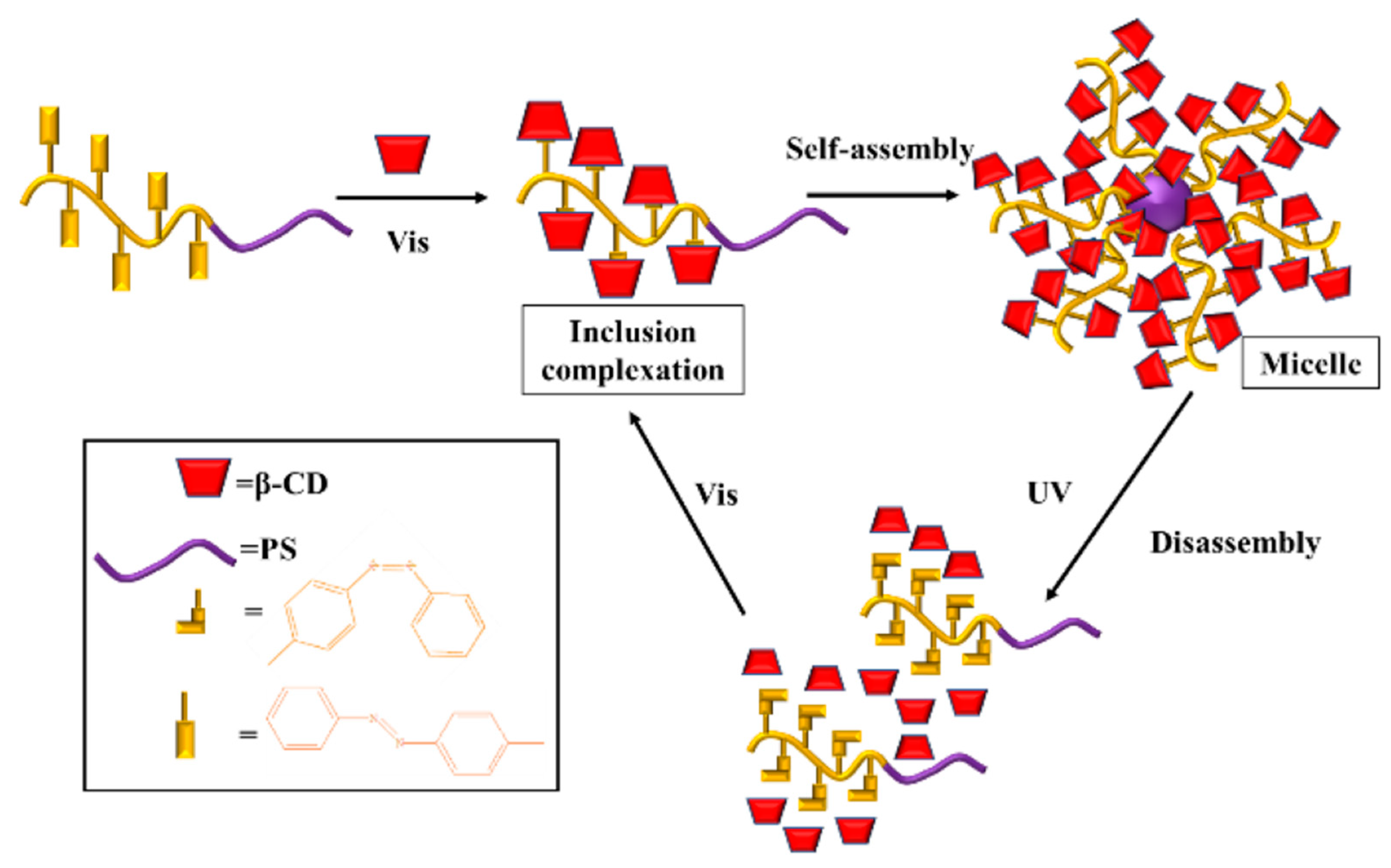
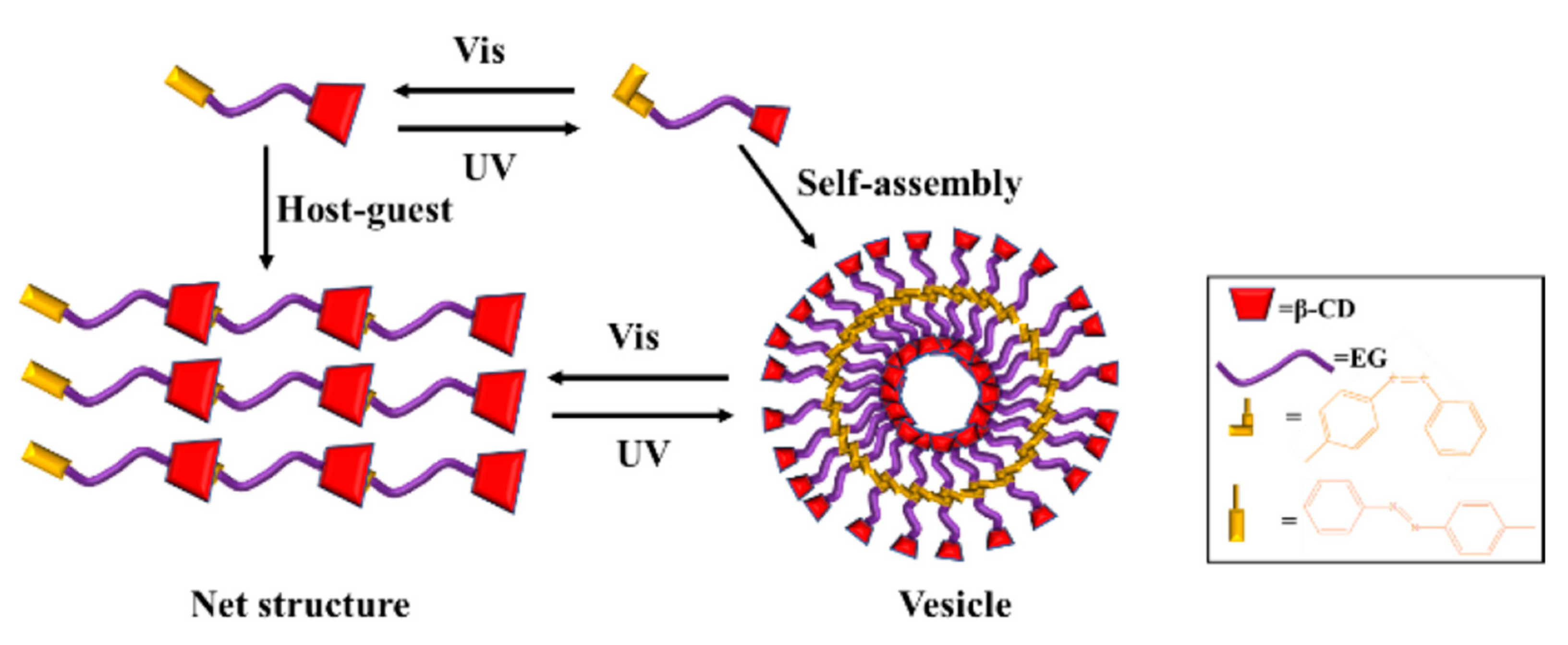
- Wang, S.; Shen, Q.; Nawaz, M.H.; Zhang, W. Photocontrolled Reversible Supramolecular Assemblies of a Diblock Azo-copolymer Based on β-cyclodextrin-Azo Host-guest Inclusion Complexation. Polym. Chem. 2013, 4, 2151–2157. [Google Scholar] [CrossRef]
- Guo, H.; Yang, J.; Zhou, J.; Zeng, L.; Zhao, L.; Xu, B. Photoresponsive self-assembly of a β-cyclodextrin derivative with an azobenzene terminal group in water. Dyes Pig. 2018, 149, 626–632. [Google Scholar] [CrossRef]
- Blasco, E.; Schmidt, B.V.K.J.; Barner-Kowollik, C.; Piñol, M.; Oriol, L. A Novel Photoresponsive Azobenzene-Containing Miktoarm Star Polymer: Self-Assembly and Photoresponse Properties. Macromolecules 2014, 47, 3693–3700. [Google Scholar] [CrossRef]
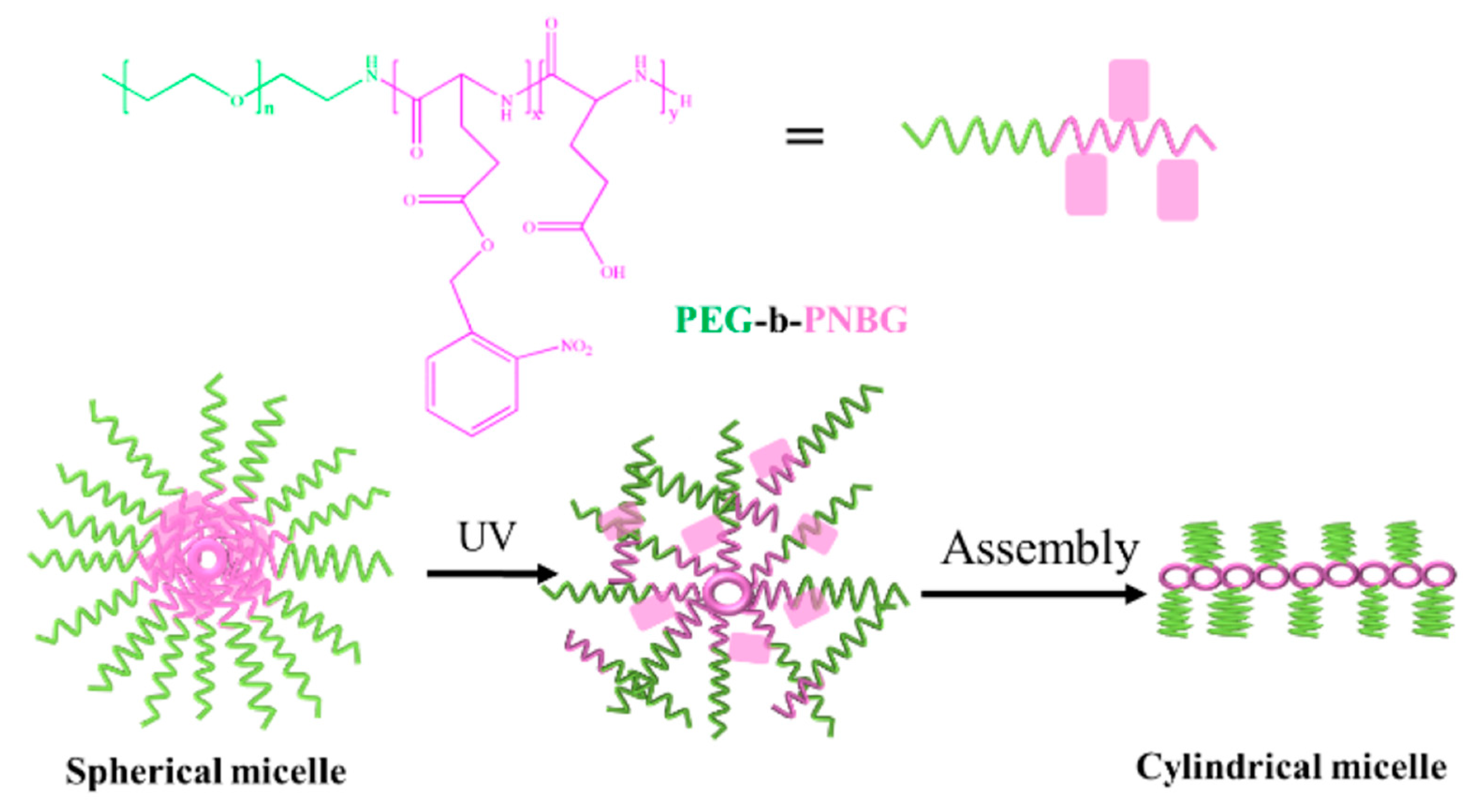
- Ji, S.; Xu, L.; Fu, X.; Sun, J.; Li, Z. Light- and Metal Ion-Induced Self-Assembly and Reassembly Based on Block Copolymers Containing a Photoresponsive Polypeptide Segment. Macromolecules 2019, 52, 4686–4693. [Google Scholar] [CrossRef]
- Floudas, G. Effects of Pressure on Systems with Intrinsic Orientational Order. Prog. Polym. Sci. 2004, 29, 1143–1171. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, J.; Chdcot, F.; Gnanou, Y.; Lecommandoux, S. Toward ‘Smart’ Nano-objects by Self-assembly of Block Copolymers in Solution. Prog. Polym. Sci. 2005, 30, 691–724. [Google Scholar] [CrossRef]
- Agut, W.; Brulet, A.; Schatz, C.; Taton, D.; Lecommandoux, S. pH and Temperature Responsive Polymeric Micelles and Polymersomes by Self-assembly of Poly [2-(dimethylamino) ethyl methacrylate]-b-poly(glutamic acid) Double Hydrophilic Block Copolymers. Langmuir 2010, 26, 10546–10554. [Google Scholar] [CrossRef]
- Roy, D.; Cambre, J.N.; Sumerlin, B.S. Sugar-responsive Block Copolymers by Direct RAFT Polymerization of Unprotected Boronic Acid Monomers. Chem. Commun. 2008, 21, 2477–2479. [Google Scholar] [CrossRef]
- Xu, Y.; He, K.; Wang, H.; Li, M.; Shen, T.; Liu, X.; Yuan, C.; Dai, L. Self-Assembly Behavior and pH-Stimuli-Responsive Property of POSS-Based Amphiphilic Block Copolymers in Solution. Micromachines 2018, 9, 258. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Bao, H.; He, J.; Dong, X. Preparation of multicompartment micelles from amphiphilic linear triblock terpolymers by pH-responsive self-assembly. Colloid Polym. Sci. 2015, 293, 3013–3024. [Google Scholar] [CrossRef]
- Liu, X.; Xie, Y.; Hu, Z.; Chen, Z.; Hu, J.; Yang, L. pH responsive self-assembly and drug release behavior of aliphatic liquid crystal block polycarbonate with pendant cholesteryl groups. J. Mol. Liq. 2018, 266, 405–412. [Google Scholar] [CrossRef]
- Rowan, S.J.; Cantrill, S.J.; Cousins, G.R.L.; Sanders, J.K.M.; Stoddart, J.F. Dynamic Covalent Chemistry. Angew. Chem. Int. Ed. 2002, 41, 898–952. [Google Scholar] [CrossRef]
- Belowich, M.E.; Stoddart, J.F. Dynamic Imine Chemistry. Chem. Soc. Rev. 2012, 41, 2003–2024. [Google Scholar] [CrossRef] [PubMed]
- Rollas, S.; Küçükgüzel, S. Biological Activities of Hydrazone Derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Springsteen, G.; Deeter, S.; Wang, B. The Relationship among pKa, pH, and Binding Constants in the Interactions between Boronic Acids and Diols-It Is Not as Simple as It Appears. Tetrahedron 2004, 60, 11205–11209. [Google Scholar] [CrossRef]
- Springsteen, G.; Wang, B. A Detailed Examination of Boronic Acid–diol Complexation. Tetrahedron 2002, 58, 5291–5300. [Google Scholar] [CrossRef]
- Jackson, A.W.; Fulton, D.A. pH Triggered Self-assembly of Core Cross-linked Star Polymers Possessing Thermoresponsive Cores. Chem. Commun. 2011, 47, 6807–6809. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Mu, J.; Li, C. Synthesis and pH/sugar/salt-sensitivity Study of Boronate Crosslinked Glycopolymer Nanoparticles. New J. Chem. 2013, 37, 796–803. [Google Scholar] [CrossRef]
- Yang, F.; Wang, J.; Cao, L.Y.; Chen, R.; Tang, L.J.; Liu, C.S. Injectable and redox-responsive hydrogel with adaptive degradation rate for bone regeneration. J. Mater. Chem. B 2014, 2, 295–304. [Google Scholar] [CrossRef]
- Yan, Q.; Yuan, J.; Cai, Z.; Xin, Y.; Kang, Y.; Yin, Y. Voltage-responsive Vesicles Based on Orthogonal Assembly of Two Homopolymers. J. Am. Chem. Soc. 2010, 132, 9268–9270. [Google Scholar] [CrossRef]
- Li, P.; Kang, H.; Che, N.; Liu, Z.; Zhang, C.; Cao, C.; Li, W.; Liu, R.; Huang, Y. Synthesis, self-assembly and redox-responsive properties of well-defined hydroxypropylcellulose-graft-poly(2-acryloyloxyethyl ferrocenecarboxylate) copolymers. Polym. Int. 2015, 64, 1015–1022. [Google Scholar] [CrossRef]
- Liu, L.; Rui, L.; Gao, Y.; Zhang, W. Self-assembly and disassembly of redox-responsive ferrocene-containing amphiphilic block copolymer for controlled release. Polym. Chem. 2015, 6, 1817–1829. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Wang, X.; Zheng, W.; Zhang, J.; Shi, F.; Liu, J. Redox-responsive self-assembly PEG nanoparticle enhanced triptolide for efficient antitumor treatment. Sci. Rep. 2018, 8, 12968. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Zhang, L.; Lin, J.; Wang, L. Self-Assembly Behavior of pH- and Thermosensitive Amphiphilic Triblock Copolymers in Solution: Experimental Studies and Self-Consistent Field Theory Simulations. J. Phys. Chem. B 2008, 112, 12666–12673. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhao, B. Tuning Micellization and Dissociation Transitions of Thermo- and pH-Sensitive Poly (ethylene oxide)-b-poly(methoxydi(ethylene glycol) methacrylate-co-methacrylic acid) in Aqueous Solution by Combining Temperature and pH Triggers. Macromolecules 2008, 41, 9366–9375. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, T.; Liu, S. Micellization Kinetics of a Novel Multi-Responsive Double Hydrophilic Diblock Copolymer Studied by Stopped-Flow pH and Temperature Jump. Macromol. Chem. Phys. 2007, 208, 2492–2501. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, C.P.; Song, X.; Zhuo, L.H.; Bu, H.T.; Tian, W. Photo- and pH- dually-responsive β-cyclodextrin-based supramolecular prodrug complexes self-assemblies for programmed drug delivery. Chem.-Asian J. 2018, 13, 3903–3911. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, J.; Li, Q.; Li, J.; He, K.; Shen, T.; Liu, X.; Yuan, C.; Zeng, B.; Dai, L. Novel azobenzene-based amphiphilic copolymers: Synthesis, self-assembly behavior and multiplestimuli-responsive properties. RSC Adv. 2018, 8, 16103–16113. [Google Scholar] [CrossRef]
- Jiang, F.; Chen, S.; Cao, Z.; Wang, G. A photo, temperature, and pH responsive spiropyran-functionalized polymer: Synthesis, self-assembly and controlled release. Polymer 2016, 83, 85–91. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Grzybowski, B. Self-Assembly at All Scales. Science 2002, 295, 2418–2421. [Google Scholar] [CrossRef]
- Chai, L.H.; Peng, X.F. Challenge from Frontier in Chemistry: Dynamic Self-Assembly. Prog. Chem. 2004, 16, 169–173. [Google Scholar]
- Xu, G.H.; Zhou, H.W.; Li, J.; Yin, L.; Zheng, Z.H.; Ding, X.B. Autonomous fluorescence regulation in responsive polymer systems driven by a chemical oscillating reaction. Polym. Chem. 2016, 7, 3211–3215. [Google Scholar] [CrossRef]
- Service, R.F. How Far Can We Push Chemical Self-Assembly. Science 2005, 309, 95. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M.; Boncheva, M. Beyond molecules: Self-assembly of Mesoscopic and Macroscopic Components. Proc. Natl. Acad. Sci. USA 2002, 99, 4769–4774. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, C.; Xu, G.; Gao, J. Stimuli-Responsive Macromolecular Self-Assembly. Sustainability 2022, 14, 11738. https://doi.org/10.3390/su141811738
Jiang C, Xu G, Gao J. Stimuli-Responsive Macromolecular Self-Assembly. Sustainability. 2022; 14(18):11738. https://doi.org/10.3390/su141811738
Chicago/Turabian StyleJiang, Chunqiang, Guohe Xu, and Jianping Gao. 2022. "Stimuli-Responsive Macromolecular Self-Assembly" Sustainability 14, no. 18: 11738. https://doi.org/10.3390/su141811738
APA StyleJiang, C., Xu, G., & Gao, J. (2022). Stimuli-Responsive Macromolecular Self-Assembly. Sustainability, 14(18), 11738. https://doi.org/10.3390/su141811738