Abstract
The environmental pollution of antibiotic resistance genes (ARGs) and antibiotic-resistant bacteria (ARB) is a growing public health concern. In the current study, de novo metagenomic assembly and bioinformatics analysis approaches were utilized to estimate the quantitative risk index of the environmental resistomes in wastewater influent (INF) and effluent (EFF) of a conventional wastewater treatment plant (WWTP) in Egypt. Furthermore, the risk indices of the local INF and EFF resistomes were compared to those calculated for the selected publicly available wastewater datasets from eight countries worldwide. Additionally, a classification framework prioritizing the public health hazard level of the discharged non-redundant highly mobilized ARGs was introduced. This integrative outline considered the estimated mobility potential percentage, host pathogenicity, and annotation category (perfect, strict, and loose) of the detected ARGs on their assembled contigs. Moreover, high-quality metagenome-assembled genomes (MAGs) were extracted and the putative genome bins with acquired ARGs were determined. The comprehensive resistome risk scores of the local WWTP showed that INF resistome had a slightly higher risk index (47.87) compared to the average score of the other examined counterparts (41.06). However, the estimated risk value of EFF resistome (26.80) was ranked within the global average (26.06) of the selected international WWTPs. Furthermore, the determination of the samples’ risk ranking showed that most of the effluent resistomes were clustered in a lower risk rank compared to the other selected samples for raw sewage, influent, and hospital wastewater, indicating the impact of the wastewater treatment process on reducing the ARG mobilization potential in downstream environments. The evaluation of the ARGs’ genetic context in their ARG-carrying contigs (ACCs) indicated that a total of 161/648 (25%) non-redundant ARGs were co-located with sequences of mobile genetic determinants on the same ACC in both the INF and EFF assemblies. These ARGs comprised the pan mobile resistome of the studied WWTP. Of them, 111 ARGs with a mobility potential percent (M%) less than 95% were grouped at the least risk level 5. The remaining 50 highly mobilized ARGs (M% ≥ 95%) were extracted and classified into four higher risk levels. Those of risk levels 1 and 2 (39 ARGs) represented the current ARG dissemination threats for further monitoring in downstream environments, where they were all carried by pathogenic hosts and annotated to the perfect and strict categories by the resistance gene identifier software (RGI). A total of 10 highly mobilized ARGs were assigned to risk rank 3, as they comprised the loose hits of the RGI analysis. Finally, the risk level 4 ARGs constituted genes that co-existed with the non-pathogenic sequence on the ACCs and were represented by one gene in the current analysis framework. The two previous categories constituted new highly mobilized ARGs of emergent threat to public health. On the other hand, a total of 35 and 118 MAGs were recovered from INF and EFF assembled metagenomes, respectively, using selection cutoff thresholds of a minimum completeness of 70% and a maximum contamination of 10%. While none of the INF MAGs carried any acquired ARGs, six EFF genome bins (5%) were associated with ten acquired ARGs, as indicated by the ResFinder software. These results suggest that potential horizontal gene transfer (HGT) events have evolved among the community members of the studied EFF samples.
1. Introduction
Antibiotic resistance is a global problem that threatens human health. The elevated levels of acquired infections among diverse communities has attracted the attention of scientific communities worldwide to to extensively monitor the development of antimicrobial resistance (AMR) in various environmental and clinical matrices [1,2].
Wastewater treatment plants were identified as hotspots for the spread of ARGs and ARB in the environment [3]. The microbial diversity within wastewater provides a large pool of genes (i.e., genetic reservoir) that assists in the transmission of mobilizable ARGs carried by mobile genetic determinants (plasmids, integrons, transposons, and integrons) among closely related species [4,5]. However, a study by Jiang et al. (2017) [6] validated the incidence of ARG transfer between evolutionary-distant phyla (Proteobacteria to Actinobacteria). This complicates the problem of ARG dissemination in various environments. Provided that environmental bacteria and human pathogens co-exist in close proximity in a WWTP, without transmission barriers, a putative exchange of resistance determinants between environmental and pathogenic bacteria through HGT might occur [7]. For example, a study demonstrated that an ARG known as CTX-M, encoding for extended-spectrum beta-lactamase (ESBL), putatively originated from a beta-lactamase gene of soil bacteria, namely, Kluyvera species [8]. Moreover, McKinnon et al. (2018) [9] indicated that commensal Escherichia coli was found to cause disease in human (i.e., urosepsis) as a consequence of acquiring a virulence plasmid carrying multidrug resistance genes associated with disease factors in one genomic cassette.
Several studies demonstrated that effluents of conventional WWTPs exhibited high pathogenic content in addition to low removal of ARGs and ARB [10,11,12]. For instance, a research work indicated that the two opportunistic pathogens Pseudomonas spp. and Aeromonas hydrophila were determined in the effluent of a conventional WWTP [13]. Furthermore, a comparative metagenomics study for ARGs in wastewater effluents and surface water in Singapore [14] concluded that many environments are impacted by clinically relevant ARGs, conferring resistance against multidrug, tetracycline, aminoglycoside, macrolide–lincosamide–streptogramine (MLS). Regardless of the applied treatment technology and the geographic location of a WWTP, it has been suggested that wastewater effluents are an important source of ARGs associated with mobile genetic elements (MGEs) such as plasmids, transposons, or integrons in the receiving water streams [15,16,17,18]. This conclusion was validated by multiple scientific reports showing that ARGs, ARB, and MGEs were found in the treated wastewater and in downstream water bodies as well [19,20,21].
The stress-inducing acquisition of a new ARG by a bacterial host can occur by two mechanisms, namely, mutation via single-nucleotide polymorphism (SNP) and/or HGT [22]. HGT was considered the major mechanism for the dissemination of ARGs and ARB in diverse environmental matrixes including WWTPs [23], for example, the mcr-1 gene, which confers resistance to the colistin antibiotic and is considered as the last resort treatment against multidrug resistant Gram-negative bacteria. Since the discovery of the first plasmid-mediated mcr-1 gene in China, in 2015 [24], the gene was isolated in more than fifty countries worldwide and it was associated with multiple different plasmids (IncI2, IncHI2, and IncX4) and providing evidence for the role of HGT in the spread of the gene [25]. ARGs, ARB, human pathogens, and MGEs in association with elevated levels of nutrients, heavy metals, and antibiotic residues in one ecological niche during the wastewater treatment process may cause a selection pressure favoring the exchange of the resistance determinants among environmental and pathogenic bacteria through HGT [26,27,28]. It was demonstrated that ARGs carried by MGEs and hosted by human pathogenic bacteria have a higher health risk due to the fact of their increased mobilization potential [29]. Several researchers have analyzed the co-occurrence of ARGs and MGEs in association with human pathogens in wastewater to assess the mobility potential of the ARGs released into the environment [30,31,32]. Although the assessment of the mobility potential of an ARG provides an indicator of the level of its health risk, it lacks a cumulative quantitative measure for the risk level at the large-scale ARG profile of a metagenomic dataset. A novel pipeline for estimating a comprehensive risk index of antibiotic resistome in an environment using metagenomic assembly was recently introduced [33]. This software tool determines the relative abundance of the total ARG-carrying contigs (ACCs) in addition to those that coexist with ARGs, MGEs, and pathogen-like sequences in the assembled metagenomic dataset. Then, the data from the previous step are used to calculate one resistome risk index for a metagenomic sample. As a result, the comparison for the values of the resistome risk among multiple environments becomes more feasible to support surveillance plans against the dissemination of ARGs and ARB in the environment.
The major drawback of the metagenomic analysis of short DNA sequences is the limited information obtained with respect to the genomic context of the recovered ARGs. Thus, the metagenomic assembly of short reads into longer contiguous sequences (contigs) can overcome this challenge and provide more detailed annotation information related to the genetic contexts of the detected ARGs [34]. Due to the low abundance of ARGs compared to other functional genes in WWTP metagenomes, deep sequencing is required to recover partial genomes and determine the co-occurrence pattern of ARGs and the associated mobile elements as plasmids [35,36]. A recent metagenomic study exploring the antibiotic resistome in 97 activated sludge samples over a 9 year sampling period at a WWTP pointed out that the optimum sequencing depth required to obtain a complete quantitative ARG profile from the whole metagenomic sequencing is 60 Gbp (giga base pairs) [37], validating the importance of deep sequencing in the retrieval of the most representative resistome profile of a WWTP microbiome.
In this study, metagenomic assembly and bioinformatic analysis approaches were applied to quantitatively evaluate the human health risks of antibiotic resistomes in wastewater influent (INF) and effluent (EFF) samples at a conventional WWTP in Egypt. These datasets were previously analyzed as unassembled paired-end reads by the same authors in a completely different prospective [38]. The current research aimed to, firstly, apply a quantitative metric to estimate the risk index associated with INF and EFF resistomes of the examined WWTP. Meanwhile, we wanted to assess the risk ranking of the local WWTP resistomes compared to those derived from globally selected wastewater metagenomes downloaded from publicly available databases. Secondly, we intended to evaluate the hazard level of the INF and EFF non-redundant ARGs in their genetic context by proposing a hierarchical ranking system based on the ARGs’ mobility potential percentage, pathogenicity, and annotation category of their homology search against the CARD database (i.e., perfect, strict, and loose). Finally, we aimed to conduct genome binning of the analyzed INF and EFF assembled metagenomes to determine the potential genomes harboring acquired ARGs as an indication of the putative HGT incidence.
2. Materials and Methods
The sampling method, DNA extraction, and operation parameters of the studied WWTP are described in our previous publication [38].
The bioinformatics analysis pipeline of the current work was shown in Figure 1. Briefly, the comprehensive resistome risk indices for the local samples of INF and EFF assembled contigs as well as the selected publicly available wastewater datasets were computed using the MetaCompare software pipeline. Then, the obtained risk scores were compared together to identify the global risk rank of INF and EFF resistomes. At the same time, MetaCompare output DNA sequences of ARG-like, MGE-like, and pathogen-like sequences were utilized for a contig-based analysis of the INF and EFF resistomes. As a result, the percentage of the ARG mobility potential at the resistance gene, drug class, and resistance mechanism levels were estimated. On the other hand, the genome-based analysis was conducted to extract high-quality MAGs of the INF and EFF datasets and to determine the genomes with acquired ARGs.
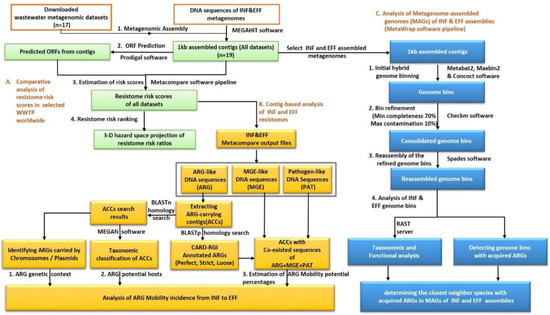
Figure 1.
The bioinformatics analysis pipeline of the wastewater influent and effluent datasets at TZ-WWTP.
2.1. Metagenomic Assembly and ORF Prediction
In the present study, four metagenomic datasets of high-quality shotgun paired-end sequences (q > 20) previously extracted from wastewater influent (INF) and effluent (EFF) samples of WWTP in Egypt [38] were involved in two assembly pipelines. To improve the depth and coverage of the performed assemblies [37], the two INF datasets were merged together and assembled as one large sample using MEGAHIT software version 1.1.3 [39] with the default parameters and using option “--min-contig-len 1000” to output contigs with a minimum size of 1 kb and only those that provided a searchable length for the possibly co-existing determinants for ARGs, MGEs, and pathogen-of-origin sequences [40]. Similarly, the two EFF datasets were incorporated into another assembly process. On the other hand, seventeen publicly available metagenomic datasets from the European Nucleotide Archives server (ENA) (https://www.ebi.ac.uk/ena/browser/view/, date accessed: 25 July 2021) were downloaded, and they are listed in Table S1. The selected metagenomes covered seven different geographic locations (i.e., Hong Kong, South Korea, Germany, United Kingdom, United States of America, Uruguay, and South Africa) and four different wastewater matrixes (Influent, effluent, raw sewage, hospital sewage). In brief, the raw reads went through adapter trimming and quality filtering using Cutadapt v1.8.1 [41] to remove the adapter sequences and retain the high-quality reads with quality scores ≥ 20. The quality-controlled datasets were assessed using FASTQC v0.11.9 [42]. The high-quality reads of the seventeen samples were incorporated into individual assembly operations using the aforementioned software and cutoff thresholds. QUality ASsessment Tool (Quast) v5.0.2 [43] was used to estimate the quality of the assembled metagenomes. The prediction of the open reading frames (ORFs) from all 1 kb assembled contigs, including those of the INF and EFF samples (n = 19), was conducted using Prodigal v2.6.3 [44]. The 1 kb assembled metagenomes were incorporated into the resistome risk analysis process.
2.2. The Comparative Analysis of the Resistome Risk Scores in the Wastewater Samples
To estimate the risk scores of the antibiotic resistomes, the nucleotide sequences of the 1 kb assembled contigs and the predicted ORFs of each sample were uploaded into the MetaCompare software [33]. MetaCompare is a three-step pipeline of homology search for the detection of ARG-like sequences, MGEs, and pathogen-like sequences of the assembled contigs. The first step was achieved by running a BLASTx search [45] of the predicted ORFs against the comprehensive antibiotic resistance database CARD v3.1.2 (23 April 2021 [46]), with the cutoff values of E-value ≤ 1e−10, identity ≥ 60%, and a minimum alignment length ≥ 25 amino acids. Then, a BLASTn search [45] was performed on the same assembled contigs of each sample against a classification of mobile genetic elements database (ACLAME v0.4) [47] using the cutoff values of E-value ≤ 1e−10 and identity > 60%. Thirdly, a BLASTn homology search of each assembly against the Pathosystems Resource Integration Center database (PATRIC v3.6.3) [48] using the cutoff values of E-value ≤ 1e−10, identity ≥ 60%, and a minimum alignment length ≥ 150 bp was conducted [33].
The results of the previous steps were involved in the calculation of three critical risk ratios representing the number of ARG-carrying contigs (ACCs) divided by the total number of assembled contigs in each sample (nARGs), the number of ACCs with ARGs and MGEs coexisted on the same contig divided by the total number of total contigs (nARGs&–MGEs), and the number of ACCs with ARGs and MGEs and associated with pathogen-like sequence on the same contig divided by the total number of contigs (nARGs&–MGEs&–PAT).
Finally, these values were incorporated into a formula to calculate the environmental resistome risk score. We optimized the Python script of the MetaCompare software in house to save all of the intermediate and final resistome risk score output files locally in the CSV text format (Supplementary Materials Script S1).
To assess the risk ranking of the INF and EFF samples associated with the current study samples compared to the datasets from other geographic locations, the three risk ratios (i.e., nARGs, nARGs&–MGEs, and nARGs&–MGEs&–PAT) were projected into a three-dimensional hazard space using the Scatterplot3d package v0.3-41 [49] in RStudio v1.3.959 (http://www.rstudio.com/accessed, 22 June 2020) with R software v4.0.2 (https://www.R-project.org, accessed on 22 June 2020)
2.3. Determination of the Risk Priority of Mobile ARGs in the Wastewater Influent and Effluent
The ACCs of the INF and EFF samples were extracted from the MetaCompare BLASTx results using the BBMap tool [50] with the command “./filterbyname.sh in=path/to/final_assembly/fasta/filenames=path/to/list/ACCs_headaers out=name/of/output/ACCs/fasta_file ow=t include=t truncateheader”. The INF and EFF ACCs were analyzed using the resistance gene identifier (RGI) software v5.2.0 with the CARD database v3.1.2, released 23 April 2021. The resistome prediction and classification was performed based on the perfect, strict, and loose paradigms of the RGI software web portal, with options for the inclusion of nudge sequences (loose hits of ≥95% identity were moved into the strict category). In brief, the RGI predicted the ORFs from the submitted ACCs using Prodigal with a cutoff selection of an ORF length ≥ 30 bp. Then, the antimicrobial resistance genes (AMR) were identified from the ORF amino acid sequences using the diamond BLASTp search against the CARD databases, with a predefined pass bitscore for the ARG classification for protein homolog models, protein variant models, and protein-overexpression models. Finally, the recovered ARGs were annotated as perfect hits with a clinical relevance if the whole query had a 100% match to the curated reference sequence in the CARD databases. On the other hand, strict and loose hits represented new variants and distant homologs of known AMR genes in the CARD database, respectively.
The potential genetic context and taxonomic lineage of the recovered INF and EFF ARGs were identified by a BLASTn homology search of ACCs against the NCBI-nt database (v5.0, July 2021) with a cutoff threshold of E-value ≤ 1e−10, identity ≥ 60%, and a minimum alignment length = 150 bp. As the genetic context of ARGs determines their mobility status, the ACC IDs in the output tables obtained from the BLASTn search against the nt and ACLAME databases were cross-mapped together to collect instances of ARG association with a sequence of mobile determinant including plasmid, transposon, integron, integrative-conjugative element (ICE), or bacteriophage [51]. The logic matrix for the classification of ACC genetic context is explained in Table S2. The ARG was annotated as mobile if it existed in co-localization with at least one MGE-like sequence on the same ACC. The mobility potential percentages (M%) of the ARGs in the INF and EFF assembled metagenomes were calculated as mentioned in (1) for the non-redundant individual resistance genes, antibiotic drug class, and resistance mechanism [52]:
where M% is the mobility potential percent; N1 is the count of ACCs associated with MGEs in each category; N2 is the total number of ACCs in this category. An ARG was considered highly mobilized if M% ≥ 95% and low-mobilized if M% ≤ 5% [52].
The output data obtained from the previous steps were utilized to classify the detected ARGs into a five-level risk ranking system based on the value of their mobility potential percentages (M%), the co-existence status of the ARG with a pathogen-of-origin sequence on the same ACC and RGI annotation category (i.e., perfect, strict, and loose) (Figure 2). Highly mobilized ARGs that coexisted with pathogen-of-origin sequences on the same contig were determined by the cross-matching of the ACCs IDs to their counterparts resulting from the BLASTn homology search against the PATRIC database.
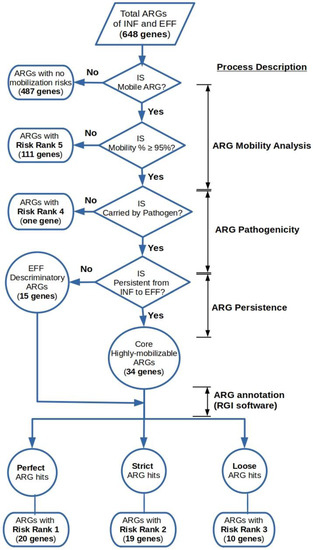
Figure 2.
Hierarchical framework for the risk ranking of highly mobilized ARGs in the wastewater’s influent and effluent metagenomes.
The ARGs that belonged to risk rank 1 constituted highly mobilized genes that co-existed with pathogen-of-origin sequences and annotated as perfect hits by the RGI tool. The criteria for the ARGs of the risk ranks 2 and 3 were the same as above, except for the RGI categorizations of strict and loose. The mobile ARGs assigned to risk rank 4 were those that were not associated with pathogenic sequences on the same ACC, indicating less dissemination potential. Moreover, the ARGs with a mobility potential percentage less than 95% were affiliated to risk rank 5, regardless of their genomic context or RGI classification. On the other hand, the ARGs not associated with an MGE determinant on the same ACC were considered as having no mobilization potential and were left unassessed.
Finally, the potential taxonomic classifications of the INF and EFF ACCs were determined by submitting BLASTn hits of the two sets into MEGAN software v6.18.0, built 28 October 2019 [53], using the default parameters.
2.4. Genome Binning of the Influent and Effluent Assembled Metagenomes
The process of genome binning and extraction of high-quality draft genomes from the INF and EFF 1 kb assembled contigs was completed using a modular pipeline for genome-resolved metagenomic data analysis software, MetaWRAP 1.2.1 [54]. Briefly, the initial binning process was performed using MaxBin2 [55], metaBAT2 [56], and CONCOCT [57] binning software using differential sequence composition and GC content percentage. Secondly, the bin refiner tool [58] was used to create hybrid bins from the previous step such that no two contigs were placed in the same bin if they were found in different bins of the original sets. Then, the high-quality consolidated genome bins were extracted using the CheckM software tool [59], with the selected cutoff threshold of a minimum genome completeness of 70% and a maximum contamination of 10% [60]. The quality filtered genome bins were incorporated into a final reassembly process using SPAdes software [61] to improve completeness of the recovered draft MAGs. Taxonomic classification and functional annotation of the extracted MAGs were completed using the online Rapid Annotation using Subsystem Technology server (RAST) (http://rast.nmpdr.org/ (accessed on 8 January 2022) [62]. The closest neighbor species for each genome bin was determined from the seed analysis of the RAST server. To infer genome bins with acquired antimicrobial resistance genes (AMRs), the nucleotide sequences of the contigs in the recovered bins (i.e., INF and EFF) were searched against the acquired antimicrobial resistance genes database ResFinder 4.1 [63] using the program defaults of a minimum percent identity of 90% and a minimum hit coverage of 60%.
3. Results
3.1. Metagenomic Assembly of the Wastewater Metagenomic Datasets
A total size of 70 Gb of data belonging to the INF (36 Gb) and EFF (34 Gb) metagenomic samples from the Tz-WWTP in Egypt were assembled using MEGAHIT software. Furthermore, a total of 180 Gb representing the seventeen globally selected metagenomes were assembled as well. The percentage of mapped reads of the local INF and EFF samples in the 1 Kb assembled datasets accounted for 73.14% and 69.00%, respectively, with an average of 71.07%. However, it ranged from 28.34% to 90.62% with an average of 56.54% for the downloaded metagenomes (Table S3). The average contig coverage for the INF and EFF assembled datasets constituted 29.55 and 21.70, respectively. While it ranged from 10.22 to 45.54 (average of 21.57) for the assembled wastewater metagenomes of the selected datasets. The above results showed a higher sequencing depth and average contig coverage of the assemblies of the samples related to the present study.
3.2. Estimation of the Comprehensive Risk Scores for the Wastewater Resistomes
The number of ACCs extracted from the MetaCompare analysis of the INF and EFF assemblies were 2409 (0.7%) and 1196 (0.2%), respectively. In addition, 4622 (1.4%) and 2955 (0.7%) MGE sequences and 138,798 (41.5%) and 78,279 (19.1%) pathogen-like sequences were derived from the INF and EFF contigs, respectively. These numbers indicate a one-fold decrease of the ACCs, MGEs, and pathogen-like sequences ratios from the INF to the EFF metagenomes. The calculated risk score of the INF resistome accounted for 47.8; however, that of the EFF resistome was 26.9. The risk ratios of the INF and EFF were compared to those derived from other wastewater datasets of the selected geographic locations to assess the risk ranking of the study samples from the Egyptian WWTP (Table 1 and Figure 3a). The MetaCompare analysis showed that the risk score of Egyptian wastewater INF resistome was ranked as the second top value after the South African INF resistome (50.3), while the EFF risk value was the third top value after the South Korean (37.9) and South African (35.5) EFF resistomes. A trend among the assessed samples was observed, where the raw sewage (RS), influent, and hospital sewage (HS) samples were ranked with the highest risk scores regardless of their geographic locations. This was validated by the 3D hazard space analysis (Figure 3b) in which the risk ratios of EFF resistomes of most samples (7/9) were clustered at lower hazard plane than INF, RS, and HS.

Table 1.
Summary of the results collected from the MetaCompare analysis pipeline for the resistome risk scores associated with the current study samples (EG-INF and EG-EFF) as well as other globally selected samples (n = 17).
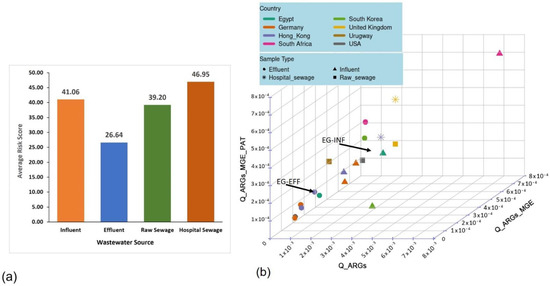
Figure 3.
Risk ranking of the antibiotic resistomes showing (a) the average risk scores of the environmental samples from the selected global wastewater sites (i.e., influent, effluent, hospital sewage, and raw sewage) and (b) a 3D hazard space diagram of the risk ratios estimated for all analyzed samples indicating the clustering of resistomes from the same sample source in closely related hazard level.
3.3. Resistome Composition of the Wastewater Influent and Effluent of the Assembled Metagenomes
The DNA sequences of the INF and EFF ACCs were extracted from the assembled metagenomes and annotated using pass-bit score cutoff values of the CARD-RGI web portal which classified the annotated ARGs into three categories, namely, perfect hits with 100% identity to manually curated known functional ARGs in CARD database, strict hits that represent variants of functional genes in the reference database, and loose hits of newly emergent ARGs that require functional validation and deviate from the cutoff threshold of CARD-RGI detection model. As a result, a total of 2386 and 1189 ACCs of INF and EFF datasets, respectively, were successfully annotated. The ACCs annotated by the RGI accounted for 99% of the MetaCompare output contigs in both samples (Table 2). Furthermore, both datasets showed the same distribution for the number of contigs carrying one ARG and those with two or more ARGs (multiple-ARG-carrying contigs) to be 60% and 40% of the total annotated ACCs, respectively. Moreover, the distribution of the ARG ORFs among the perfect, strict, and loose categories exhibited slight differences in their relative abundances before treatment (INF) and after treatment (EFF). For the INF ACCs, a total number of 64 (2%) perfect, 415 (11%) strict, and 3412 (87%) loose ARG sequences were detected. However, 56 (3%) perfect, 174 (9%) strict, and 705 (88%) loose ARG hits were derived from the EFF ACCs (Table 2). A total number of 648 non-redundant ARGs were recovered from the INF and EFF assembled metagenomes belonging to 26 different drug classes (Table S4). Of these ARGs, only 30% could not be detected in the effluent compartment, 17% were found only in the EFF ACCs, and 53% of the total non-redundant ARGs were shared by both sites (core resistome) implicating insufficient removal of these genes through the treatment process (Figure 4a). Five drug classes for multidrug, beta-lactam, macrolide–lincosamide–streptogramin (MLS), aminoglycoside, and tetracycline accounted for approximately 71% of the detected ARGs (Figure 4b).
Table 2.
Summary of the ARG annotations using the CARD resistance genes identifier (RGI) software for the ARG-carrying contigs (ACCs) of the local influent and effluent assemblies in the Tz-WWTP.
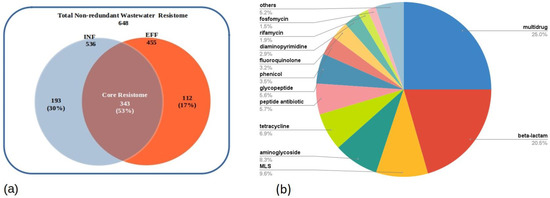
Figure 4.
Resistome composition of the assembled metagenomes showing (a) the non-redundant ARGs annotated in both INF and EFF and (b) the abundance percentage of ARGs per drug class. (The drug classes with abundance percentages less than 1% are not listed in the Pie chart and included in the “others” category).
To identify potential ARG hosts, the BLASTn search results of the INF and EFF ACCs were submitted into MEGAN software to assign each contig to the lowest common taxonomic rank. The classification of the INF and EFF ACCs at the phylum level showed that Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria represented the most abundant phyla with total ACC percentages of 94% and 84% in the INF and EFF, respectively (Figure S1). Although 97% and 93% of the ACCs in the INF and EFF samples were successfully assigned to a taxonomic name at any level, only 82% and 63% could be classified at the genus level, respectively (Figure S2). The genera for Pseudomonas, Aeromonas, and Klebsiella were the most prevalent potential ARG hosts with relative abundances of 22%, 11%, and 6% in the INF ACCs and 26%, 9%, and 4% in the EFF ACCs, respectively (Figure 5).
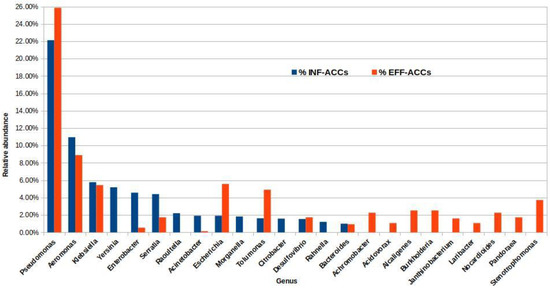
Figure 5.
Percentage of the ARG-carrying contigs (ACCs) extracted from the influent and effluent assembled metagenomes at the genus level only.
However, the analysis showed variations in the taxonomic assignments of some other genera from INF to EFF. For example, nine genera, namely, Achromabacter, Acidovorax, Alcaligenes, Burkhulderia, Janthinobacterium, Laribacter, Nocardioides, Pandoraea, and Stenotrophomonas were detected as potential ARG carriers in the EFF ACCs with relative abundance above 1%, while for INF ACCs, they were below the detection limit. In contrast, Yersinia, Raoultella, Morganella, and Rahnella were annotated as potential ARG hosts in the INF ACCs only (Figure 5). Remarkably, the relative abundance of the Escherichia genus, as an indicator for the fecal contamination of the discharged effluent, was enriched from almost 2% in the INF ACCs to approximately 6% of the treated EFF ACCs. Table S5 summarizes the potential ACC hosts at the genus level and the corresponding number of contigs in each drug class associated with the annotated host.
3.4. Evaluation of the Mobility Incidence Percentage of the Antibiotic Resistomes in the WWTP
The co-occurrence pattern analysis of the ARGs and MGEs (plasmids, transposons, integrons, ICEs, and bacteriophages) on ACCs was conducted to assess the mobility incidence in the INF and EFF assembled datasets (Tables S6 and S7). A total number of 117 (5%) and 58 (5%) ACCs were found to carry 163 and 92 mobile ARGs that coexisted with an MGE-like sequence on the same ACC of the INF and EFF, respectively (Table S8). Of those ACCs, 96 (82%) and 51 (88%) were associated with pathogen-like sequences in the INF and EFF, respectively. These results showed that the majority of the ARG mobility incidence of the studied datasets took place in pathogenic hosts before and after wastewater treatment. The mobile ARGs of the INF ACCs comprised 47%, 12%, and 2% of the total perfect, strict, and loose annotated hits, respectively. Similarly, the percentages of 46% perfect, 16% strict, and 5% loose hits were determined from the EFF ACCs.
The mobility potential percentages (M%) of non-redundant ARGs in both datasets were calculated to assess the treatment impact on the ARG mobilization. The total mobile resistome in the studied WWTP comprised 161 ARGs that existed in 14 drug classes accounting for 25% of the total non-redundant ARGs of INF and EFF ACCs (648 genes) (Figure 6a and Table S9). The distribution of the percentage of ARG mobility incidence among drug classes is shown in Figure 6bx. The percentages of the mobility that decreased from INF to EFF of the ARGs were present in the drug classes for sulfonamide (40–9%), nucleoside (30–0%), diaminopyrimidine (19–9%), and tetracycline (14–7%). However, the mobility incidence percentages showed an increase from INF to EFF in the drug classes for aminoglycoside (14–20%), beta-lactam (13–20%), fluoroquinolone (3–19%), phenicol (5–11%), and disinfecting agents and intercalating dyes (9% to 33%) (Table S10a). With respect to the percentage of the mobility of the ARGs per resistance mechanism, it was found that antibiotic inactivation (19%), antibiotic target alteration (14%), and antibiotic target replacement (21%) were the most abundant mechanisms in the INF. While antibiotic inactivation (23%), antibiotic target protection (17%), and reduced permeability to antibiotic (9%) resistance mechanisms were the dominant mechanisms used by mobile ARGs of the EFF wastewater (Figure 6c and Table S10b). The above described results indicate the existence of shifts in the drug classes and resistance mechanisms of mobile ARGs from INF to EFF as a result of the treatment process.
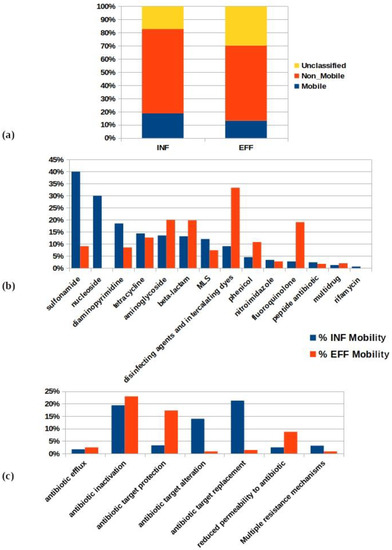
Figure 6.
Analysis of the ARG mobility potential in the WWTP showing (a) the percentage mobility incidence of the total non-redundant ARGs in both treatment compartments; (b) a comparison of the ARG mobility potential percentages in the influent and effluent per drug class; (c) the per resistance mechanism (MLS: macrolide–lincosamide–streptogramin drug class).
3.5. Risk Priority Classification of the Wastewater Mobile Resistome
The proposed ranking system determines the priority level for further surveillance of the mobile ARGs in a downstream environment. The extracted mobile ARGs (161 genes) were initially divided into two groups according to the value of M% assigned to each gene. The first group constituted 111 genes (17% of the total 648 non-redundant ARGs) with M% < 95% and, hence, were classified as risk level 5 representing the least hazards for potential ARG mobilization (Table S11). The other 50 highly mobilized ARGs (8% of the total ARGs) with M% ≥ 95% were assigned to risk levels from 1 to 4 with respect to ARG pathogenicity and annotation cutoff class (Table 3). Risk level 4 ARGs comprised only one transposon-encoded highly mobilized ARG from beta-lactam drug class OXA-347 which was hosted on non-pathogen-of-origin ACC. The remaining 49 genes were all hosted by potential pathogens and classified due to their BLASTP pass bitscore in RGI analysis. A total of 20 ARGs annotated as perfect hits by RGI software were allocated in risk level 1 representing the highest priority ARGs of 100% homology to functional resistance genes in CARD database. Those genes conferred resistance to clinically-relevant antibiotics namely aminoglycoside (AAC(6′)-Il, AAC(6′)-IIa, aadA2, aadA6, and ANT(2″)-Ia), beta-lactam (CfxA2, CMY-4, GES-2, OXA-2, PER-3, TEM-181, and VEB-9), diaminopyrimidine (dfrB5), fluoroquinolone (QnrS1, QnrS2, and QnrVC4), MLS (mphE), peptide (MCR-5.1), and phenicol (catI and cmlA5). Risk level 2 constituted 19 ARGs belonged to RGI strict hits forming a group of highly mobilized ARGs for unknown variants of functional resistance genes in CARD database.
Table 3.
Risk priority classification of highly mobilized ARGs in the effluent (ARG count = 50).
The ARGs of this class comprised genes for resistance against aminoglycoside (AAC(6′)-Ib9, aadA, aadA10, aadA15, aadA16, aadA16, and ANT(3″)-IIa), beta-lactam (NPS-1, OXA-1, OXA-129, and OXA-663), diaminopyrimidine (dfrA1 and dfrA1), MLS (ErmB, lnuB, and lnuF), phenicol (floR), and tetracycline (tetM and tetX). Lastly, a total of 10 ARGs associated with the RGI loose hits were ranked as risk level 3, indicating new emergent threats for ARG mobilization that require further functional validation. The list of genes belonging to this group comprised ARGs encoded for resistance to aminoglycoside (ANT(9)-Ia and aadA6/aadA10), beta-lactam (TLA-3, OXA-15, and OXA-898), fluoroquinolone (norB), MLS (lsaB and mef(D)), multidrug (efpA), and tetracycline (tetA(P)) antibiotics (Figure S3).
3.6. Genome-Based Analysis of the INF and EFF Samples
A total of 35 and 118 high-quality draft genomes were recovered from the INF and EFF assembled metagenomes, respectively, using MetaWRAP software with bin refinement cutoff thresholds of a minimum completeness of 70% and a maximum contamination of 10% (Figure S3). The consolidated high-quality draft genomes extracted by MetaWRAP constituted approximately 10% and 23% of the total size of INF and EFF assembled contigs respectively (Table 4). The heatmaps in Figure S5 show the level of completeness and contamination in each reassembled genomic bin of the INF and EFF metagenomes. To determine the potential ARG-carrying genomes (ACGs), the contig IDs of INF and EFF ACCs were cross-matched to those of MetaWRAP genome bins. A total of 30/35 and 50/118 INF and EFF ACGs contained 90 and 200 ACCs, respectively. The RAST seed analysis for the closest neighbor species resulted in the assignment of 26 and 79 non-redundant bacterial species belonging to 25 and 70 genera to the recovered draft genomes of the INF and EFF samples, respectively (Table S12).
Table 4.
Summary of the genome binning results for the influent and effluent assembled contigs using a hybrid bin extraction algorithm in the MetaWRAP software.
A total of 10 species belonging to nine genera were shared by the INF and EFF genome bins, indicating the potential persistence of these bacteria throughout the treatment process. The persistent genera accounted for Acidovorax, Akkermansia, Bacteroides, Eubacterium, Faecalibacterium, Stenotrophomonas, Thauera, Thermincola, and Tolumonas (Table S13). An opportunistic pathogen, Bacteroides fragilis, was among the persistent species assigned to one bin in the INF (bin1) and four bins in the EFF genomes (i.e., bin 37, bin 65, bin 111, and bin 116). Although we could not recover any genomes on the WHO list for high-priority human pathogens in the INF bins, two EFF genome bins were assigned to Salmonella enterica (bin101) and Shigella dysenteriae (bin 11). In addition, two EFF genome bins (i.e., bin 40 and bin 95) were assigned to the Gram-negative bacillus Laribacter hongkongensis. Moreover, an emerging human pathogen species, namely, Arcobacter butzleri, was assigned to EFF bin 32.
The ResFinder search showed that none of the INF genome bins harbored any acquired ARGs; however, six EFF genome bins were encoded for ten acquired ARGs (Table 5). The identity percentage of the acquired ARGs to the reference sequences from ResFinder database ranged from 97 to 100%. The genome bins with acquired ARGs comprised three genomes carrying more than one ARG and the other three with one resistance gene each. Bin 88 (closest-neighbor species, Bacteroides thetaiotaomicron) was encoded for two ARGs against MLS (lnuB and lsaE), bin 94 (closest-neighbor species, Anoxybacillus flavithermus) harbored genes against beta-lactam (cfxA6) and tetracycline (tetQ), and bin 75 (closest-neighbor species, Acinetobacter calcoaceticus PHEA-2) with two genes against MLS (mphE and msrE) and one against tetracycline (tet39) (Table S14). Furthermore, three bins, namely, bin 3 (Janibacter sp. HTCC2649), bin 82 (Bacteroides vulgatus ATCC 8482), and bin 101 (Salmonella enterica subsp. enterica serovar Typhi Ty2), carried a single ARG against beta-lactam (blaGES-14), nitroimidazole (nimD), and MLS (mdfA). Strikingly, it was observed that the tet(39) gene was flanked by two MGE sequences, namely, the transposase and resolvase genes, on its carrying contig in bin 75 (contig# k141_59336_length_24223_cov_13.4223), suggesting a transposon-mediated ARG mobility event within the genomic region of the recovered MAG (Figure 7).
Table 5.
List of acquired ARGs detected by ResFinder software in EFF genome bins along with their annotations.
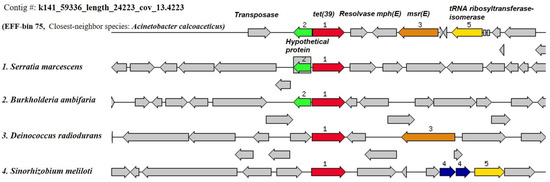
Figure 7.
Example of the functional annotation by the RAST subsystems for the chromosomal region with acquired ARGs as determined by the ResFinder software in EFF MAG bin 75 (Contig#: k141_59336_length_24223_cov_13.4223, starting base # 1 and ending base # 8616). Sets of the acquired ARGs with a focus on tet(39) were compared to similar organisms to show the conserved gene patterns. Homologous genes were labeled by the same number and color.
The RAST functional analysis of the resistance to antibiotics and toxic compounds category showed that genes encoding for antibiotic resistance against fluoroquinolone, beta-lactamase, multidrug resistance efflux pumps, tetracycline, vancomycin, fosfomycin, streptothricin, and multiple antibiotic resistance (MAR) locus existed in the INF and EFF genome bins. In addition, genes in the metal resistance category, including copper homeostasis, cobalt–zinc–cadmium resistance, zinc resistance, mercuric reductase, mercury resistance operon, arsenic resistance, resistance to chromium compounds, and cadmium resistance were also present in the recovered genomes (Table S15).
4. Discussion
This study utilized the same datasets as in our previous publication [38] in a completely different prospective. Although we previously analyzed the microbiome composition and ARG abundance profiles of the nucleotide sequences extracted from the wastewater influent, effluent, and activated sludge samples of a full-scale WWTP in Egypt, we could not examine the ARGs in their genomic context due to the limitations of short read lengths which resulted from the Illumina shotgun metagenomics sequencing (150 bp). In the present study, the metagenomic assembly and bioinformatic analysis approaches were utilized to quantitatively assess the potential human health risks of environmental resistomes and to estimate the ARG mobility potential percentage of the influent and effluent datasets at the same WWTP. In addition, high-quality draft metagenome-assembled genomes (MAGs) of the INF and EFF samples were recovered and investigated for conferring acquired ARGs as evidence for the incidence of horizontal gene transfer events during wastewater treatment. Our analysis provided insight into the predicted impact of conventional wastewater treatment on the ARG mobilization among the microbial communities of the receiving environments.
The application of a quantitative metric to evaluate the environmental resistomes in WWTPs elaborated the role of conventional wastewater treatment to reduce ARG dissemination risks in the receiving water streams. For example, the comparison of the INF and EFF risk ratios to the selected samples from various geographic locations revealed the existence of a global trend in the risk ranking, indicating that wastewater influent, raw sewage, and hospital sewage have higher risk ranks than effluent worldwide. These results are consistent with a recent study [64] that demonstrated that the INF samples had a higher risk index than the secondary effluent because of the removal of the ARGs and pathogenic bacteria during the wastewater treatment.
The contig-based analysis of the INF and EFF ACCs showed that a large portion of the detected ARGs (53%) persisted throughout the treatment process representing the core resistome of the examined WWTP.
The calculated average percentage of ARG mobility potential in both INF and EFF ACCs (5%) was comparable to those estimated by Ju et al. (2019) [52] (5.4%) and Sentchilo et al. (2013) [65] in two WWTPs (2.5% and 4%) suggesting a common pattern of ARG mobility incidence in WWTPs regardless of their geographical location.
We integrated the quantitative method for estimating the ARG mobility potential [52] with the ARG annotation results obtained from the RGI analysis. This enabled us to identify ARGs associated with high mobility potential and clinical significance at the same time for further surveillance in downstream environments. The proposed classification framework in the current study focused on the 50 highly mobilized ARGs with an M% in EFF greater than 95% out of the total mobile ARGs (161 genes), since this group included ARGs with the greatest potential of dissemination in the microbiomes of the receiving environments. Theses ARGs either persisted throughout the treatment process from INF to EFF (35 genes) or were detected in EFF only (15 genes). Most of the selected ARGs (49/50 genes) were found to be associated with pathogenic sequences on their ACCs, raising a concern regarding their potential health risk in the receiving environment and calling for further investigation. Highly mobilized ARGs related to risk levels 1 (20 genes), with a perfect match to known functional ARGs in the CARD database, and level 2 (19 genes) for new variants of known ARGs accounted for the resistance genes posing current health threats because of their clinical relevance. An example of a risk level 1 ARG is a plasmid-mediated phosphoethanolamine transferase gene, MCR-5.1, from the peptide antibiotics drug class conferring resistance to colistin, which is one of the last resort antibiotics for the treatment of human infections [66]. Another group of highly mobilized ARGs that encode for resistance to clinically important antibiotics from class-A, -C, -D beta-lactamases, including VEB-9 and TEM-181, multiple genes from the OXA-family, CMY-4, and CfxA2 were among the highest risk levels 1 and 2, according to our classification.
The current analysis framework enabled us to extract a group of ARGs (10 genes) representing the emergent threats (risk level 3) that belonged to the loose category of the RGI cutoff threshold. A phenotype analysis for the genes of this group is needed to validate their potential risks. Only one ARG (OXA-347) of the highly mobilized group (50 genes) conferring resistance to the beta-lactam class (carbapenem, cephalosporin, penam) was categorized as risk level 4, since it was located on non-pathogenic ACCs of the EFF samples. The remaining group of non-redundant mobile ARGs (111 genes) detected in both the INF and EFF ACCs with an M% less than 95%, regardless of their potential hosts and the RGI classification cutoff, were grouped in the least risk level 5. This class of genes was considered as a lower risk priority due to the fact of their low mobility potential (from 0% to 63%) or complete removal from the discharged effluent. The proposed ranking outline provided an initial step towards combining the results of the quantitative metric for the ARG mobility potential (M%) with the knowledge for host pathogenicity and the homology search results to the curated protein database of the functional AMRs in CARD. The classified mobile ARGs by our approach were consistent with those detected from the quantitative analysis of the ARG mobility potential from 12 WWTPs in China [52]. In addition, the high-priority ARGs detected in the current research were comparable to the results obtained from a recent study for a multihabitat ARG health risk ranking based on ARG enrichment in human-impacted environments, mobility potential, and host pathogenicity [67].
The MEGAN taxonomic analysis of the INF and EFF ACCs at the genus level indicated that the diversity of the potential ARG hosts increased from INF to EFF, as 82% of the analyzed INF ACCs were assigned to 121 genera, while only 62% of the EFF ACCs were allocated in 125 genera. The previous observation can be interpreted as some of the non-ARG hosts in the influent possibly acquired ARGs during the treatment process [68]. Similarly, it was demonstrated that the diversity of recipient phyla for the IncP-1 conjugative plasmid pKJK5 in the effluent wastewater microbiome was broader than that of the influent [69].
The application of the modular hybrid genome binning pipeline of the MetaWRAP software allowed for improving the percentages of genome completion and minimizing the level of contamination in the recovered high-quality draft genomes, as explained in Figure S4. However, the majority of the total size of the assembled contigs (90% in the influent and 77% in the effluent) were below the selected bin refinement thresholds for the extracted MAGs. This could happen due to the known metagenomic assembly challenges of non-uniform contig coverage caused by the microbial complexity and diversity of the environmental samples [70] as well as unresolved interspecies conserved repeat sequences either of plasmidic or chromosomal origins which, in turn, resulted in the unsuccessful resolution of most genomes from the metagenomics samples [71]. Despite the previous challenges, we could highlight some remarkable observations in the recovered MAGs regarding the impact of conventional wastewater treatment on the reduction of biomass content and indirectly the ARG concentration from influent to effluent. For example, the percentage of ACGs in the recovered high-quality draft MAGs declined from 86% (30/35 bins) in the INF bins into 42% (50/118 bins) in the EFF genomes emphasizing the removal of 50% of the ARB content from the discharged effluent. Furthermore, the number of the extracted draft MAGs in EFF (118 genome bins) was higher compared to those determined in the effluent of a WWTP in Singapore containing a membrane bioreactor (101 genome bins) [60], suggesting more biomass discharged into the environment from the conventional WWTP in our study.
Although we could not recover any high-quality genomes associated with E. coli as an indicator for fecal coliform contamination in our samples, two species belonged to the WHO’s high-priority human pathogens list [72], accounting for the closest neighbor species Salmonella enterica subsp. Enterica serovar Typhi Ty2 and Shigella dysenteriae, which were assigned to the EFF genome bins 101 and 11, respectively, as indicated by the RAST analysis. This observation suggests the need for an in-depth functional investigation to assess the regrowth possibility for the two organisms in the receiving water currents which might have further implications for both human and environmental health.
The ResFinder analysis indicated no acquired ARGs in the INF MAGs. However, EFF genome bin 101 (Salmonella enterica) was found to exhibit a multidrug resistance capacity by encoding the acquired ARGs against beta-lactam (blaGES-14), nitroimidazole (nimD), and MLS (mdfA) antibiotics, indicating the potential mobilization of these genes among EFF microbial community members. Furthermore, the acquired mobile ARGs cassette detected by ResFinder in bin 75 was previously determined to be plasmid-mediated in the human pathogen Acinetobacter baumannii [73]. While we could locate genes associated with this resistance cassette (i.e., tet39, mphE, msrE, resolvase, and transposase) encoded in EFF bin 75 (closest neighbor species Acinetobacter calcoaceticus), suggesting the possible incidence of HGT events among the microbes of the examined WWTP. A recent study [74] compared the reconstructed genomes of 82 multidrug-resistant bacteria isolated from WWTPs to their closest neighbors of human/animal-associated bacteria in the publicly available databases. The results showed that WWTP-associated bacteria had ARG-carrying plasmids with a distinctive structure that mediated the potential HGT of multidrug resistance determinants between WWTP bacteria and those of human/animal origin. It is worth mentioning here that the extent of the HGT incidents in WWTPs, with respect to host range, frequency of occurrence, and the environmental factors affecting them, is still debatable [75].
A recent study demonstrated that the examined Archobacter genomes were associated with high levels of virulence genes proposing an elevated pathogenicity potential of community members affiliated to this genus [76]. As a result, the recovered EFF genome of bin 32 assigned to Arcobacter butzleri RM4018 might pose a concern as an emerging human pathogen and more detailed targeted studies are recommended to explore metabolic activities associated with this genome. Similarly, we were able to recover two EFF MAGs (i.e., bin 40 and bin 95) assigned to a closest neighbor species, namely, Laribacter hongkongensis HLHK9, which exists extensively in fresh water fish [75], and a strain HLGZ1, which was recently isolated from clinical samples in Hong Kong [77]. These findings necessitate an in-depth functional research and long-term surveillance plans to explore the potential interactions of the above-mentioned organisms within downstream environments.
The current study applied three analysis approaches to assess the assembled metagenomic datasets for potential public health risks of wastewater resistomes. Firstly, a comparative quantitative method using MetaCompare software pipeline was applied to evaluate resistome risk ranking of local and international wastewater samples. This step validated the role of wastewater treatment process in the reduction of ARG spread risks at the downstream environments. Secondly, a contig-based analysis for the local INF and EFF assembled metagenomes was conducted to compare the ARG mobility potential percentage before and after treatment in the examined WWTP. As a result, we proposed a hierarchical framework to select the ARGs with a high mobility potential, carried by pathogenic host, persisting from INF to EFF, and annotated as perfect or strict protein hits by the RGI software. The detected ARGs using our method are considered of the highest health hazard, which require further monitoring efforts in downstream environments. Thirdly, we applied a software pipeline (MetaWRAP) to recover high-quality MAGs from the INF and EFF assemblies. The extracted MAGs were searched for acquired ARGs on their contigs, which indicates the potential incidence of HGT and confirms the possibility of ARG mobility during wastewater treatment. Six genome bins of the EFF MAGs were found to have 10 acquired ARGs. Finally, the analysis showed the persistence of pathogens belonging to the WHO high-priority list such as Salmonella enterica and Shigella dysenteriae in the wastewater effluent. The above-mentioned findings suggest the need for long-term surveillance plans by decision makers to minimize the potential hazards of the transfer of ARGs into pathogens in receiving environments.
5. Conclusions
To our knowledge, this is the first study in Arab countries to assess the possible risks of environmental resistomes in the influent and effluent of a conventional WWTP using a metagenomic assembly approach. The current study provided a comprehensive risk analysis of wastewater resistomes in a full-scale WWTP in Egypt. The risk scores of the wastewater influent and effluent resistomes belonging to the study samples were compared to their counterparts of other selected wastewater metagenomes from the publicly available databases to determine the risk ranking of the global environmental resistomes. The quantitative estimation for the resistome risk ranking by this study will help to improve surveillance and monitor trends of resistance genes released to receiving environments.
We proposed an integrative framework to identify the risk priority of unique ARGs on ACCs based on the mobility potential percentage, host pathogenicity, and ARG annotation cutoff category in RGI software with the CARD reference database. The proposed risk ranking system with five levels provided insight into the highest priority ARGs with clinical relevance (levels 1 and 2) and emergent threats (levels 3, 4, and 5) for further follow up in receiving environments. The study sheds light on the potential role of horizontal gene transfer in the spread of ARGs as a result of the co-occurrence of ARGs and MGEs in human pathogenic bacterial hosts in wastewater metagenomes. Despite the low abundance of mobile ARGs and pathogenic bacteria in the WWTP effluent, further investigations for the functional activities and regrowth possibilities of the released ARB is recommended to evaluate their impact on environmental and public health. Furthermore, future studies that explore the development of microbial community and ARGs dynamics in downstream environment are required to assess the implications of mixing treated wastewater with receiving water bodies. Additionally, we recommend conducting larger scale metagenomic studies either locally or internationally, especially in low-income countries to serve in building a comprehensive profile of ARGs and ARB along with their correlation to the operation parameters of WWTPs to help improve surveillance protocols and mitigation strategies to minimize the potential mobilization of ARGs in environmental microbiomes.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/su142114292/s1, Figure S1: The relative abundance percentage of ARG-carrying contigs (ACCs) at phylum level of INF and EFF assembled metagenomes using MEGAN analysis; Figure S2: The percentage of ARG-carrying contigs (ACCs) at all taxonomic ranks of the influent and effluent assembled metagenomes; Figure S3: The risk priority classification of the total mobile ARGs (161 genes) detected in influent and effluent ACCs (link to full resolution); Figure S4: Comparison of the genome binning results of the wastewater influent and effluent assembled metagenomes showing the improvement in the bin completion and contamination percentages at the selected cutoff thresholds as a result of using hybrid binning approach by MetaWRAP software; Figure S5: Heatmap shows the level of completeness and contamination of the metagenomic-assembled genomes (MAGs) extracted from INF and EFF datasets; Table S1: List of the selected publicly available wastewater datasets from the European Nucleotide Archive server (ENA) for the resistome risk analysis; Table S2: The logic matrix for predicting the ARG genetic context and mobility status of INF and EFF assembled contigs; Table S3: Statistics of the assembly results for the current study samples (EG_INF and EG_EFF) as well as the seventeen wastewater metagenomes selected from the ENA server; Table S4: the detailed description of the non-redundant ARGs detected in ACCs of both INF and EFF assembled metagenomes from the local WWTP; Table S5: Summary of the ACCs potential hosts at genus level with minimum abundance of 1% in any sample and the number of ACCs in each drug class of the annotated host; Table S6: The co-occurrence analysis of mobile genetic determinants and pathogen-like sequences on ACCs of the influent metagenome; Table S7: The co-occurrence analysis of mobile genetic determinants and pathogen-like sequences on the ACCs of the Effluent metagenome; Table S8: Summary of ACCs mobility analysis in assembled metagenomes of wastewater influent and effluent samples; Table S9: Composition of total mobile resistome composition in the wastewater influent and effluent metagenomes; Table S10: comparison of the total ARG mobility potential percentage per (a) drug classes and (b) resistance mechanisms in influent and effluent samples; Table S11. Total Mobile ARGs in INF and EFF with mobility potential percentage less than 95% in the discharged wastewater effluent streams; Table S12: Statistics of the genome binning results by MetaWrap pipeline and the closest neighbor species assigned by RAST server of INF and EFF assembled metagenomes; Table S13: Relative abundance of the genera detected in INF and EFF MAGs by RAST server; Table S14: Example of the functional annotation for the chromosomal region encoding for acquired ARGs in EFF-MAG (bin 75) at contig# k141_59336_length_24223_cov_13.4223 to explore flanking genes that indicate a potential ARG mobilization events; Table S15: Functional annotation of the resistance to antibiotic and toxic compounds category in the INF and EFF genome bins using Rast subsystems servers.; Script S1: modifications made in the MetaCompare software script to save all the risk analysis results into CSV files instead of displaying them on the computer screen.
Author Contributions
Data curation, O.S.A.; Formal analysis, A.A.O.; Funding acquisition, A.S.A., R.A. and B.A.; Investigation, A.K. and B.A.; Methodology, O.S.A.; Resources, A.A.O.; Supervision, W.G.H. and H.M.M.; Validation, A.A.O.; Visualization, A.K., R.A. and H.M.M.; Writing—original draft, O.S.A.; Writing—review & editing, S.E. and A.A.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Please refer to suggested Data Availability Statements in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics. You might choose to exclude this statement if the study did not report any data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO. Global Action Plan on Antimicrobial Resistance; WHO: Geneva, Switzerland, 2015; Volume 10. [Google Scholar]
- Hofer, U. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Rev. Microbiol. 2019, 17, 1124. [Google Scholar]
- Pazda, M.; Kumirska, J.; Stepnowski, P.; Mulkiewicz, E. Antibiotic resistance genes identified in wastewater treatment plant systems—A review. Sci. Total Environ. 2019, 697, 134023. [Google Scholar] [CrossRef] [PubMed]
- Fouz, N.; Pangesti, K.N.A.; Yasir, M.; Al-Malki, A.L.; Azhar, E.I.; Hill-Cawthorne, G.A.; El Ghany, M.A. The Contribution of Wastewater to the Transmission of Antimicrobial Resistance in the Environment: Implications of Mass Gathering Settings. Trop. Med. Infect. Dis. 2020, 5, 33. [Google Scholar] [CrossRef]
- Metcalf, W.W.; Griffin, B.M.; Cicchillo, R.M.; Gao, J.; Janga, S.C.; Cooke, H.A.; Circello, B.T.; Evans, B.S.; Martens-Habbena, W.; Stahl, D.A.; et al. Synthesis of Methylphosphonic Acid by Marine Microbes: A Source for Methane in the Aerobic Ocean. Science 2012, 337, 1104–1107. [Google Scholar] [CrossRef]
- Jiang, X.; Ellabaan, M.M.H.; Charusanti, P.; Munck, C.; Blin, K.; Tong, Y.; Weber, T.; Sommer, M.O.A.; Lee, S.Y. Dissemination of antibiotic resistance genes from antibiotic producers to pathogens. Nat. Commun. 2017, 8, 15784. [Google Scholar] [CrossRef] [PubMed]
- Larsson, D.G.J.; Flach, C.-F. Antibiotic resistance in the environment. Nat. Rev. Genet. 2021, 20, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Cantón, R.; González-Alba, J.M.; Galán, J.C. CTX-M Enzymes: Origin and Diffusion. Front. Microbiol. 2012, 3, 110. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, J.; Chowdhury, P.R.; Djordjevic, S.P. Genomic analysis of multidrug-resistant Escherichia coli ST58 causing urosepsis. Int. J. Antimicrob. Agents 2018, 52, 430–435. [Google Scholar] [CrossRef]
- de Souza, M.C.R.; Marques, C.T.; Dore, C.M.G.; da Silva, F.R.F.; Rocha, H.A.O.; Leite, E.L. Antioxidant activities of sulfated polysaccharides from brown and red seaweeds. J. Appl. Phycol. 2006, 19, 153–160. [Google Scholar] [CrossRef]
- Łuczkiewicz, A.; Jankowska, K.; Fudala-Książek, S.; Olańczuk-Neyman, K. Antimicrobial resistance of fecal indicators in municipal wastewater treatment plant. Water Res. 2010, 44, 5089–5097. [Google Scholar] [CrossRef]
- da Costa, P.M.; Vaz-Pires, P.; Bernardo, F. Antimicrobial resistance in Enterococcus spp. isolated in inflow, effluent and sludge from municipal sewage water treatment plants. Water Res. 2006, 40, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- Al-Jassim, N.; Ansari, M.I.; Harb, M.; Hong, P.-Y.Y. Removal of bacterial contaminants and antibiotic resistance genes by conventional wastewater treatment processes in Saudi Arabia: Is the treated wastewater safe to reuse for agricultural irrigation? Water Res. 2015, 73, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.; Tay, M.; Tan, B.; Le, T.-H.; Haller, L.; Chen, H.; Koh, T.H.; Barkham, T.M.S.; Thompson, J.R.; Gin, K.Y.-H. Characterization of Metagenomes in Urban Aquatic Compartments Reveals High Prevalence of Clinically Relevant Antibiotic Resistance Genes in Wastewaters. Front. Microbiol. 2017, 8, 2200. [Google Scholar] [CrossRef] [PubMed]
- Cacace, D.; Fatta-Kassinos, D.; Manaia, C.M.; Cytryn, E.; Kreuzinger, N.; Rizzo, L.; Karaolia, P.; Schwartz, T.; Alexander, J.; Merlin, C.; et al. Antibiotic resistance genes in treated wastewater and in the receiving water bodies: A pan-European survey of urban settings. Water Res. 2019, 162, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.N.; Scriver, L.; Neudorf, K.D.; Hansen, L.T.; Jamieson, R.C.; Yost, C. Antimicrobial resistance gene surveillance in the receiving waters of an upgraded wastewater treatment plant. FACETS 2018, 3, 128–138. [Google Scholar] [CrossRef]
- Karkman, A.; Do, T.T.; Walsh, F.; Virta, M.P. Antibiotic-Resistance Genes in Waste Water. Trends Microbiol. 2018, 26, 220–228. [Google Scholar] [CrossRef]
- Liu, S.-S.; Qu, H.-M.; Yang, D.; Hu, H.; Liu, W.-L.; Qiu, Z.-G.; Hou, A.-M.; Guo, J.; Li, J.-W.; Shen, Z.-Q.; et al. Chlorine disinfection increases both intracellular and extracellular antibiotic resistance genes in a full-scale wastewater treatment plant. Water Res. 2018, 136, 131–136. [Google Scholar] [CrossRef]
- Magnusdottir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2016, 35, 81–89. [Google Scholar] [CrossRef]
- LaPara, T.M.; Burch, T.R.; McNamara, P.J.; Tan, D.T.; Yan, M.; Eichmiller, J.J. Tertiary-Treated Municipal Wastewater is a Significant Point Source of Antibiotic Resistance Genes into Duluth-Superior Harbor. Environ. Sci. Technol. 2011, 45, 9543–9549. [Google Scholar] [CrossRef]
- Pruden, A.; Arabi, M.; Storteboom, H.N. Correlation Between Upstream Human Activities and Riverine Antibiotic Resistance Genes. Environ. Sci. Technol. 2012, 46, 11541–11549. [Google Scholar] [CrossRef]
- Jäger, T.; Hembach, N.; Elpers, C.; Wieland, A.; Alexander, J.; Hiller, C.; Krauter, G.; Schwartz, T. Reduction of Antibiotic Resistant Bacteria During Conventional and Advanced Wastewater Treatment, and the Disseminated Loads Released to the Environment. Front. Microbiol. 2018, 9, 2599. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.Q.; Vu, H.P.; Nguyen, L.N.; Wang, Q.; Djordjevic, S.P.; Donner, E.; Yin, H.; Nghiem, L.D. Monitoring antibiotic resistance genes in wastewater treatment: Current strategies and future challenges. Sci. Total Environ. 2021, 783, 146964. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Wang, Y.; Walsh, T.R.; Yi, L.-X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Matamoros, S.; van Hattem, J.M.; Arcilla, M.S.; Willemse, N.; Melles, D.C.; Penders, J.; Vinh, T.N.; Hoa, N.T.; Bootsma, M.C.J.; van Genderen, P.J.; et al. Global phylogenetic analysis of Escherichia coli and plasmids carrying the mcr-1 gene indicates bacterial diversity but plasmid restriction. Sci. Rep. 2017, 7, 15364. [Google Scholar] [CrossRef]
- Aminov, R.I. Horizontal Gene Exchange in Environmental Microbiota. Front. Microbiol. 2011, 2, 158. [Google Scholar] [CrossRef]
- Michael, I.; Rizzo, L.; McArdell, C.S.; Manaia, C.M.; Merlin, C.; Schwartz, T.; Dagot, C.; Fatta-Kassinos, D. Urban wastewater treatment plants as hotspots for the release of antibiotics in the environment: A review. Water Res. 2013, 47, 957–995. [Google Scholar] [CrossRef]
- Watkinson, A.; Murby, E.; Costanzo, S. Removal of antibiotics in conventional and advanced wastewater treatment: Implications for environmental discharge and wastewater recycling. Water Res. 2007, 41, 4164–4176. [Google Scholar] [CrossRef]
- Martínez, J.L.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Genet. 2014, 13, 116–123. [Google Scholar] [CrossRef]
- Li, A.-D.; Li, L.-G.; Zhang, T. Exploring antibiotic resistance genes and metal resistance genes in plasmid metagenomes from wastewater treatment plants. Front. Microbiol. 2015, 6, 1025. [Google Scholar] [CrossRef]
- Parsley, L.C.; Consuegra, E.J.; Kakirde, K.S.; Land, A.M.; Harper, W.F.; Liles, M.R. Identification of Diverse Antimicrobial Resistance Determinants Carried on Bacterial, Plasmid, or Viral Metagenomes from an Activated Sludge Microbial Assemblage. Appl. Environ. Microbiol. 2010, 76, 3753–3757. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, X.-X.; Ye, L. Plasmid Metagenome Reveals High Levels of Antibiotic Resistance Genes and Mobile Genetic Elements in Activated Sludge. PLoS ONE 2011, 6, e26041. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Pruden, A.; Chen, C.; Heath, L.S.; Xia, K.; Zhang, L. MetaCompare: A computational pipeline for prioritizing environmental resistome risk. FEMS Microbiol. Ecol. 2018, 94, fiy079. [Google Scholar] [CrossRef] [PubMed]
- Boolchandani, M.; D’Souza, A.W.; Dantas, G. Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet. 2019, 20, 356–370. [Google Scholar] [CrossRef]
- Albertsen, M.; Hugenholtz, P.; Skarshewski, A.; Nielsen, K.L.; Tyson, G.; Nielsen, P.H. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 2013, 31, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Hultman, J.; Waldrop, M.; Mackelprang, R.; David, M.M.; McFarland, J.W.; Blazewicz, S.J.; Harden, J.W.; Turetsky, M.R.; McGuire, A.D.; Shah, M.B.; et al. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 2015, 521, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Deng, Y.; Ma, L.; Wang, Y.; Chan, L.Y.; Zhang, T. Exploration of the antibiotic resistome in a wastewater treatment plant by a nine-year longitudinal metagenomic study. Environ. Int. 2019, 133, 105270. [Google Scholar] [CrossRef]
- Ali, O.S.; Hozayen, W.G.; Almutairi, A.S.; Edris, S.A.; Abulfaraj, A.A.; Ouf, A.A.; Mahmoud, H.M. Metagenomic Analysis Reveals the Fate of Antibiotic Resistance Genes in a Full-Scale Wastewater Treatment Plant in Egypt. Sustainability 2021, 13, 11131. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Fresia, P.; Antelo, V.; Salazar, C.; Giménez, M.; D’Alessandro, B.; Afshinnekoo, E.; Mason, C.; Gonnet, G.H.; Iraola, G. Urban metagenomics uncover antibiotic resistance reservoirs in coastal beach and sewage waters. Microbiome 2019, 7, 35. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FASTQC. A Quality Control Tool for High Throughput Sequence Data 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 16 September 2022).
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Leplae, R.; Lima-Mendez, G.; Toussaint, A. ACLAME: A CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res. 2010, 38, D57–D61. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2016, 45, D535–D542. [Google Scholar] [CrossRef] [PubMed]
- Ligges, U.; Maechler, M. scatterplot3d-AnRPackage for Visualizing Multivariate Data. J. Stat. Softw. 2003, 8, 1–20. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Zhao, R.; Yu, K.; Zhang, J.; Zhang, G.; Huang, J.; Ma, L.; Deng, C.; Li, X.; Li, B. Deciphering the mobility and bacterial hosts of antibiotic resistance genes under antibiotic selection pressure by metagenomic assembly and binning approaches. Water Res. 2020, 186, 116318. [Google Scholar] [CrossRef]
- Ju, F.; Beck, K.; Yin, X.; Maccagnan, A.; McArdell, C.S.; Singer, H.P.; Johnson, D.R.; Zhang, T.; Bürgmann, H. Wastewater treatment plant resistomes are shaped by bacterial composition, genetic exchange, and upregulated expression in the effluent microbiomes. ISME J. 2019, 13, 346–360. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition—Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-W.; Tang, Y.-H.; Tringe, S.G.; Simmons, A.B.; Singer, S.W. MaxBin: An automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2014, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [PubMed]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Loman, N.J.; Andersson, A.F.; Quince, C. CONCOCT: Clustering contigs on coverage and composition. arXiv 2013. [Google Scholar] [CrossRef]
- Song, W.-Z.; Thomas, T. Binning_refiner: Improving genome bins through the combination of different binning programs. Bioinformatics 2017, 33, 1873–1875. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Ng, C.; Tan, B.; Jiang, X.-T.; Gu, X.; Chen, H.; Schmitz, B.W.; Haller, L.; Charles, F.R.; Zhang, T.; Gin, K. Metagenomic and Resistome Analysis of a Full-Scale Municipal Wastewater Treatment Plant in Singapore Containing Membrane Bioreactors. Front. Microbiol. 2019, 10, 172. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Majeed, H.J.; Riquelme, M.V.; Davis, B.C.; Gupta, S.; Angeles, L.; Aga, D.S.; Garner, E.; Pruden, A.; Vikesland, P.J. Evaluation of Metagenomic-Enabled Antibiotic Resistance Surveillance at a Conventional Wastewater Treatment Plant. Front. Microbiol. 2021, 12, 657954. [Google Scholar] [CrossRef] [PubMed]
- Sentchilo, V.; Mayer, A.P.; Guy, L.; Miyazaki, R.; Tringe, S.; Barry, K.; Malfatti, S.; Goessmann, A.; Robinson-Rechavi, M.; van der Meer, J.R. Community-wide plasmid gene mobilization and selection. ISME J. 2013, 7, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Jaiswal, S.; Sodhi, K.K.; Shree, P.; Singh, D.K.; Agrawal, P.K.; Shukla, P. Antibiotics bioremediation: Perspectives on its ecotoxicity and resistance. Environ. Int. 2019, 124, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.-N.; Gaston, J.M.; Dai, C.L.; Zhao, S.; Poyet, M.; Groussin, M.; Yin, X.; Li, L.-G.; van Loosdrecht, M.C.M.; Topp, E.; et al. An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat. Commun. 2021, 12, 4765. [Google Scholar] [CrossRef] [PubMed]
- Hultman, J.; Tamminen, M.; Pärnänen, K.; Cairns, J.; Karkman, A.; Virta, M. Host range of antibiotic resistance genes in wastewater treatment plant influent and effluent. FEMS Microbiol. Ecol. 2018, 94, fiy038. [Google Scholar] [CrossRef] [PubMed]
- Jacquiod, S.; Brejnrod, A.; Morberg, S.M.; Abu Al-Soud, W.; Sørensen, S.J.; Riber, L. Deciphering conjugative plasmid permissiveness in wastewater microbiomes. Mol. Ecol. 2017, 26, 3556–3571. [Google Scholar] [CrossRef]
- Lapidus, A.L.; Korobeynikov, A.I. Metagenomic Data Assembly—The Way of Decoding Unknown Microorganisms. Front. Microbiol. 2021, 12, 613791. [Google Scholar] [CrossRef]
- Antipov, D.; Raiko, M.; Lapidus, A.; Pevzner, P.A. Plasmid detection and assembly in genomic and metagenomic data sets. Genome Res. 2019, 29, 961–968. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Blackwell, G.A.; Hall, R.M. Mobilisation of a small Acinetobacter plasmid carrying an oriT transfer origin by conjugative RepAci6 plasmids. Plasmid 2019, 103, 36–44. [Google Scholar] [CrossRef]
- Che, Y.; Xu, X.; Yang, Y.; Břinda, K.; Hanage, W.; Yang, C.; Zhang, T. High-resolution genomic surveillance elucidates a multilayered hierarchical transfer of resistance between WWTP- and human/animal-associated bacteria. Microbiome 2022, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Moralez, J.; Szenkiel, K.; Hamilton, K.; Pruden, A.; Lopatkin, A.J. Quantitative analysis of horizontal gene transfer in complex systems. Curr. Opin. Microbiol. 2021, 62, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Šilha, D.; Vacková, B.; Šilhová, L. Occurrence of virulence-associated genes in Arcobacter butzleri and Arcobacter cryaerophilus isolates from foodstuff, water, and clinical samples within the Czech Republic. Folia Microbiol. 2018, 64, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-K.; Chen, J.-H.; Yang, L.; Li, A.-R.; Su, D.-H.; Lin, Y.-P.; Chen, D.-Q. Emergence and genomic analysis of MDR Laribacter hongkongensis strain HLGZ1 from Guangzhou, China. J. Antimicrob. Chemother. 2017, 73, 643–647. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).