Abstract
Macromolecular self-assembly has great potential for application in the field of the design of molecular machines, in molecular regulation, for biological tissue, and in biomedicine for the optical, electrical, and biological characteristics that the assembly unit does not possess. In this paper, the progress in macromolecular self-assembly is systematically reviewed, including its conception, processes and mechanisms, with a focus on macromolecular self-assembly by stimuli. According to the difference in stimuli, macromolecular self-assembly can be classified into temperature-responsive self-assembly, light-responsive self-assembly, pH-responsive self-assembly, redox-responsive self-assembly, and multi-responsive self-assembly. A preliminary study on constructing dynamic macromolecular self-assembly based on a chemical self-oscillating reaction is described. Furthermore, the problems of macromolecular self-assembly research, such as the extremely simple structure of artificial self-assembly and the low degree of overlap between macromolecular self-assembly and life sciences, are analyzed. The future development of stimuli-responsive macromolecular self-assembly should imitate the complex structures, processes and functions in nature and incorporate the chemical-oscillation reaction to realize dynamic self-assembly.
1. Introduction
As a universal phenomenon in nature, self-assembly is a spontaneous process from disorder to order, and simple to complex. For example, the creation of living things, the development and growth of embryos, and the formation of orderly bodies such as trees, rivers and landforms that surround us are all self-assemblies. Usually, these self-assemblies have complex morphologies, highly ordered microstructures, and some specific functions [1]. Therefore, self-assembly has received a great level of attention from scientists and has become a research hotspot.
In 1981, Drexle et al. introduced the assembly mode of living organisms into the discipline of chemistry, resulting in chemical self-assembly [2]. As the core of supramolecular chemistry, self-assembly is the spontaneous formation process of supramolecular structures or aggregates with specific structures and functions [3,4,5,6,7,8,9,10] through non-covalent bond interactions, such as van der Waals forces, hydrogen bonds, π-π interactions, electrostatic interaction, hydrophobic interaction, metal coordination bonds and other non-covalent bonding interactions [11,12,13]. The self-assembly process is driven by these non-covalent bond interactions, which can also maintain the stability and integrity of self-assembly structures. Ultimately, the resulting self-assemblies can exhibit properties that the assembled units do not possess, such as optical, electrical, and biological properties, which have great potential applications in molecular machines, molecular regulation, and controlled-release drug delivery systems [14,15,16].
According to the size of building units, self-assembly can be divided into small molecular self-assembly and macromolecular self-assembly [17,18,19]. Among them, the self-assembly behaviors of small molecules have received attention and have been extensively studied, e.g., in relation to the association of small molecular surfactants in solution. The macromolecular self-assembled units are rich and varied, can be a variety of linear polymers or nonlinear polymers, and can also be biological macromolecules. As a result, macromolecular self-assembly is more complex than small molecular self-assembly. Moreover, since macromolecules have better processing and mechanical properties than smaller molecules, functional materials formed by macromolecular self-assembly have a wider range of applications [20,21,22].
After decades of development, much progress in macromolecular self-assembly has been achieved. Additionally, the self-assembly of macromolecules has become a hot topic in many interdisciplinary fields involving chemistry, physics, materials science, biology, and nanoscience [23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39]. For example, B. Mahesh et al. reported self-assembly driven by the hydrogen bond between the hydroxyl group of hydroxypropylmethylcellulose (HPMC) and the carbonyl functional group of the elastin-based polypeptide poly(GVGIP), and revealed the changes in the wavenumber and intensity of the functional group by FTIR. The research holds promise as an alternative material for a variety of biomedical applications. [40] The study of macromolecular self-assembly evolved from the traditional focus on block copolymer-based self-assembly to the intelligent macromolecular self-assembly that can respond to external stimuli, such as light, temperature, pH, redox, etc. [41,42,43,44,45,46,47]), and is developing towards multiple stimuli-responsive macromolecular self-assembly [48,49,50].
Studies on stimuli-responsive peptide self-assembly were previously summarized by our group [51]. This paper focuses on the latest research results of stimuli-responsive synthetic macromolecular self-assembly. The concepts, processes, and mechanisms of stimuli-responsive macromolecular self-assembly are reviewed, and the characteristics and research progress of stimuli-responsive macromolecular self-assembly based on different stimuli are summarized in detail. Finally, the development of stimuli-responsive macromolecular self-assembly is discussed.
2. Mechanism of Stimuli-Responsive Macromolecular Self-Assembly
The principle of self-assembly is to form a molecular aggregate with a specific arrangement by the non-covalent interaction between one part and another part of a molecule or two molecules [52]. Not all molecules can self-assemble. Self-assembly requires the following two conditions: the driving force and the orienting effect. The driving forces of self-assembly include the van der Waals force, hydrogen bond, electrostatic force, π-π force, hydrophobic force, dynamic covalent bond, surface tension, capillary force and other non-covalent interactions [53,54,55,56,57]. These non-covalent interactions are directional and selective, and most of them show additive and synergistic effects. Although their binding force is not as strong as chemical bonds, they can still provide power for self-assembly and maintain the stability of the assembly. The orienting effect of self-assembly refers to molecular recognition. Molecular recognition here does not simply refer to the mutual recognition between molecules, but also refers to the mutual recognition between various components of the assembly. The recognition, which results in the lowest free energy of the final assembly, contains the following two aspects: one is the spatial complementarity of size and shape between molecules (or modules); the second is the recognition of non-covalent bond interactions such as intermolecular hydrogen bond, electrostatic interaction and π-π interaction [58,59,60]. In conclusion, the self-assembly process is the embodiment of the interaction and combination of various driving forces, and the molecular orienting effect is the key to the formation of high-level ordered assembly.
The specific process of stimuli-responsive macromolecular self-assembly is as follows: In external stimuli (such as pH, temperature, light, oxidation reduction, etc.), some non-covalent interactions (such as hydrogen bonding, hydrophobic forces, π-π interactions, electrostatic interactions, metal coordination bond, etc.) in macromolecules (such as block copolymers [61,62,63,64,65,66,67], random copolymers [68,69,70,71,72,73,74,75,76,77,78], etc.) change, resulting in certain parts of the macromolecular chain segment changing, causing the whole molecule to have different physical and chemical properties. For example, one segment crystallizes easily and another does not, or some segments are well dissolved in a solvent while others are not, and thus microphase separation occurs [79]. However, the different chain segments are still linked by chemical bonds, and the microphase separation is limited, which leads to self-assembly into highly regular supramolecular structures, such as spherical micelles, vesicles, rods, layers, columns, and reticular structures, as shown in Figure 1.
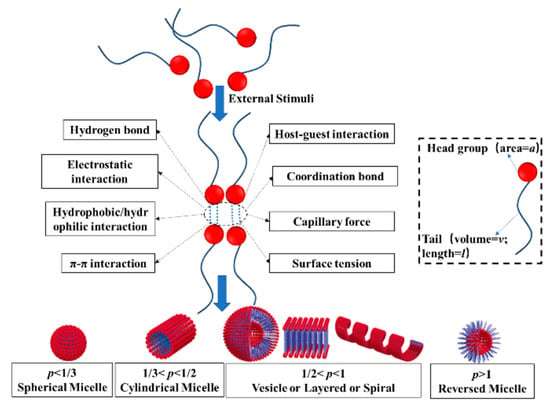
Figure 1.
The mechanism of macromolecular self-assembly [80,81,82].
The final structure of assembly can be predicted by the critical packing parameter theory [80,81,82]. The formula of the theory is p = V/(A·l) (where p is the critical packing parameter, V is the volume of the hydrophobic part of the molecule, A is the cross-sectional area of the hydrophilic part, and l is the average length of the hydrophobic chain segment). According to this theory, as shown in Figure 1, when p < 1/3, the assembly is conical and it is easy to form spherical micelles. When 1/3 < p < 1/2, the assembly is conical with the top part off, and larger ellipsoidal micelles or rod-shaped micelles are easily formed. When 1/2 < p < 1, the assembly is close to the column shape, and it is easy to form a bilayer structure such as a vesicle, lamellar or spiral. When p > 1, it is easy to form reverse structures (e.g., reverse micelles).
3. Research Progress of Stimuli-Responsive Macromolecular Self-Assembly
With the development of macromolecular self-assembly, it is hoped that the structure, morphology and function of self-assembly can be controlled manually. Therefore, some intelligent self-assembly systems that respond to environmental stimuli (such as changes in temperature, pH, light, etc.) have gradually attracted the attention of researchers. According to the different nature of stimuli, it is mainly divided into temperature-responsive self-assembly, light-responsive self-assembly, pH-responsive self-assembly, redox-responsive self-assembly and multi-responsive self-assembly.
3.1. Temperature-Responsive Macromolecular Self-Assembly
Temperature is the most commonly used stimulus, because the change in temperature is easy to control and operates both inside and outside of the organism. Generally speaking, in aqueous solution, macromolecules can respond to temperature stimuli. When the temperature changes in a certain range, a chain segment in the molecule changes from a dissolved to an insoluble phase [83]. The phase transition process of the chain segment is usually divided into two situations [84]: (1) When the temperature rises to a certain temperature, a chain segment changes from a dissolved state to an insoluble state, which is called the Lower Critical Solution Temperature (LCST). (2) When the temperature rises to a certain temperature, a chain segment changes from the insoluble state to the dissolved state, which is called the Upper Critical Solution Temperature (UCST). At present, there are more kinds of macromolecules with LCST.
The common polymer segments used to construct temperature-responsive macromolecular self-assembly are shown in Figure 2, such as poly (N-isopropyl acrylamide) (PNIPAM), poly (N, N-diethyl acrylamide) (PDEAM), poly (N-isopropyl meth acrylamide) (PNIPMAM), polymethyl ethylene ether (PVME), polyvinyl caprolactam (PVCL), etc.
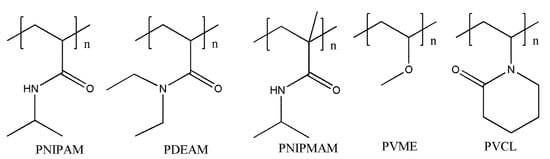
Figure 2.
The temperature-responsive polymer chain segments.
Among them, poly N-isopropyl acrylamide (PNIPAM) is the most frequently researched and applied, and has an LCST (≈32 °C) in water [84], which is just lower than the physiological temperature (~37 °C). The LCST of PNIPAM can be adjusted to the desired temperature by controlling the molecular weight of the polymer or selecting suitable hydrophilic or hydrophobic monomers to copolymerize with it [85,86]. PNIPAM has a hydrophilic amide group and hydrophobic isopropyl group, and the molecular chain shows hydrophilic and hydrophobic changes in a certain temperature range. Therefore, PNIPAM aqueous solution is evidently temperature-responsive. When the temperature was set to lower than its LCST, the intermolecular hydrogen bonding between the PNIPAM chain and water molecule was dominant, and the PNIPAM chain was soluble in aqueous solution. When the temperature was higher than the LCST, the intramolecular hydrogen bonds formed by C=O and N-H on PNIPAM played a dominant role, leading to the collapse of the PNIPAM chain due to hydrophobicity.
Talsma et al. introduced PNIPAM into polyethylene glycol (PEG) to prepare PNIPAM-b-PEG. They found that when the temperature is 25 °C, which is lower than the LCST of PNIPAM blocks, both blocks are fully hydrophilic and can be dissolved in water [87]. However, when the temperature increases to 45 °C, which is higher than the LCST of PNIPAM blocks, PNIPAM blocks become hydrophobic. The block copolymer turns from fully hydrophilic to amphiphilic in aqueous solution; therefore, a micellar structure with PNIPAM as the core and PEG as the shell can be self-assembled, as shown in Figure 3.
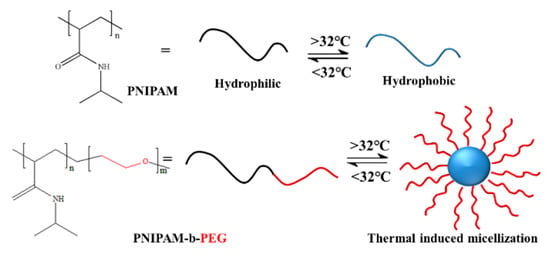
Figure 3.
The temperature-responsive self-assembly of block copolymer PNIPAM-b-PEG [88].
Chen et al. studied the temperature-responsive self-assembly of amphoteric gold nanoparticles (AuNPs) molecules (Au@PEG/PNIPAM) modified by PEG and PNIPAM. As shown in Figure 4, when T < Tc (e.g., at 25 °C), both PEG and PNIPAM are hydrophilic, and Au@PEG/PNIPAM is independently dispersed in aqueous solution. When T > Tc (e.g., at 50 °C), the PNIPAM chain becomes hydrophobic and aggregates at the junction of AuNPs to form a local hydrophobic region [89]. The PNIPAM chain and PEG chain self-assemble into oligomers by phase separation.
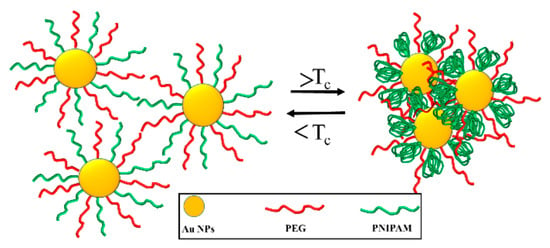
Figure 4.
The temperature-responsive self-assembly based on Au@PEG/PNIPAM [89].
Yoshida et al. studied the temperature-responsive self-assembly of a zwitterionic block copolymer (PNIPAM-b-PMPDSAH) composed of PNIPAM and N, N-dimethyl-N-(3-(methyl acrylamide group) propyl group) amino propane sulfonate [90]. When the temperature was lower than 35 °C, PNIPAM-b-PMPDSAH formed vesicles in aqueous solution. When the temperature of the solution increased (20–40 °C), the vesicles grew from small to large and then from large to small, and gradually disappeared. When the temperature of the solution increased to about 55 °C, macroaggregates (large micelles with PNIPAM as nucleus and PMPDSAH as shell) were formed. As the solution temperature continued to rise, the turbidity of the solution became clear, and a small spherical micelle with PNIPAM as the nucleus and PMPDSAH as the shell was formed, as shown in Figure 5. PNIPAM-b-PMPDSAH has triple thermal response characteristics. The first transition is the UCST-type transition at 20–37 °C. The increase in temperature leads to the destruction of the force between the ions in the copolymer chain. The disintegration of the ionic network leads to the destruction of the vesicle structure. The second transition is the LCST type transition at 37–47 °C. The PNIPAM segment changes from soluble to insoluble, and the polymer recombines into larger aggregates. As the temperature continues to increase, the repulsion between the PMPDSAH chains and PNIPAM spherical aggregates increases; thus, the aggregates depolymerize to form small spherical micelles with a core–shell structure.
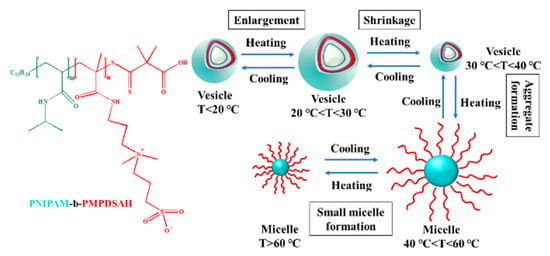
Figure 5.
The temperature-responsive self-assembly based on PNIPAM-b-PMPDSAH [90].
Paradossi et al. reported the temperature-responsive self-assembly of an amphiphilic random copolymer (PEGMA-co-FA) prepared by the atom transfer radical polymerization (ATRP) of hydrophilic polyethylene glycol methacrylate (PEGMA) and hydrophobic perfluorohexyl ethyl acrylate (FA) as raw materials [91]. The copolymer formed a spherical ellipsoid with the FA segment as the core and PEGMA segment as the shell in aqueous solution. When the temperature of the aqueous solution increased to 59 °C, the copolymer formed multi-chain aggregates. The random copolymers containing PEGMA side chains with a lower LCST in solution are soluble in water and partially organic solvents. When the temperature of the aqueous solution is higher than LCST, the interaction between the hydrophilic chains of the copolymer is destroyed. At this time, the hydrophobic force is dominant and water is discharged from the shell, resulting in the merging of collapsed chains to form multi-chain aggregates.
Ma et al. prepared a kind of temperature-responsive cooperative self-assembly system based on poly(2-(2-methoxyethoxy) ethyl methacrylate-co-octadecyl methacrylate) (P(MEO2MA94-co-ODA6)), monomethoxy polyethylene glycol-b-poly (D, L-lactide-co-ethyl lactide) block polymer (mPEG13-b-PLGA5-3) and monomethoxy polyethylene glycol-b-(D, L-lactide) (mPEG45-b-PLA28) [92]. As shown in Figure 6, spherical micelles with poly (2-(2-methoxyethoxy) methacrylate, poly (D, L-lactide-co-ethyl lactide) and poly (D, L-lactide) chain segments as shells and poly (ethylene glycol) and poly (octadecyl methacrylate chain segments as the core can be formed by self-assembly of the system in aqueous solution. When the temperature is higher than 18 °C, the micelles undergo phase transformation to release the encapsulated pyrethrin, which can realize the intelligent release of insecticides. Research shows that P(MeO2MA94-co-ODA6) is temperature-sensitive and has an LCST (≈17.9 °C). When the temperature is higher than LCST, the polyocracy methacrylate chain segment changes from hydrophilic to hydrophobic, and the previous hydration is destroyed, resulting in the destruction of the mixed-mice structure and the release of the contained pyrethroid.
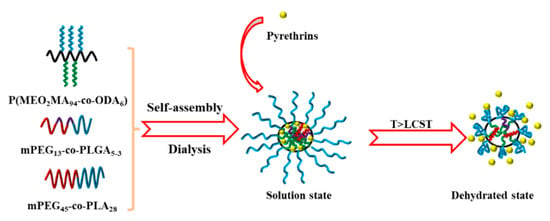
Figure 6.
The temperature-responsive self-assembly based on P(MEO2MA94-co-ODA6), mPEG13-b-PLGA5-3 and mPEG45-b-PLA28 [92].
3.2. Light-Responsive Macromolecule Self-Assembly
Light is a clean and controllable external stimulus. If it is used in the control of macromolecular self-assembly, it has important practical significance for the design of new polymer self-assembly systems and related applications. It is widely used to design and synthesize light-responsive self-assembly systems by introducing light-responsive groups into the main chain or side chain of macromolecules. There are many types of light-responsive groups, such as diazo or azide light-responsive groups (e.g., o-azo sulfonyl, azobenzene), photodimeric light-responsive groups (e.g., coumarin, cinnamate), acrylic groups and light-responsive groups with special functions (e.g., photocatalysis, photochromic and photoconductive groups), etc. These groups are sensitive to light. When the molecules absorb light energy, the corresponding physical properties will change with the help of the absorbed energy (such as solubility, color and conductivity, etc.) or chemical reactions (such as polymerization, dimerization, isomerization, photolysis, etc.), reversible dimerization (such as coumarins, cinnamates, etc.) [93,94,95,96] a reversible dipole change (e.g., cyanine, azobenzene, etc.) [97,98,99] or unidirectional dissociation occurs (such as benzyl esters) [100,101]. Thus, the microstructure of the self-assembly would be transformed or even mutated.
Zhang et al. studied the synthesis of block copolymer PS-b-PAzo containing polystyrene (PS) and azobenzene (Azo) segments [102]. As shown in Figure 7, under visible light (Vis) irradiation, azobenzene exhibited a trans structure, and became hydrophilic by inclusion with β-cyclodextrin (β-CD), which is assembled into micelles with PS as the core and β-CD as the shell. However, under UV irradiation, azobenzene changed from a trans to cis structure, resulting in the separation of Azo from β-CD cavity, and the chain segment changed from hydrophilic to hydrophobic, so that the micelle reassembled into the original polymer chain.
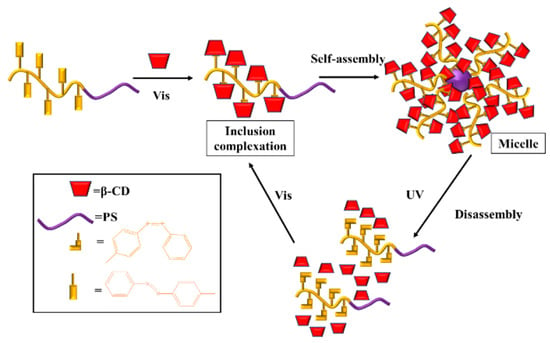
Figure 7.
The UV/Vis-responsive self-assembly of PAzo-b-PS with β-CD [102].
Xu et al. reported the light-responsive macromolecular self-assembly of a supramolecular polymer (Azo-EG-β-CD) synthesized from a β-CD derivative containing an azobenzene terminal group and oligomer ethylene glycol [103]. As shown in Figure 8, Trans-Azo-EG-β-CD forms intermolecular host–guest complexes in the aqueous solution and self-assembles to form a network structure. The conversion of trans azobenzene to cis azobenzene can be induced by ultraviolet light, while the reverse can be induced by visible light. When the Azo-EG-β-CD solution is irradiated with ultraviolet light, the host–guest complex is converted into a single cis-Azo-EG-β-CD molecule; then, the cis-Azo-EG-β-CD molecule self-assembles in water to form a vesicle structure. When irradiated with visible light, it can undergo reverse transformation to form an intermolecular host–guest complex.
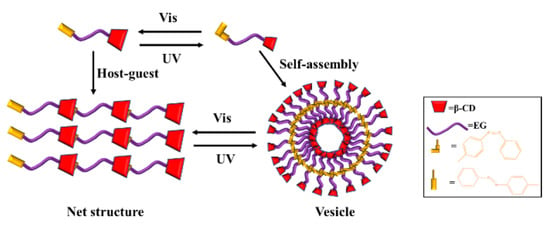
Figure 8.
The UV/Vis-responsive self-assembly of Azo-EG-β-CD [103].
Oriol et al. prepared a method of light-responsive macromolecular self-assembly based on a novel star hetero polymer (PAZO17) (PEG12)3 containing azo phenyl groups [104]. The polymer is made of azobenzene poly(methacrylate) as the light-responsive arm and three poly(N, N-diethyl acrylamide) through an ATRP and azide alkyne cycloaddition reaction. Due to the amphiphilic nature of the polymer, it can self-assemble to form vesicles in water. Under low intensity UV irradiation, the self-assembly of the vesicles is destroyed due to the isomerization of azo phenyl groups (from trans to cis) on the vesicle membrane, leading to the deformation of the membrane. This facilitates the release of molecular probes and delivery drugs.
Li et al. prepared light-responsive self-assembly of PEG-b-poly (o-nitro benzyl-L-type glutamic acid) (PEG-b-PNBG) diblock copolymers with ring-opening polymerization [105]. As shown in Figure 9, after adding deionized water dropwise into THF solvent and removing THF using the dialysis method, PEG-b-PNBG self-assembled into spherical micelles with hydrophobic PNBG as the core and hydrophilic PEG as the shell. After two hours of UV irradiation, it was found that the solution changed from transparent to light yellow and the spherical micelles changed to cylindrical micelles. This is due to the decomposition of o-nitrobenzyl groups under UV irradiation and conversion to o-nitrobenzaldehyde and hydrophilic polyglycolic acid (PGA); the amphiphilicity of block copolymers is destroyed, resulting in the change of the solution’s color and micelle’s morphology.
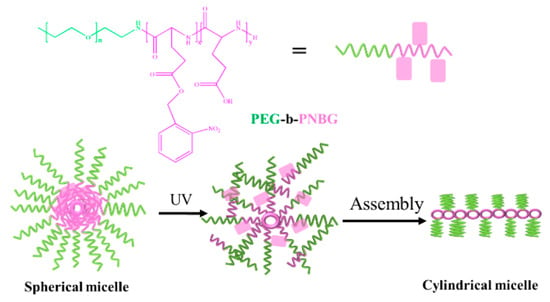
Figure 9.
The light-responsive self-assembly of PEG-b-PNBG [105].
3.3. pH-Responsive Macromolecule Self-Assembly
pH is a very widely used external stimulus. Therefore, pH-responsive macromolecular self-assembly is the most widely studied form of intelligent responsive self-assembly at present. In general, such self-assembled macromolecules usually contain pH-sensitive groups (such as amine, carboxyl, and pyridine groups). As the pH value of the system approaches a certain value, the pH-sensitive segments undergo a reversible conformational transition (sometimes accompanied by a phase transition) [106,107], which causes hydrogen bonding or electrostatic complexation between different blocks or different polymers and changes their solubility. Such macromolecules are generally classified into two types: (1) macromolecules with uncharged basic units (such as amines) that are hydrophilic at low pH and protonate into hydrophobic chain segments at a high pH; (2) macromolecules of charged acidic units (such as carboxyl groups) that are protonated at a low pH and electronegative at a high pH. Common macromolecules with pH responsiveness are as follows: polyacrylic acid (PAA), polyvinyl pyridine (PVPy), polymethacrylic acid (PMAA), polymethylacrylate-N, N-dimethyl aminoethyl ester (PDMAEMA), polyallylamine hydrochloride (PAH), polymethacrylate-2-(diethylamino) ethyl ester (PDEA), etc., as shown in Figure 10.
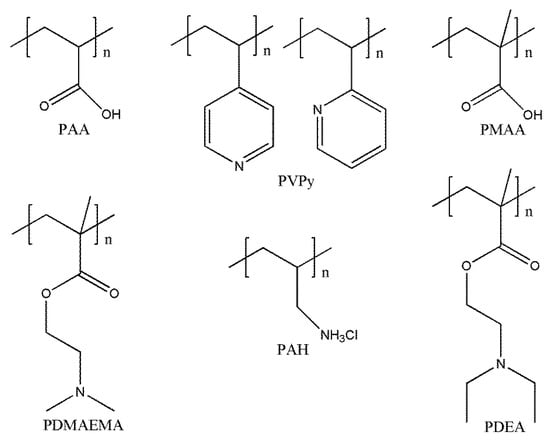
Figure 10.
The common pH-responsive polymers.
Agut et al. synthesized a poly (methacrylate)-N, N-dimethyl aminoethyl ester-B-poly (glutamic acid) copolymer (PDMAEMA-b-PGA) and studied its self-assembly behavior under different pH conditions [108]. The results showed that PDMAEMA85-b-PGA186 and PDMAEMA85-b-PGA77 were self-assembled into nanoscale vesicles at pH 6.2 and 4–6. As calculated using the Henderson–Hasselbalch law, in this environment, more than 90% of the amino groups are protonated, while almost all of the PGA blocks are deprotonated; thus, the formation of the vesicle membrane can be explained by electrostatic interaction. This electrostatic action comes from a large number of carboxyl salts on the PGA block of the oppositely charged groups and protonated amino groups on the PDMAEMA block.
Sumerlin et al. synthesized a PDMA-b-PAPBA block copolymer containing phenylboric acid (PBA) and N, N-dimethylacrylamide (DMA) by using the reversible addition–fragmentation chain transfer polymerization (RAFT) technique and made use of the characteristic that phenylboric acid groups can form a hydrophilic tetrahedral structure with a negative charge under alkaline conditions. The reversible conversion of micelles and polymer chains was realized by changing the pH value, as shown in Figure 11 [109].
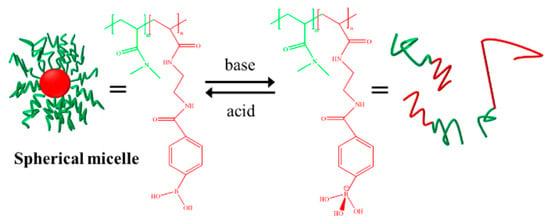
Figure 11.
The pH-responsive self-assembly of PDMA-b-PAPBA [109].
In their work, Xu et al. used methoxy polyethylene glycol (MEPEG), isobutyl methacrylate polyhedral oligosiloxane (MAPOSS), and 2-(diisopropylamino) ethyl methacrylate (DPA). As a raw material, a POSS-modified amphiphilic block copolymer MePEG-b-P(MAPOSS-co-DPA) was prepared for pH-responsive macromolecule self-assembly [110]. As shown in Figure 12, the block copolymer self-assembled in ultra-pure water at room temperature to form a micelle with the hydrophobic block MAPOSS-co-DPA as the core and the hydrophilic block MEPEG as the shell. The micelles formed were pH-responsive. When the pH changes from 3 to 2, the matrix formation of tertiary amines in DPA becomes hydrophilic and diffuses into the shell. At the same time, there is a strong electrostatic repulsion between the protonated tertiary amines, which leads to the greater attraction of DPA to water molecules; thus, the hydrodynamic size of the micelles increases and the micelles increase in size. However, when the pH drops to below 2, the POSS cage detaches and the micelles disassemble, resulting in a decrease in the size of the micelles. When the pH ranges from 3 to 12, the tertiary amine groups in DPA are deprotonated to hydrophobic, which causes the micelles to shrink and the micelles to become smaller.
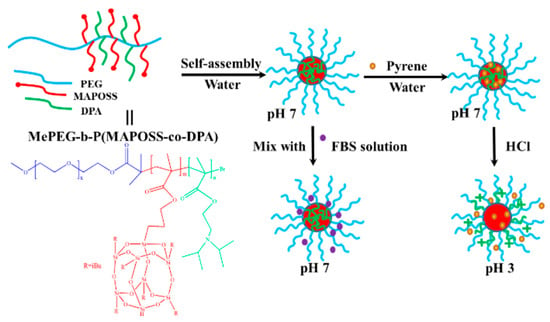
Figure 12.
The pH-responsive self-assembly of MePEG-b-P(MAPOSS-co-DPA) [110].
Xia et al. prepared a triblock copolymer polymethyl methacrylate-b-2-(cinnamoyl) methyl methacrylate block-b-dimethyl aminoethyl methacrylate block segment (PMMA-b-PCEMA-b-PDMAEM) that was pH-responsive with macromolecular self-assembly [111]. In a mixed solution of acetonitrile and isopropanol, the copolymer can form micelles with PCEMA as the core and PMMA and PDMAEM as the shell. After the formed micelles were dialyzed in aqueous solution with pH = 6.86, the PCEMA chain segment cut the core of the micelles to form multi-compartment micelles, and the formed structure changed depending on the proportion of hydrophobic segments. For example, PMMA266-b-PCEMA121-b-PDMAEMA269 was cut from two sides, while PMMA319-b-PCEMA162-b-PDMAEMA316 was cut in the middle. When pH = 4, the amino group in the PCEMA segment is protonated, and the micellar core contracts under the electrostatic force. When pH = 9.16, the amino groups in the PCEMA segment are deprotonated and hydrophobic and shrink, and the micelles become worm-like polymers.
Yang et al. prepared pH-responsive macromolecular self-assembly based on the polysanymethyl carbonate (HTMC)-ethylene glycol (MPEG) block copolymer MPEG43-b-P[(TMC-C)28-HTMC3] containing a 6-cholesterol-oxy-oxanocaproic acid side chain [112]. The block copolymers can self-assemble into spherical micelles with MPEG and HTMC shells, and TMC-C chains as the core in aqueous solution at room temperature. The micelle size increases with the increase in pH at pH < 7, reaches the maximum at pH = 7 and decreases with the further increase in pH. When the pH is less than 7, the hydroxyl group in the HTMC chain segment is protonated, the hydrogen bond between the copolymer and the water molecule is weakened, the force of the hydrophobic segment increases and the micelle shrinks, and the size becomes smaller. When pH > 7, the hydroxyl group ionizes and the hydrophobicity is also enhanced. In addition, the negative charge generated by ionization causes electrostatic repulsion between molecules, which also leads to the decrease in micelle size. Based on this feature, the micelle can be used as a drug carrier to carry the drug doxorubicin (DOX). The experimental results show that the release of the drug DOX can be regulated by controlling the pH value, because the hydrogen bonding forces of DOX and the copolymer are strongest at pH 8.4. When the pH is less than 8.4, partial hydrogen bond forces are reduced by hydroxyl protonation and DOX release is accelerated.
With the development of self-assembly, pH-responsive macromolecule self-assembly is no longer limited to the macromolecule systems containing protonated or deprotonated groups. Some dynamic covalent bonds [113] that can reversibly “form” and “break” with the change of pH have gradually been introduced into the macromolecule self-assembly system. A dynamic covalent bond is a special kind of covalent bond, which has the stability of a covalent bond and the characteristics of reversible “formation” and “fracture” of a non-covalent bond, meaning that the system has strong stability and stimulus responsiveness. Common pH-responsive dynamic covalent bonds are as follows (Figure 13): (a) imine bonds formed by the reaction of primary amines with a carbonyl or aldehyde group [114]; (b) arylhydrazone bonds formed by the reaction of acyl hydrazide group with aldehyde group or ketone [115]; (c) the reaction of a phenyl borate group with a dihydroxyl group to form a phenylboronate bond [116,117].
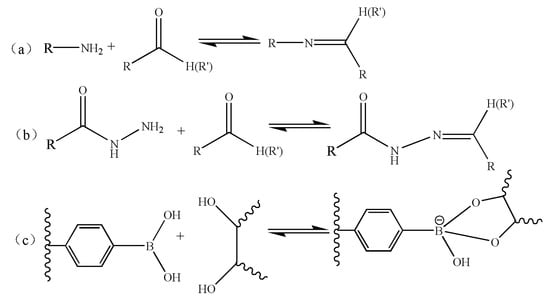
Figure 13.
The common pH-responsive dynamic covalent bonds. (a) Imine [114], (b) arylhydrazone bond [115], (c) phenylboronate bond [116,117].
Fulton et al. synthesized block polymers P1 and P2 containing benzaldehyde and amine groups, respectively (Figure 14) [118]. When the pH of the system is 11, the aldehyde and amine groups will form imine bonds, and the system will form micelles with imine bond chain segments as cores and other chain segments as shells. When the pH of the system decreases to 5.5, the imine bonds break and the micelles dissociate.
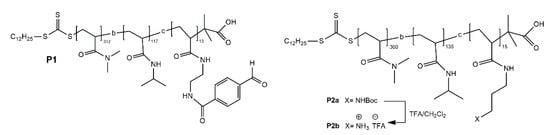
Figure 14.
The structure of P1 and P2 [118].
Zhang et al. synthesized a homopolymer containing phenyl boric acid (PMAPBA) and a block copolymer containing galactose (PEG-b-PAEG) by performing RAFT polymerization and ATRP polymerization, respectively [119]. In an alkaline solution, the phenylboronate group in PMAPBA will form a micelle with PMAPBA and phenyl borate as the core, and PEG chain segments as the shell. When the pH of the solution becomes acidic, the phenyl borate bond breaks and, as a result, the micelles formed dissociate again, as shown in Figure 15.
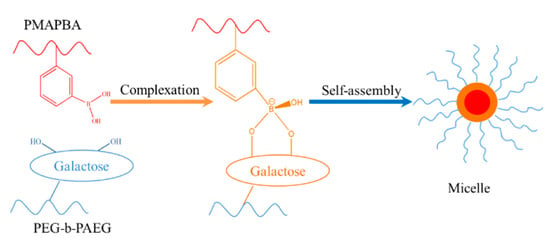
Figure 15.
pH-responsive self-assembly based on phenylboronate bond [119].
3.4. Redox-Responsive Macromolecule Self-Assembly
Redox-responsive macromolecule self-assembly mainly realizes the reversible assembly and disassembly of the macromolecule system by applying an electric field or adding an oxidizing or reducing agent. In general, redox-responsive macromolecule self-assembly systems often contain ferrocene (Fc), a disulfide bond and other groups. For Fc, when in an oxidizing environment, it is oxidized to Fc+, changing from hydrophobic to hydrophilic. When in a reducing environment, Fc+ is reduced to Fc, changing from hydrophilic to hydrophobic. For a disulfide bond, when in an oxidizing environment, the disulfide bond exists stably; when in a reducing environment, the disulfide bond breaks and is reduced to sulfhydryl group (-SH) [120]. These characteristics can be used to regulate system self-assembly. At present, redox-responsive macromolecular self-assembly systems have been rarely studied.
Yan et al. modified β-CD on one end of hydrophobic PS and Fc on one end of hydrophilic poly (ethylene oxide) (PEO) [121]. Based on the host–guest recognition between β-CD and Fc groups, PS-β-CD and PEO-Fc were connected together. A novel amphiphilic block polymer with a controllable structure was prepared. As shown in Figure 16, vesicles can self-assemble in aqueous solution. When applied at 1.5 V, Fc was oxidized to Fc+, and PEO-Fc+ separated from the CD cavity of PS-β-CD, resulting in the dissociation of the vesicles. When the dissociated system was subjected to a voltage of −1.5 V, Fc+ reduced to Fc, which was repackaged with PS-β-CD to form vesicles again. The system can be used for controlled drug release system after drug loading, and controlled drug release can be achieved by regulating vesicle dissociation.
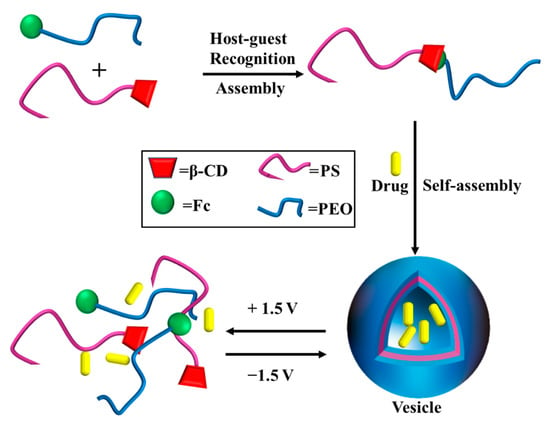
Figure 16.
The redox-responsive self-assembly of PS-β-CD/PEO-Fc [121].
Huang et al. prepared macromolecular self-assembly based on amphiphilic graft copolymer polyhydroxypropylcellulose-graft-2-acryloyloxyethyl ferrocene carboxylate (HPC-g-PAEFC) [122]. The copolymer can self-assemble into spherical micelles in aqueous solution to form spherical micelles with a hydrophilic HPC segment as the shell and a hydrophobic PAEFC segment as the core. When oxidizing substances are added, Fc is oxidized to positively charged Fc+, which results in the change of the ferrocene group from hydrophobic to hydrophilic, and the size of micelles increases due to electrostatic repulsion. When reducing substances are added, Fc+ is reduces to Fc again, the ferrocene group changes from hydrophilic to hydrophobic again, and micelle sizes decrease. However, it cannot fully recover, because a micelle structure formed by graft copolymer prevents the charge transfer and diffusion of reducing agents. As a result, the Fc+ cannot be completely reduced, and the size of the micelle cannot be fully recovered.
Zhang et al. prepared amphiphilic block copolymer polyethylene glycol-b-poly-2-(methylacrylyl oxy) ferrocene carboxylate (PEG-b-PMAEFc) using the ATRP method [123]. As shown in Figure 17, the block copolymer spontaneously assembles in aqueous solution to form vesicles with a redox response. After adding H2O2, Fc was oxidized to Fc+, the ferrocene group changed from hydrophobic to hydrophilic, the vesicle structure was destroyed, and large aggregates were formed. However, the destroyed vesicles could not be reduced after the addition of antioxidant acid, because the oxidized ferrocene was confined to the dense, unoxidized ferrocene structural domain, which impeded the reduction. However, the destroyed vesicles could not be reduced after the addition of antioxidant acid, because the oxidized ferrocene was confined to the dense, unoxidized ferrocene structural domain, which impeded the reduction. Since the structure of the vesicle formed by this self-assembled system is affected by oxidants, it can be used as a drug carrier, which can destroy the vesicle structure to release drugs (e.g., Rhodamine B) in an oxidizing environment.
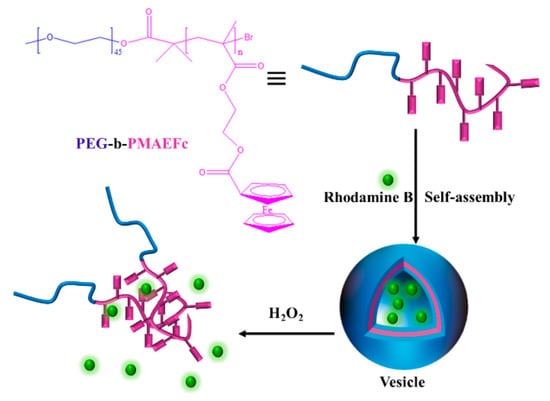
Figure 17.
The redox-responsive self-assembly of PEG-b-PMAEFc [123].
Wang et al. used dithiodiglycolic acid as the coupling agent (SS), and triptolide (TP) and vitamin E (VE) as the main body to form a coupled prodrug (TP-SS-VE) [124]. TP-SS-VE and polyethylene glycol-linoleic acid (PEG200-LD) self-assemble in distilled water to form redox-responsive drug-carrying nanoparticles, which can be used in the treatment of tumor cells. The environment of tumor cells is oxidized due to the large amount of glutathione (GSH) and reactive oxygen species (ROS) produced in tumor cells. Therefore, in the oxidized environment of tumor cells, the disulfide bond in TP-SS-VE is rapidly broken into free sulfhydryl groups, which causes the release of TP to act on tumor cells.
3.5. Multiple-Responsive Self-Assembly
Self-assembly gradually develops from a single stimulus response to multiple stimulus responsiveness. In some special applications, people often require a multiple-responsive ability of self-assembly. It is of great importance to develop multiple-responsive self-assembly. Appropriate stimulation methods can be selected according to the different environments so as to meet actual needs. For example, combined with external changes such as temperature, pH or ionic strength, fully hydrophilic macromolecules can also exhibit interesting micellization behavior in aqueous solutions. Among them, there are more studies on systems with a dual responsiveness of temperature and pH [125,126].
Zhang et al. synthesized a self-assembled block copolymer (PNIPAM232-b-PDEA106) with both temperature and pH responsiveness using ATRP method [127]. The results show that reversible micellar formation and micellar dissociation can be observed under different “temperature-pH” combinations by changing the temperature and pH values of the system (Figure 18), which effectively realizes the dual control of micellar morphology.
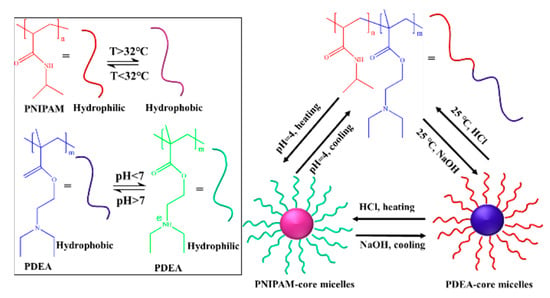
Figure 18.
The temperature- and pH-responsive self-assembly of PNIPAM232-b-PDEA106 [127].
Bai Yang et al. study light/pH supramolecular prodrug complexes (SPCs) based on β-cyclodextrin (β-CD)-aclhydrazone-doxorubicin (DOX) (β-CD-AH-DOX) and the targeting of azobenzene-terminated poly (2-(dimethylamino) ethyl methacrylate) (Azo-PDMA-FA) (β-CD-AH-DOX), as shown in Figure 19 [128]. After adding water dropwise into the DMF solution of SPCS and removing DMF by performing dialysis, SPCS self-assembled and formed multi-compartment vesicles, with a slow-release rate of DOX. This is due to the amphipathicity of SPCs, and the formation of multicompartment vesicles is caused by the aggregation of small vesicles with weak hydrophobicity. When the solution is irradiated with UV, azobenzene changes from a trans configuration to a cis configuration, resulting in the host–guest interaction with β-CD being destroyed. Then, the inclusion complex is dissociated, and multi-compartment vesicles transfer composite micelles. Therefore, the DOX release speed is accelerated. When the solution was irradiated with visible light, azobenzene changed from a cis configuration to trans configuration, and formed an inclusion complex with β-CD-AH-Dox again due to host and guest interaction. The composite micelles were able to change back to multi-compartment vesicles again, and the release rate of DOX slowed down again. When the pH of the adjusting solution was gradually reduced from 7.4 to 5, the acylhydrazone bond was partially broken, and a large amount of hydrophobic DOX was released. The research is expected to lead to the development of cancer treatments.
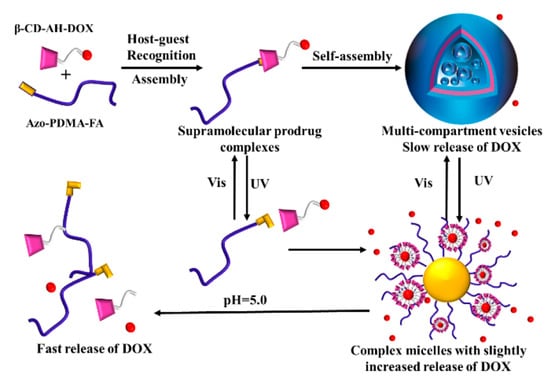
Figure 19.
The Light/pH-responsive self-assembly of β-CD-AH-DOX and Azo-PDMA-FA [128].
Xu et al. prepared a light/pH/redox-triple responsive self-assembly system based on amphiphilic random copolymers P(POSSMA-co-AZOMA-co-DMAEMA) (Figure 20) [129]. The amphiphilic random copolymers have a combination of photoreactive and reductive azobenzene groups, pH-responsive tertiary amine groups and superhydrophobic POSS parts with multiple-responsive characteristics. In aqueous solution, P(POSSMA-co-AZOMA-co-DMAEMA) self-assembled to form spherical micelles with hydrophobic POSSMA and AZOMA as cores and hydrophilic DMAEMA as shells. Under UV irradiation, azobenzene groups and azophenyl groups undergo isomerization from trans configuration to cis configuration, resulting in a reduction in the micellar molecular size and the formation of more compact aggregates in the micellar core. When the pH of the solution decreases from 6.9 to 6.0, the size of the micelles increases significantly. When the pH is 6.5, the spherical structure of the micelles is destroyed and irregular polymer aggregates are formed. When the pH is further reduced to 5.5, the structure of the micelles is destroyed. The variation of micelles with pH is attributed to the protonation of tertiary amine groups in DMAEMA and AZOMA. When sodium disulfite is added to the solution, the azobenzene side chains with a reduction response are completely decomposed and disappear into more hydrophilic aniline, which results in the destruction of the hydrophilic–hydrophobic balance of the copolymer in the solution and the deformation of the polymer micelles into irregular nanometer aggregates.
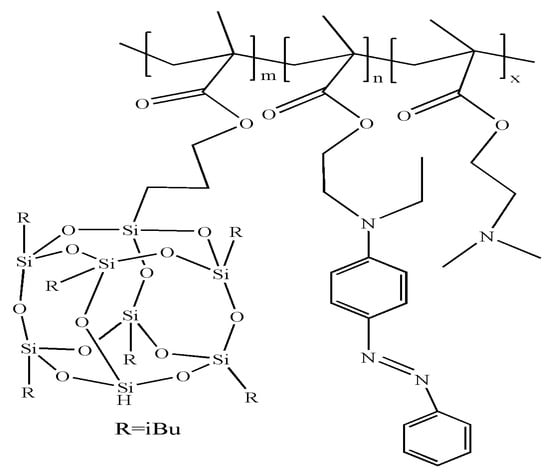
Figure 20.
The structure of P(POSSMA-co-AZOMA-co-DMAEMA) [129].
Wang et al. synthesized temperature-, pH- and light-responsive self-assembly copolymer P(DMAEMA-SP) using spiropyran derivative (SPN-Cl) and poly (dimethyl aminoethyl methacrylate) (PDMAEMA) as raw materials [130]. P(DMAEMA-SP) was self-assembled in aqueous solution to form nanoparticles with SPN-Cl as the core and PDMAEMA as the shell. When the nanoparticles are irradiated with ultraviolet light, the SP part changes from hydrophobic to hydrophilic, the hydrophilic–hydrophobic balance of the copolymer is destroyed, and the self-assembled nanoparticles are dissociated. When the nanoparticles irradiated by ultraviolet light are exposed to visible light, the SP part changes from hydrophilic to hydrophobic, and the nanoparticles recover. When the temperature is higher than the LCST, the hydrogen bonds between PDMAEMA and water break, the hydrophobicity increases, and the polymer nanoparticles shrink. Under acidic conditions, due to the protonation of the tertiary amine group, the hydrophilicity of the PDMAEMA chain segment increased and the polymer nanoparticles expanded. Under alkaline conditions, due to the deprotonation of tertiary amine groups, the hydrophobicity of PDMAEMA nanoparticles increases, which causes the polymer nanoparticles to shrink.
4. Dynamic Self-Assembly of Macromolecules
According to the background conditions of the occurrence of the self-assembly phenomenon, the self-assembly phenomenon can be divided into static self-assembly and dynamic self-assembly [131]. Static self-assembly refers to self-assembly which can be completed and maintained without energy consumption under the condition of equilibrium and does not change with time. Dynamic self-assembly refers to self-assembly for which the system must be completed and maintained under the condition of consuming energy or material flow, and the structure often changes dynamically with time. At present, most self-assembly studies (including the above macromolecular self-assembly systems) focus on static self-assembly. However, the birth of cells, the development of organisms, the formation of trees, rivers, streets, clouds, landforms and other natural ordered bodies with a wider sense of self-assembly all belong to the category of dynamic self-assembly. Obviously, the study of dynamic self-assembly is a more attractive and challenging frontier problem [132]. However, dynamic self-assembly is still in the primary stage of research, and the research results are few.
In our group, based on a poly (N, N-dimethylacrylamide)-b-poly (N-(4-ethyl formate phenyl borate))-allylamine (PDMA33-b-PAPBA17/ARS) and H2O2-Na2S2O4 (HPD) pH oscillation reaction, a macromolecular dynamic self-assembly system with the function of “firefly” was constructed [133]. As shown in Figure 21, by using the independent, reversible and cyclic change of pH value of the pH oscillating reaction in a continuous flow stirred reactor (CSTR), the reversible and controllable dynamic covalent bonds can be driven to “form” and “break” independently, reversibly and cyclically, thus achieving the function of the system to produce the fluorescent light–dark change similar to that of “firefly”. When in a neutral environment, the PBA group and ARS can assemble into a complex through the dynamic covalent bond of the phenyl boronic acid ester group to produce fluorescence. Under the acidic condition (pH < 4) or alkaline condition (pH > 9), the dynamic covalent bond of the phenyl borate ester group dissociates; then, the assembled complex disassembles; and finally, the fluorescence disappears. Due to the continuous characteristics of CSTR, the pH oscillation can continue, thereby ensuring the continuous, dynamic and reversible self-assembly process.
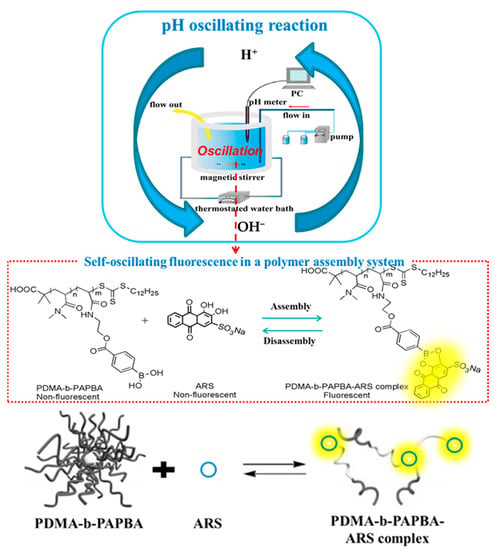
Figure 21.
The dynamic self-assembly based on PDMA33-b-PAPBA17/ARS and pH oscillating-reaction [133].
5. Conclusions and Prospects
Macromolecular self-assembly research has made great progress, such as in the development of macromolecular self-assembly with different structures or morphologies, which include self-assembled structures with a controllable and adjustable morphology. Additionally, preliminary explorations of the application of molecular self-assembly (such as drug-controlled release) have been performed. It is now possible to control molecular self-assembly to a certain extent to produce ordered assemblies of slightly more complex structures or functions.
Nevertheless, the current research on macromolecular self-assembly still requires development for the field to reach maturity, mainly in the following two aspects: (1) compared with the highly ordered and complex self-assembly structures in nature, the current research on chemical self-assembly is too simple. For example, in early July 2005, on the occasion of its 125th anniversary, the Science journal proposed “How far can we push forward chemical self-assembly” [134], pointing out that future studies on self-assembly should be more complex and take inspiration from nature; (2) at present, there is a low degree of crossover between self-assembly research and the life sciences field. Whitesides [135], as a famous scholar in the field of self-assembly, believe that one of the ultimate goals of self-assembly research is to understand and simulate some biological processes in nature.
It can be seen that in order to develop the self-assembly of macromolecules, it is necessary to strengthen the intersection of self-assembly research and the field of life sciences, imitating the self-assembled structure, and precise assembly law seen in nature. Then, a certain function of living organisms in nature will be achieved.
However, at present, the realization of a self-assembly structure or function is mostly a process of system perception and response to some external stimulus, and tends towards a new and relatively stable state. In general, this process is controlled by equilibrium thermodynamics. This means that the responsive process is temporary, and the system can only make a response or show a specific state. Therefore, in order for the self-assembly system to undergo a state change or exhibit a new responsive behavior, the external “on/off” stimulation is still an indispensable condition. Because of this dependence, it is difficult for the system to realize autonomous, cyclic and reversible self-assembly behavior similar to that seen in some self-assembly bodies in nature (such as the heart beating at a certain frequency, brainwave, clock flower opening and closing, etc.). If a kind of independent, continuous, circular source of external stimuli (such as a chemical oscillating reaction) was developed to replace the “on/off” stimulation currently required, a similar self-assembly system with the characteristics of oscillation behavior, independence, circulation, and a reversible response may be feasible. This will undoubtedly open up a new pathway in the research and development of macromolecular self-assembly.
Author Contributions
Writing—original draft preparation, C.J.; investigation, J.G.; writing—review and editing, funding acquisition, G.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Science and Technology Research Project of Hebei Education Department of China (QN2020265), Hebei Province Graduate Student Innovation Ability Training Project of China (CXZZSS2022101), Natural Science Foundation of Hebei Province (No. C2019204010), Specialized Research Fund for the Doctoral Program of Hebei Agricultural University (ZD201708).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, J.M.; Jiang, J.H.; Lin, S.; Cornel, E.J.; Li, C.; Du, J.Z. Polymersomes: From Macromolecular Self-Assembly to Particle Assembly. Chin. J. Chem. 2022, 40, 1842–1855. [Google Scholar] [CrossRef]
- Drexler, K.E. Molecular engineering: An approach to the development of general capabilities for molecular manipulation. Proc. Natl. Acad. Sci. USA 1981, 78, 5275–5278. [Google Scholar] [CrossRef] [PubMed]
- Li, D.H.; Wu, M.J.; Chen, X.F.; Liu, J.H.; Sun, Y.; Huang, J.; Zou, Y.L.; Wang, X.P.; Chen, D.Y.; Zhang, K.K. Boosting Organic Afterglow Performance via a Two-Component Design Strategy Extracted from Macromolecular Self-Assembly. J. Phys. Chem. Lett. 2022, 13, 5030–5039. [Google Scholar] [CrossRef]
- Theato, P.; Sumerlin, B.S.; O’Reilly, R.K. Stimuli responsive materials. Chem. Sov. Rev. 2013, 42, 7055–7056. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Shao, Y.G.; Yan, F.Z.; Zhang, Z.B.; Li, S.J. Construction of Supramolecular Polymers with Different Topologies by Orthogonal Self-Assembly of Cryptand-Paraquat Recognition and Metal Coordination. Molecules 2021, 26, 952. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.J.; Pashuck, E.T. (Macro)molecular self-assembly for hydrogel drug delivery. Adv. Drug Deliv. Rev. 2021, 172, 275–295. [Google Scholar] [CrossRef]
- Xing, P.; Chen, H.; Bai, L.; Hao, A.; Zhao, Y. Superstructure Formation and Topological Evolution Achieved by Self-Organization of a Highly Adaptive Dynamer. ACS Nano 2016, 10, 2716–2727. [Google Scholar] [CrossRef]
- Ke, D.; Zhan, C.; Xu, S.; Ding, X.; Peng, A.; Sun, J.; He, S.; Li, A.D.; Yao, J. Self-assembled hollow nanospheres strongly enhance photoluminescence. J. Am. Chem. Soc. 2011, 133, 11022–11025. [Google Scholar] [CrossRef]
- Li, L.Y.; Raghupathi, K.; Song, C.; Prasad, P.; Thayumanavan, S. Self-assembly of random copolymers. Chem. Commun. 2014, 50, 13417–13432. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.J.; Lou, Z.C. Recent Advancements in MOF/Biomass and Bio-MOF Multifunctional Materials: A Review. Sustainability 2022, 14, 5768. [Google Scholar] [CrossRef]
- Liu, D.D.; Yang, S.Q.; Peng, S.J.; Chen, Y.; Zhang, L.; Tan, J.B. Simultaneous Synthesis and Self-Assembly of Bottlebrush Block Copolymers at Room Temperature via Photoinitiated RAFT Dispersion Polymerization. Macromol. Rapid Commun. 2022, 43, 2100921. [Google Scholar] [CrossRef] [PubMed]
- Dergham, M.; Lin, S.M.; Geng, J. Supramolecular Self-Assembly in Living Cells. Angew. Chem. Int. 2022, 61, e202114267. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Zhen, F.H.; Chao, W.; Lei, Z.; Qian, Y.X.; Su, Y.W.; Yan, H.W.; Duo, P.; Bin, B.D.; Ren, B.W.; et al. The recent progress of synergistic supramolecular polymers: Preparation, properties and applications. Chem. Commun. 2021, 57, 1413–1429. [Google Scholar] [CrossRef]
- Zhang, Z.K.; Ma, R.J.; Shi, L.Q. Cooperative macromolecular self-assembly toward polymeric assemblies with multiple and bioactive functions. Acc. Chem. Res. 2014, 47, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.; Steed, J.W. Supramolecular Gel Phase Crystallization: Orthogonal Self-assembly under Non-equilibrium Conditions. Chem. Soc. Rev. 2014, 43, 2080–2088. [Google Scholar] [CrossRef]
- Mishra, R.K.; Das, S.; Vedhanarayanan, B.; Das, G.; Ajayaghosh, A. Chapter 7: Stimuli-responsive Supramolecular Gels. In Molecular Gels: Structure and Dynamics; Royal Society of Chemistry: London, UK, 2018; pp. 190–226. [Google Scholar] [CrossRef]
- Zhang, D.W.; Zhao, X.; Hou, J.L.; Li, Z.T. Aromatic Amide Foldamers: Structures, Properties, and Functions. Chem. Rev. 2012, 112, 5271–5316. [Google Scholar] [CrossRef]
- Cheng, X.X.; Miao, T.F.; Qian, Y.L.; Zhang, Z.B.; Zhang, W.; Zhu, X.L. Supramolecular Chirality in Azobenzene-Containing Polymer System: Traditional Postpolymerization Self-Assembly Versus In Situ Supramolecular Self-Assembly Strategy. Int. J. Mol. Sci. 2020, 21, 6186. [Google Scholar] [CrossRef]
- Tao, H.; Galati, E.; Kumacheva, E. Temperature-Responsive Self-Assembly of Nanoparticles Grafted with UCST Polymer Ligands. Macromolecules 2018, 51, 6021–6027. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, Y.; Wu, G.; Liu, J.R.; Cao, N.; Wang, L.; Wang, Y.; Li, X.; Hong, X.; Yang, C.; et al. Temperature-dependent Self-Assembly of a Purely Organic Cage in Water. Chem. Commun. 2018, 54, 3138–3141. [Google Scholar] [CrossRef]
- Pan, Q.; Zong, Z.; Shen, J.; Xue, H.; Pu, Y. Synthesis, self-assembly, and pH-responsive Drug Release of PHMEMA-PEG-PHMEMA ABA triblock Copolymers. J. Macromol. Sci. A 2018, 55, 691–697. [Google Scholar] [CrossRef]
- Wang, T.P.; Shen, L.Y.; Lin, F.Y.; Forrester, M.; Torres, S.W.; Robison, T.W.; Lee, T.H.; Goyal, S.; Cochran, E.W. Standalone Block Copolymer Nanoballoons: Decoupling Self-Assembly from Implementation in Nanomanufacturing. ACS Appl. Polym. Mater. 2022, 4, 5134–5143. [Google Scholar] [CrossRef]
- Yi, M.H.; Guo, J.Q.; He, H.J.; Tan, W.Y.; Harmon, N.; Ghebreyessus, K.; Xu, B. Phosphobisaromatic motifs enable rapid enzymatic self-assembly and hydrogelation of short peptides. Soft Matter. 2021, 17, 8590–8594. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, K.; Kelderman, J.; Coleman, H.; Aguilar, M.I.; Parkington, H.; Del Borgo, M. Self-assembly of trifunctional tripeptides to form neural scaffolds. J. Mater. Chem. B 2021, 9, 4475–4479. [Google Scholar] [CrossRef] [PubMed]
- Percec, V.; Peterca, M.; Tadjiev, T.; Zeng, X.; Ungar, G.; Leowanawat, P.; Aqad, E.; Imam, M.R.; Rosen, B.M.; Akbey, U.; et al. Self-Assembly of Dendronized Perylene Bisimides into Complex Helical Columns. J. Am. Chem. Soc. 2011, 133, 12197–12219. [Google Scholar] [CrossRef] [PubMed]
- Avestro, A.J.; Belowich, M.E.; Stoddart, J.F. Cooperative Self-assembly: Producing Synthetic Polymers with Precise and Concise Primary Structures. Chem. Soc. Rev. 2012, 41, 5881–5895. [Google Scholar] [CrossRef]
- Forgan, R.S.; Gassensmith, J.J.; Cordes, D.B.; Boyle, M.M.; Hartlieb, K.J.; Friedman, D.C.; Slawin, A.M.Z.; Stoddart, J.F. Self-Assembly of a 2 Pseudorota 3 Catenane in Water. J. Am. Chem. Soc. 2012, 134, 17007–17010. [Google Scholar] [CrossRef]
- Gassensmith, J.J.; Erne, P.M.; Paxton, W.F.; Frasconi, M.; Donakowski, M.D.; Stoddart, J.F. Patterned Assembly of Quantum Dots onto Surfaces Modified with Click Microcontact Printing. Adv. Mater. 2013, 25, 223–226. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, Y.; Jiang, M.; Chen, D. Cascade Molecule-Particle-Molecule Self-Assemblies for Fabricating Narrowly Size-Distributed Polymeric Superparticles with a Bicontinuous Nanostructure. ACS Macro Lett. 2012, 1, 1312–1316. [Google Scholar] [CrossRef]
- Wei, K.; Li, J.; Chen, G.; Jiang, M. Dual Molecular Recognition Leading to a Protein-Polymer Conjugate and Further Self-Assembly. ACS Macro Lett. 2013, 2, 278–283. [Google Scholar] [CrossRef]
- Zhang, K.; Jiang, M.; Chen, D. DNA/Polymeric Micelle Self-Assembly Mimicking Chromatin Compaction. Angew. Chem. Int. Ed. 2012, 51, 8744–8747. [Google Scholar] [CrossRef]
- Zhang, K.; Jiang, M.; Chen, D. Self-assembly of Particles: The regulatory role of Particle Flexibility. Prog. Polym. Sci. 2012, 37, 445–486. [Google Scholar] [CrossRef]
- Guo, W.; Wang, X.P.; Wang, X.P.; Zhang, K.K. Achieving Purely-Organic Room-Temperature Aqueous Phosphorescence via a Two-Component Macromolecular Self-Assembly Strategy. Chem. Asian J. 2020, 15, 3469–3474. [Google Scholar] [CrossRef]
- Tang, Y.; Zhou, L.P.; Li, J.; Luo, Q.A.; Huang, X.; Wu, P.; Wang, Y.G.; Xu, J.Y.; Shen, J.C.; Liu, J.Q. Giant Nanotubes Loaded with Artificial Peroxidase Centers: Self-Assembly of Supramolecular Amphiphiles as a Tool To Functionalize Nanotubes. Angew. Chem. Int. Ed. 2010, 49, 3920–3924. [Google Scholar] [CrossRef]
- Wang, G.; Wang, C.; Wang, Z.; Zhang, X. Bolaform Superamphiphile Based on A Dynamic Covalent Bond and Its Self-Assembly in Water. Langmuir 2011, 27, 12375–12380. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, G.T.; Wang, Z.Q.; Zhang, X. A pH-Responsive Superamphiphile Based on Dynamic Covalent Bonds. Chem. Eur. J. 2011, 17, 3322–3325. [Google Scholar] [CrossRef] [PubMed]
- Huan, X.Y.; Wang, D.L.; Dong, R.J.; Tu, C.L.; Zhu, B.S.; Yan, D.Y.; Zhu, X.Y. Supramolecular ABC Miktoarm Star Terpolymer Based on Host-Guest Inclusion Complexation. Macromolecules 2012, 45, 5941–5947. [Google Scholar] [CrossRef]
- Sun, W.Q.; He, X.H.; Gao, C.Y.; Liao, X.J.; Xie, M.R.; Lin, S.L.; Yan, D.Y. Novel Amphiphilic and Photo-responsive ABC 3-miktoarm Star Terpolymers: Synthesis, Self-assembly and Photo-responsive behavior. Polym. Chem. 2013, 4, 1939–1949. [Google Scholar] [CrossRef]
- Tao, W.; Liu, Y.; Jiang, B.; Yu, S.; Huang, W.; Zhou, Y.; Yan, D. A Linear-Hyperbranched Supramolecular Amphiphilic and Its Self-Assembly into Vesicles with Great Ductility. J. Am. Chem. Soc. 2012, 134, 762–764. [Google Scholar] [CrossRef]
- Kathyayani, D.; Mahesh, B.; Chamaraja, N.A.; Madhukar, B.S.; Channe Gowda, D. Synthesis and structural characterization of elastin-based polypentapeptide/hydroxypropylmethylcellulose blend films: Assessment of miscibility, thermal stability and surfacecharacteristics. Colloids Surf. A 2022, 649, 129503–129517. [Google Scholar] [CrossRef]
- Zhou, J.; He, R.; Ma, J. RAFT-Mediated Polymerization-Induced Self-Assembly of Poly(Acrylic Acid)-b-Poly(Hexafluorobutyl Acrylate): Effect of the pH on the Synthesis of Self-Stabilized Particles. Polymers 2016, 8, 207. [Google Scholar] [CrossRef]
- Xiao, D.; Jia, H.Z.; Ma, N.; Zhuo, R.X.; Zhang, X.Z. A Redox-Responsive Mesoporous Silica Nanoparticle Capped with Amphiphilic Peptieds by Self-assembly for Cancer Targeting Drug Delivery. Nanoscale 2015, 7, 10071–10077. [Google Scholar] [CrossRef] [PubMed]
- Chong, D.; Tan, J.; Zhang, J.; Zhou, Y.; Wan, X.; Zhang, J. Dual electrical switching permeability of vesicles via redox-responsive self-assembly of amphiphilic block copolymers and polyoxometalates. Chem. Commun. 2018, 54, 7838–7841. [Google Scholar] [CrossRef]
- Picco, A.S.; Yameen, B.; Knoll, W.; Ceolín, M.R.; Azzaroni, O. Temperature-driven self-assembly of self-limiting uniform supraparticles from non-uniform unimolecular micelles. J. Colloid Interface Sci. 2016, 471, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yuan, D.; Yuan, T.; Dong, J.; Feng, N.; Han, G. A Visible Light Responsive Azobenzene-Functionalized Polymer: Synthesis, Self-Assembly, and Photoresponsive Properties. J. Polym. Sci. A 2015, 53, 2768–2775. [Google Scholar] [CrossRef]
- Wu, A.; Lu, F.; Sun, P.; Gao, X.; Shi, L.; Zheng, L.Q. Photo-Responsive Self-Assembly of Surface Active Ionic Liquid. Langmuir 2016, 32, 8163–8170. [Google Scholar] [CrossRef]
- Shao, L.; Hua, B.; Sun, J.; Li, Q.; Yang, J.; Yu, G. A cucurbit[7]uril-based supra-amphiphile: Photo-responsive self-assembly and application in controlled release. Tetrahedron Lett. 2017, 58, 1863–1867. [Google Scholar] [CrossRef]
- Jiang, Y.; Geng, T.; Li, Q.; Li, G.; Ju, H. Influences of temperature, pH and salinity on the surface property and self-assembly of 1:1 salt-free catanionic surfactant. J. Mol. Liq. 2014, 199, 1–6. [Google Scholar] [CrossRef]
- Chung, C.Y.S.; Yam, V.W.W. Dual pH- and Temperature-Responsive Metallosupramolecular Block Copolymers with Tunable Critical Micelle Temperature by Modulation of the Self-Assembly of NIR-Emissive Alkynylplatinum(II) Complexes Induced by Changes in Hydrophilicity and Electrostatic Effects. Chem. Eur. J. 2013, 19, 13182–13192. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, W.G.; Li, J.W.; Hong, C.Y.; Zhang, W.J.; You, Y.Z. Preparation of pH- and reductive-responsive prodrug nanoparticles via polymerization-induced self-assembly. Sci. China Chem. 2018, 61, 1159–1166. [Google Scholar] [CrossRef]
- Guo, J.; Lu, Y.; Bi, C.; Fan, J.; Xu, G.H.; Ma, J. Stimuli-Responsive Peptides Self-Assembly and Its Application. Prog. Chem. 2019, 31, 83–93. [Google Scholar] [CrossRef]
- Dong, S.; Luo, Y.; Yan, X.; Zheng, B.; Ding, X.; Yu, Y.; Ma, Z.; Zhao, Q.; Huang, F. A dual-responsive supramolecular polymer gel formed by crown ether based molecular recognition. Angew. Chem. Int. Ed. 2011, 50, 1905–1909. [Google Scholar] [CrossRef] [PubMed]
- Beves, J.E.; Campbell, C.J.D.; Leigh, A.; Pritchard, R.G. Tetrameric Cyclic Double Helicates as a Scaffold for a Molecular Solomon Link. Angew. Chem. Int. Ed. 2013, 52, 6464–6467. [Google Scholar] [CrossRef] [PubMed]
- Rodler, F.; Linders, J.; Fenske, T.; Rehm, T.; Mayer, C.; Schmuck, C. pH-Switchable Vesicles from a Serine-Derived Guanidiniocabonyl Pyrrole Carboxylate Zwitterion in DMSO. Angew. Chem. Int. Ed. 2010, 49, 8747–8750. [Google Scholar] [CrossRef] [PubMed]
- Gröger, G.; Meyer-Zaika, W.; Böttcher, C.; Gröhn, F.; Ruthard, C.; Schmuck, C. Switchable Supramolecular Polymers from the Self-assembly of a Small Monomer with Two Orthogonal Binding Interactions. J. Am. Chem. Soc. 2011, 133, 8961–8971. [Google Scholar] [CrossRef] [PubMed]
- Yuen, F.; Tam, K.C. Cyclodextrin-Assisted Assembly of Stimuli-Responsive Polymers in Aqueous Media. Soft Matter 2010, 6, 4613–4630. [Google Scholar] [CrossRef]
- Xu, J.F.; Chenm, Y.Z.; Wu, L.Z.; Tung, C.H.; Yang, Q.Z. Dynamic Covalent Bond Based on Reversible Photo [4+4]cycloaddition of Anthracene for Construction of Double-Dynamic Polymers. Org. Lett. 2013, 15, 6148–6151. [Google Scholar] [CrossRef]
- Qi, Z.; Schalley, C.A. Exploring Macrocycles in Functional Supramolecular Gels: From Stimuli Responsiveness to Systems Chemistry. Acc. Chem. Res. 2014, 47, 2222–2233. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, D.; Yan, X.; Chen, J.; Dong, S.; Zheng, B.; Huang, F. Self-Healing Supramolecular Gels Formed by Crown Ether Based Host-Guest Interactions. Angew. Chem. Int. Ed. 2012, 51, 7011–7015. [Google Scholar] [CrossRef]
- Harada, A.; Kobayashi, R.; Takashima, Y.; Hashidzume, A.; Yamaguchi, H. Macroscopic Self-Assembly through Molecular Recognition. Nat. Chem. 2011, 3, 34–37. [Google Scholar] [CrossRef]
- Liu, G.; Ma, R.J.; Ren, J.; Li, Z.; Zhang, H.X.; Zhang, Z.K.; An, Y.L.; Shi, L.Q. A glucose-responsive complex polymeric micelle enabling repeated on–off release and insulin protection. Soft Matter 2013, 9, 1636–1644. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, C.M.; Dong, H.Y.; Chen, D.Y. Solution-based fabrication of narrow-disperse ABC three-segment and Θ-shaped nanoparticles. Angew. Chem. Int. Ed. 2016, 128, 6290–6294. [Google Scholar] [CrossRef]
- Lee, H.J.; Koo, A.N.; Lee, S.W.; Lee, M.H.; Lee, S.C. Catechol-functionalized adhesive polymer nanoparticles for controlled local release of bone morphogenetic protein-2 from titanium surface. J. Control. Release 2013, 170, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Authimoolam, S.P.; Vasilakes, A.L.; Shah, N.M.; Puleo, D.A.; Dziubla, T.D. Synthetic oral mucin mimic from polymer micelle networks. Biomacromolecules 2014, 15, 3099–3111. [Google Scholar] [CrossRef] [PubMed]
- Authimoolam, S.P.; Lakes, A.L.; Puleo, D.A.; Dziubla, T.D. Layer-by-layers of polymeric micelles as a biomimetic drug-releasing network. Macromol. Biosci. 2016, 16, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Kong, J.H.; Sun, J.T.; Li, H.; He, C.B. Stable superhydrophobic porous coatings from hybrid ABC triblock copolymers and their anticorrosive performance. ACS Appl. Mater. Interfaces 2017, 9, 30056–30063. [Google Scholar] [CrossRef] [PubMed]
- Tambunlertchai, S.; Srisang, S.; Nasongkla, N. Development of antimicrobial coating by later-by-layer dip coating of chlorhexidine-loaded micelles. J. Mater. Sci. Mater. Med. 2017, 28, 90. [Google Scholar] [CrossRef]
- Yang, Y.Q.; Yi, C.L.; Luo, J.; Liu, R.; Liu, J.K.; Jiang, J.Q.; Liu, X.Y. Glucose sensors based on electrodeposition of molecularly imprinted polymeric micelles: A novel strategy for MIP sensors. Biosens. Bioelectron. 2011, 26, 2607–2612. [Google Scholar] [CrossRef]
- Zhu, X.W.; Liu, M.H. Self-assembly and morphology control of new L-glutamic acid-based amphiphilic random copolymers: Giant vesicles, vesicles, spheres, and honeycomb film. Langmuir 2011, 27, 12844–12850. [Google Scholar] [CrossRef]
- Falentin-Daudré, C.; Faure, E.; Svaldo-Lanero, T.; Farina, F.; Jerome, C.; Van, de.; Weerdt, C.; Martial, J.; Duwez, A.S.; Detrembleur, C. Antibacterial polyelectrolyte micelles for coating stainless steel. Langmuir 2012, 28, 7233–7241. [Google Scholar] [CrossRef]
- Sun, Y.X.; Ren, K.F.; Zhao, Y.X.; Liu, X.S.; Chang, G.X.; Ji, J. Construction of redox-active multilayer film for electrochemically controlled release. Langmuir 2013, 29, 11163–11168. [Google Scholar] [CrossRef]
- Wang, F.; Yang, X.W.; Zhang, L.L.; Xu, W.S.; Liu, H. Novel macromolecular emulsifiers as coatings with water-tolerant antifogging properties based on coumarin-containing copolymeric micelles. Macromol. Mater. Eng. 2017, 302, 1700173. [Google Scholar] [CrossRef]
- Sun, J.D.; Zhu, Y.; Meng, L.; Chen, P.; Shi, T.T.; Liu, X.Y.; Zheng, Y.F. Electrophoretic deposition of colloidal particles on Mg with cytocompatibility, antibacterial performance, and corrosion resistance. Acta Biomater. 2016, 45, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Wang, D.Y.; Lin, X.; Politakos, N.; Tuninetti, J.S.; Moya, S.E.; Gao, C.Y. Fabrication of UV responsive micelles-containing multilayers and their influence on cell adhesion. Sci. China Chem. 2018, 61, 54–63. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Y.H.; Yuan, X.Y. Facile preparation of superhydrophobic coating by spraying a fluorinated acrylic random copolymer micelle solution. Soft Matter 2013, 9, 1005–1009. [Google Scholar] [CrossRef]
- Rabnawaz, M.; Liu, G.J. Graft-copolymer-based approach to clear, durable, and anti-smudge polyurethane coatings. Angew. Chem. Int. Ed. 2015, 54, 6516–6520. [Google Scholar] [CrossRef]
- Rabnawaz, M.; Liu, G.J.; Hu, H. Fluorine-free anti-smudge polyurethane coatings. Angew. Chem. Int. Ed. 2015, 54, 12722–12727. [Google Scholar] [CrossRef]
- Iqbal, S.; Zhang, Y.J.; Liu, Y.T.; Akram, R.; Lv, S.S. Polymer micelles as building blocks for layer-by-layer assembly of multilayers under a high-gravity field. Chem. Eng. J. 2016, 293, 302–310. [Google Scholar] [CrossRef]
- Klok, H.A.; Lecommandoux, S. Supramolecular Materials via Block Copolymer Self-assembly. Adv. Mater. 2001, 13, 1217–1229. [Google Scholar] [CrossRef]
- Vriezema, D.M.; Aragonès, M.C.; Elemans, J.A.A.W.; Cornelissen, J.J.L.M.; Rowan, A.E.; Nolte, R.J.M. Self-Assembled Nanoreactors. Chem. Rev. 2005, 705, 1445–1490. [Google Scholar] [CrossRef]
- Blanazs, S.; Armes, S.P.; Ryan, A.J. Self-assembled block copolymer aggregates: From micelles to vesicles and their biological applications. Macromol. Rapid Commun. 2009, 30, 267–277. [Google Scholar] [CrossRef]
- Warren, N.J.; Armes, S.P. Polymerization-induced self-assembly of block copolymer nano-objects via RAFT aqueous dispersion polymerization. J. Am. Chem. Soc. 2014, 136, 10174–10185. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Xu, X.; Kirkland-York, S.E.; Savin, D.A.; McCormick, C.L. “Schizophrenic” Self-assembly of Block Copolymers Synthesized via Aqueous RAFT Polymerization: From Micelles to Vesicles in a Series on Water-soluble Polymers. Macromolecules 2010, 43, 1210–1217. [Google Scholar] [CrossRef]
- Latridi, Z.; Tsitsilianis, C. Water-soluble Stimuli Responsive Star-shaped Segmented Macromolecules. Polymers 2011, 3, 1911–1933. [Google Scholar] [CrossRef]
- Li, G.; Shi, L.; Ma, R.; An, Y.; Huang, N. Formation of Complex Micelles with Double-responsive Channels from Self-assembly of Two Diblock Copolymers. Angew. Chem. Int. Ed. 2006, 45, 4959–4962. [Google Scholar] [CrossRef]
- Kujawa, P.; Segui, F.; Shaban, S.; Diab, C.; Okada, Y.; Tanaka, F.; Winnik, F.M. Impact of End-group Association and Main-chain Hydration on the Thermosensitive Properties of Hydrophobically Modified Telechelic Poly (N-isopropylacrylamides) in Water. Macromolecules 2005, 39, 341–348. [Google Scholar] [CrossRef]
- Xia, Y.; Yin, X.; Burke, N.A.D.; Stover, H.D.H. Thermal Response of Narrow-disperse Poly (N-isopropylacrylamide) Prepared by Atom Transfer Radical Polymerization. Macromolecules 2005, 38, 5937–5943. [Google Scholar] [CrossRef]
- Topp, M.D.C.; Dijkstra, P.J.; Talsma, H.; Feijen, J. Thermosensitive Micelle-Forming Block Copolymers of Poly (ethylene glycol) and Poly (N-isopropylacrylamide). Macromolecules 1997, 30, 8518–8520. [Google Scholar] [CrossRef]
- Dai, L.; Liu, Z.; Zhang, L.; Rong, Y.; Huang, Y.; Chen, T. Temperature-Dependent Self-Assembly/Disassembly of Gold Nanoparticles Oligomers. J. Nanosci. Nanotechnol. 2016, 16, 5826–5832. [Google Scholar] [CrossRef]
- Jiménez, Z.A.; Yoshida, R. Temperature Driven Self-Assembly of a Zwitterionic Block Copolymer That Exhibits Triple Thermoresponsivity and pH Sensitivity. Macromolecules 2015, 48, 4599–4606. [Google Scholar] [CrossRef]
- Martinelli, E.; Guazzelli, E.; Galli, G.; Telling, M.T.F.; Poggetto, G.D.; Immirzi, B.; Domenici, F.; Paradossi, G. Prolate and Temperature-Responsive Self-Assemblies of Amphiphilic Random Copolymers with Perfluoroalkyl and Polyoxyethylene Side Chains in Solution. Macromol. Chem. Phys. 2018, 219, 1800210. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, W.; Jing, M.; Liu, S.; Feng, J.; Wu, H.; Zhou, Y.; Zhang, X.; Ma, Z. Self-assembled mixed micelle loaded with natural pyrethrins as an intelligent nano-insecticide with a novel temperature-responsive release mode. Chem. Eng. J. 2019, 361, 1381–1391. [Google Scholar] [CrossRef]
- Trenor, S.R.; Shultz, A.R.; Love, B.J.; Long, T.E. Coumarous in Polymers: From Light Harvesting to Photo-cross-linkable Tissue Scaffolds. Chem. Rev. 2004, 104, 3059–3078. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.; Liu, G. Hollow Nanospheres from Polyisoprene-b-poly (2-cinnamoylethyl methacrylate)-b-poly (tert-butyl acrylate). Chem. Mater. 1999, 11, 1048–1054. [Google Scholar] [CrossRef]
- Yan, X.; Liu, G.; Li, Z. Preparation and Phase Segregation of Block Copolymer Nanotube MuItiblocks. J. Am. Chem. Soc. 2004, 126, 10059–10066. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Kreiling, S.; Greiner, A.; Hampp, N. Two-photon-induced Cycloreversion Reaction of Coumarin Photodimers. Chem. Phys. Lett. 2003, 372, 899–903. [Google Scholar] [CrossRef]
- Li, Y.; Deng, Y.; Tong, X.; Wang, X. Formation of Photoresponsive Uniform Colloidal Spheres from an Amphiphilic Azobenzene-containing Random Copolymer. Macromolecules 2006, 39, 1108–1115. [Google Scholar] [CrossRef]
- Wang, G.; Tong, X.; Zhao, Y. Preparation of Azobenzene-containing Amphiphilic Diblock Copolymers for Light-responsive Micellar Aggregates. Macromolecules 2004, 37, 8911–8917. [Google Scholar] [CrossRef]
- Junge, D.M.; Mcgrath, D.V. Photoresponsive Azobenzene-containing Dendrimers with Multiple Discrete States. J. Am. Chem. Soc. 1999, 121, 4912–4913. [Google Scholar] [CrossRef]
- Goodwin, A.P.; Mynar, J.L.; Ma, Y.; Fleming, G.R.; Fréchet, J.M.J. Synthetic Micelle Sensitive to IR Light via a Two-photon Process. J. Am. Chem. Soc. 2005, 127, 9952–9953. [Google Scholar] [CrossRef]
- Jiang, J.; Tong, X.; Morris, D.; Zhao, Y. Toward Photoconlrolled Release Using Light-dissociable Block Copolymer Micelles. Macromolecules 2006, 39, 4633–4640. [Google Scholar] [CrossRef]
- Wang, S.; Shen, Q.; Nawaz, M.H.; Zhang, W. Photocontrolled Reversible Supramolecular Assemblies of a Diblock Azo-copolymer Based on β-cyclodextrin-Azo Host-guest Inclusion Complexation. Polym. Chem. 2013, 4, 2151–2157. [Google Scholar] [CrossRef]
- Guo, H.; Yang, J.; Zhou, J.; Zeng, L.; Zhao, L.; Xu, B. Photoresponsive self-assembly of a β-cyclodextrin derivative with an azobenzene terminal group in water. Dyes Pig. 2018, 149, 626–632. [Google Scholar] [CrossRef]
- Blasco, E.; Schmidt, B.V.K.J.; Barner-Kowollik, C.; Piñol, M.; Oriol, L. A Novel Photoresponsive Azobenzene-Containing Miktoarm Star Polymer: Self-Assembly and Photoresponse Properties. Macromolecules 2014, 47, 3693–3700. [Google Scholar] [CrossRef]
- Ji, S.; Xu, L.; Fu, X.; Sun, J.; Li, Z. Light- and Metal Ion-Induced Self-Assembly and Reassembly Based on Block Copolymers Containing a Photoresponsive Polypeptide Segment. Macromolecules 2019, 52, 4686–4693. [Google Scholar] [CrossRef]
- Floudas, G. Effects of Pressure on Systems with Intrinsic Orientational Order. Prog. Polym. Sci. 2004, 29, 1143–1171. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, J.; Chdcot, F.; Gnanou, Y.; Lecommandoux, S. Toward ‘Smart’ Nano-objects by Self-assembly of Block Copolymers in Solution. Prog. Polym. Sci. 2005, 30, 691–724. [Google Scholar] [CrossRef]
- Agut, W.; Brulet, A.; Schatz, C.; Taton, D.; Lecommandoux, S. pH and Temperature Responsive Polymeric Micelles and Polymersomes by Self-assembly of Poly [2-(dimethylamino) ethyl methacrylate]-b-poly(glutamic acid) Double Hydrophilic Block Copolymers. Langmuir 2010, 26, 10546–10554. [Google Scholar] [CrossRef]
- Roy, D.; Cambre, J.N.; Sumerlin, B.S. Sugar-responsive Block Copolymers by Direct RAFT Polymerization of Unprotected Boronic Acid Monomers. Chem. Commun. 2008, 21, 2477–2479. [Google Scholar] [CrossRef]
- Xu, Y.; He, K.; Wang, H.; Li, M.; Shen, T.; Liu, X.; Yuan, C.; Dai, L. Self-Assembly Behavior and pH-Stimuli-Responsive Property of POSS-Based Amphiphilic Block Copolymers in Solution. Micromachines 2018, 9, 258. [Google Scholar] [CrossRef]
- Zhang, W.; Bao, H.; He, J.; Dong, X. Preparation of multicompartment micelles from amphiphilic linear triblock terpolymers by pH-responsive self-assembly. Colloid Polym. Sci. 2015, 293, 3013–3024. [Google Scholar] [CrossRef]
- Liu, X.; Xie, Y.; Hu, Z.; Chen, Z.; Hu, J.; Yang, L. pH responsive self-assembly and drug release behavior of aliphatic liquid crystal block polycarbonate with pendant cholesteryl groups. J. Mol. Liq. 2018, 266, 405–412. [Google Scholar] [CrossRef]
- Rowan, S.J.; Cantrill, S.J.; Cousins, G.R.L.; Sanders, J.K.M.; Stoddart, J.F. Dynamic Covalent Chemistry. Angew. Chem. Int. Ed. 2002, 41, 898–952. [Google Scholar] [CrossRef]
- Belowich, M.E.; Stoddart, J.F. Dynamic Imine Chemistry. Chem. Soc. Rev. 2012, 41, 2003–2024. [Google Scholar] [CrossRef] [PubMed]
- Rollas, S.; Küçükgüzel, S. Biological Activities of Hydrazone Derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Springsteen, G.; Deeter, S.; Wang, B. The Relationship among pKa, pH, and Binding Constants in the Interactions between Boronic Acids and Diols-It Is Not as Simple as It Appears. Tetrahedron 2004, 60, 11205–11209. [Google Scholar] [CrossRef]
- Springsteen, G.; Wang, B. A Detailed Examination of Boronic Acid–diol Complexation. Tetrahedron 2002, 58, 5291–5300. [Google Scholar] [CrossRef]
- Jackson, A.W.; Fulton, D.A. pH Triggered Self-assembly of Core Cross-linked Star Polymers Possessing Thermoresponsive Cores. Chem. Commun. 2011, 47, 6807–6809. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Mu, J.; Li, C. Synthesis and pH/sugar/salt-sensitivity Study of Boronate Crosslinked Glycopolymer Nanoparticles. New J. Chem. 2013, 37, 796–803. [Google Scholar] [CrossRef]
- Yang, F.; Wang, J.; Cao, L.Y.; Chen, R.; Tang, L.J.; Liu, C.S. Injectable and redox-responsive hydrogel with adaptive degradation rate for bone regeneration. J. Mater. Chem. B 2014, 2, 295–304. [Google Scholar] [CrossRef]
- Yan, Q.; Yuan, J.; Cai, Z.; Xin, Y.; Kang, Y.; Yin, Y. Voltage-responsive Vesicles Based on Orthogonal Assembly of Two Homopolymers. J. Am. Chem. Soc. 2010, 132, 9268–9270. [Google Scholar] [CrossRef]
- Li, P.; Kang, H.; Che, N.; Liu, Z.; Zhang, C.; Cao, C.; Li, W.; Liu, R.; Huang, Y. Synthesis, self-assembly and redox-responsive properties of well-defined hydroxypropylcellulose-graft-poly(2-acryloyloxyethyl ferrocenecarboxylate) copolymers. Polym. Int. 2015, 64, 1015–1022. [Google Scholar] [CrossRef]
- Liu, L.; Rui, L.; Gao, Y.; Zhang, W. Self-assembly and disassembly of redox-responsive ferrocene-containing amphiphilic block copolymer for controlled release. Polym. Chem. 2015, 6, 1817–1829. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Wang, X.; Zheng, W.; Zhang, J.; Shi, F.; Liu, J. Redox-responsive self-assembly PEG nanoparticle enhanced triptolide for efficient antitumor treatment. Sci. Rep. 2018, 8, 12968. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Zhang, L.; Lin, J.; Wang, L. Self-Assembly Behavior of pH- and Thermosensitive Amphiphilic Triblock Copolymers in Solution: Experimental Studies and Self-Consistent Field Theory Simulations. J. Phys. Chem. B 2008, 112, 12666–12673. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhao, B. Tuning Micellization and Dissociation Transitions of Thermo- and pH-Sensitive Poly (ethylene oxide)-b-poly(methoxydi(ethylene glycol) methacrylate-co-methacrylic acid) in Aqueous Solution by Combining Temperature and pH Triggers. Macromolecules 2008, 41, 9366–9375. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, T.; Liu, S. Micellization Kinetics of a Novel Multi-Responsive Double Hydrophilic Diblock Copolymer Studied by Stopped-Flow pH and Temperature Jump. Macromol. Chem. Phys. 2007, 208, 2492–2501. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, C.P.; Song, X.; Zhuo, L.H.; Bu, H.T.; Tian, W. Photo- and pH- dually-responsive β-cyclodextrin-based supramolecular prodrug complexes self-assemblies for programmed drug delivery. Chem.-Asian J. 2018, 13, 3903–3911. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, J.; Li, Q.; Li, J.; He, K.; Shen, T.; Liu, X.; Yuan, C.; Zeng, B.; Dai, L. Novel azobenzene-based amphiphilic copolymers: Synthesis, self-assembly behavior and multiplestimuli-responsive properties. RSC Adv. 2018, 8, 16103–16113. [Google Scholar] [CrossRef]
- Jiang, F.; Chen, S.; Cao, Z.; Wang, G. A photo, temperature, and pH responsive spiropyran-functionalized polymer: Synthesis, self-assembly and controlled release. Polymer 2016, 83, 85–91. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Grzybowski, B. Self-Assembly at All Scales. Science 2002, 295, 2418–2421. [Google Scholar] [CrossRef]
- Chai, L.H.; Peng, X.F. Challenge from Frontier in Chemistry: Dynamic Self-Assembly. Prog. Chem. 2004, 16, 169–173. [Google Scholar]
- Xu, G.H.; Zhou, H.W.; Li, J.; Yin, L.; Zheng, Z.H.; Ding, X.B. Autonomous fluorescence regulation in responsive polymer systems driven by a chemical oscillating reaction. Polym. Chem. 2016, 7, 3211–3215. [Google Scholar] [CrossRef]
- Service, R.F. How Far Can We Push Chemical Self-Assembly. Science 2005, 309, 95. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M.; Boncheva, M. Beyond molecules: Self-assembly of Mesoscopic and Macroscopic Components. Proc. Natl. Acad. Sci. USA 2002, 99, 4769–4774. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).