
A Comprehensive Profile of Antibiotic Resistance Genes in the Water Column of a Shallow-Sea Hydrothermal Vent Ecosystem

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
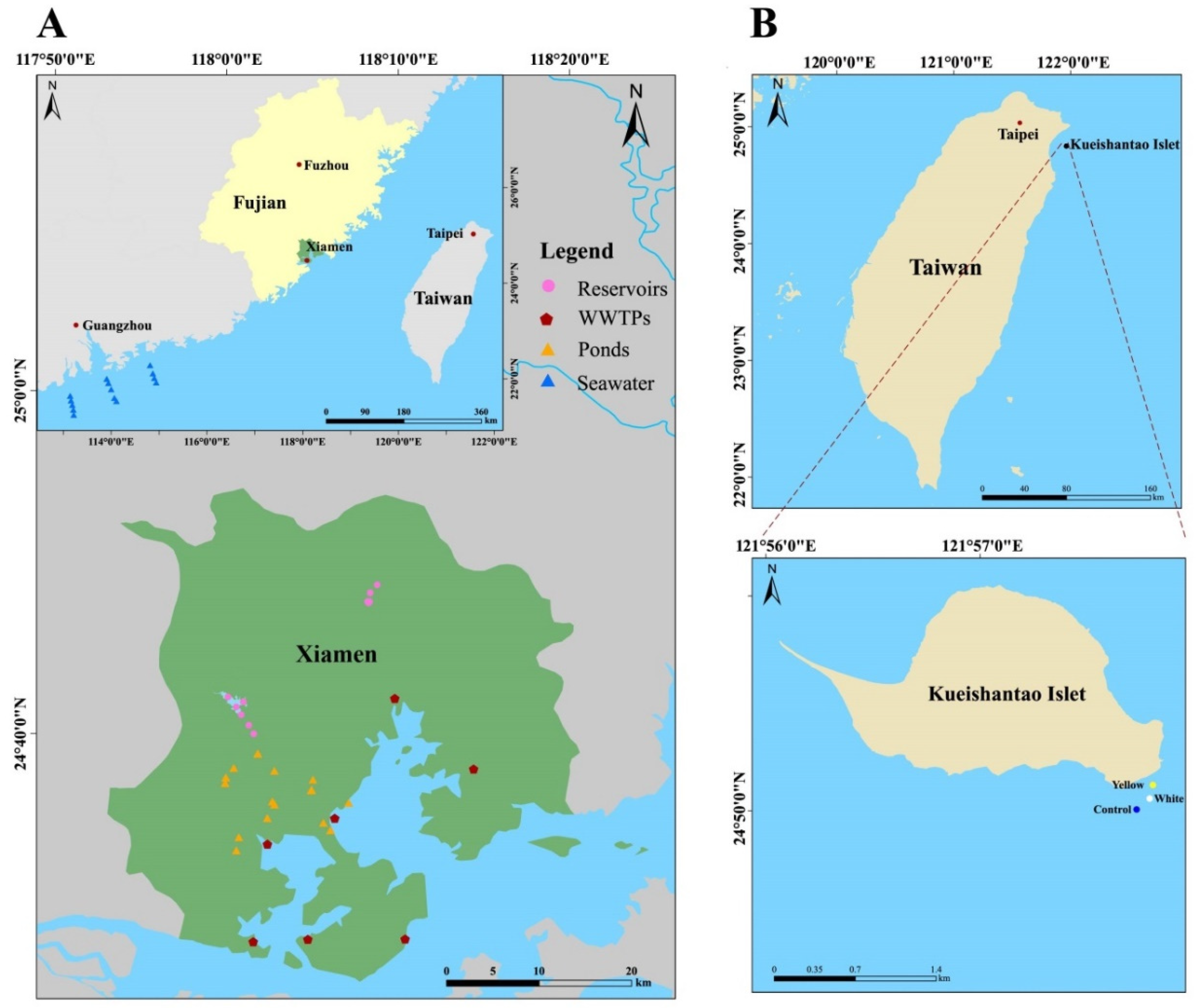
2.1. Sampling Design, Collection and Physicochemical Analysis
2.2. DNA Extraction, 16S rRNA Gene Full-Length High-Throughput Sequencing and Sequence Analysis
2.3. HT-qPCR Analysis of ARGs
2.4. Statistical Analysis
3. Results and Discussion
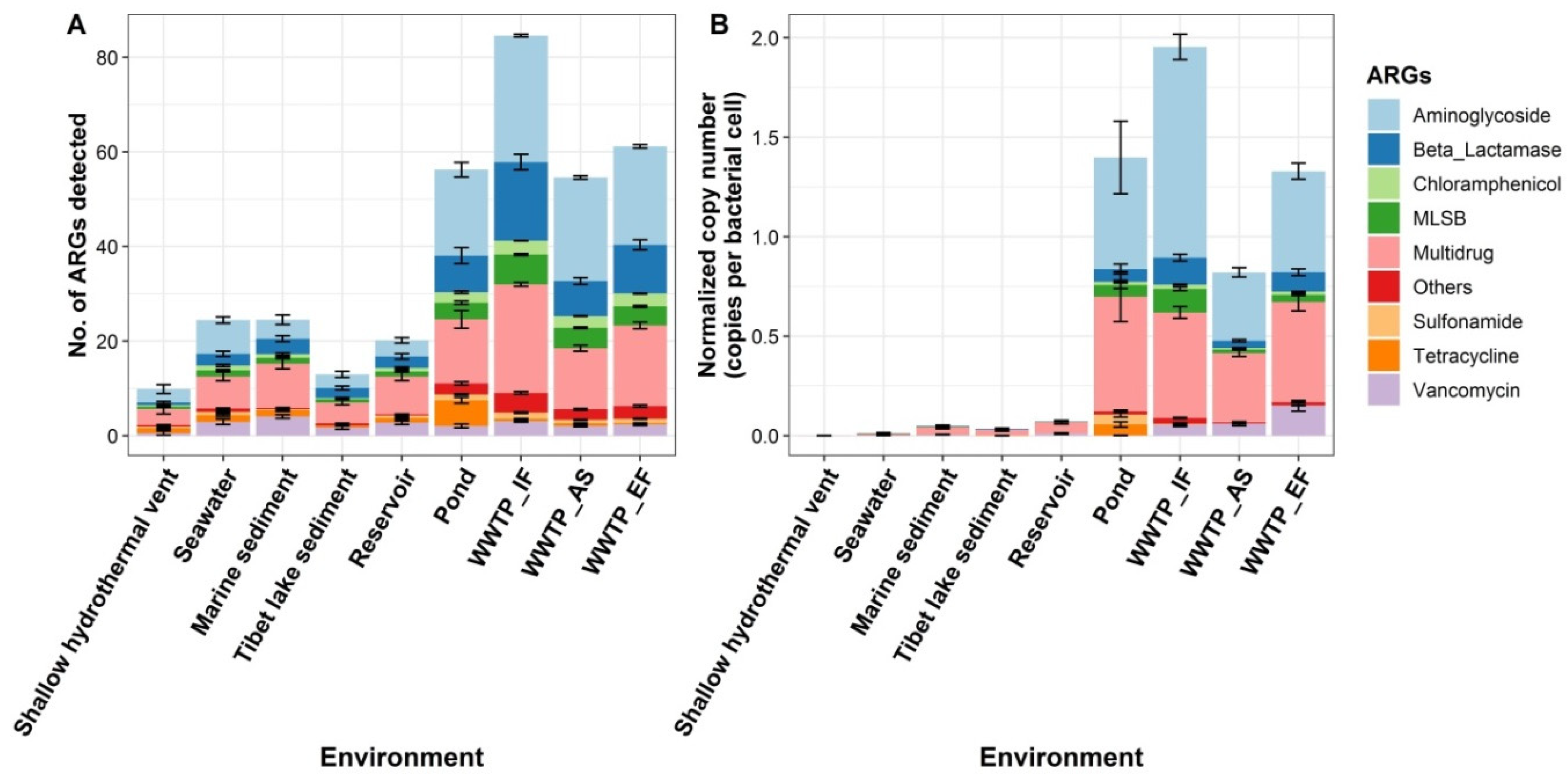
3.1. Profiling ARGs in Multiple Pristine and Anthropogenic-Disturbed Environments
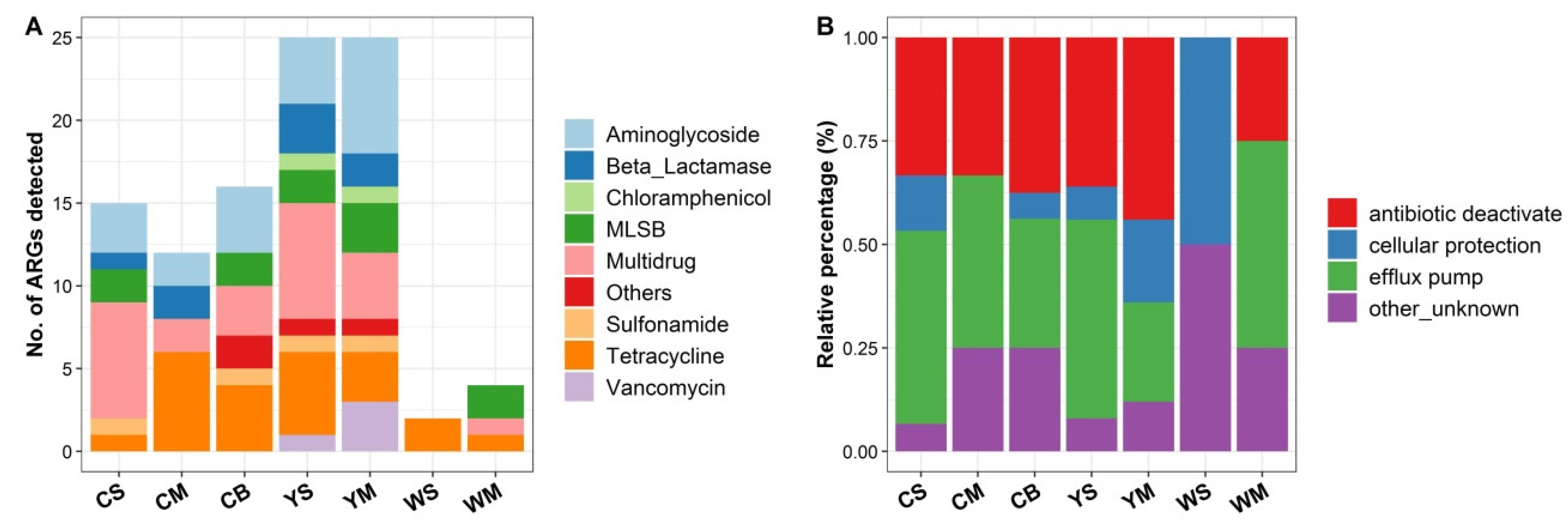
3.2. Diversity and Abundance of ARGs in the Shallow-Sea Hydrothermal Vent Ecosystem
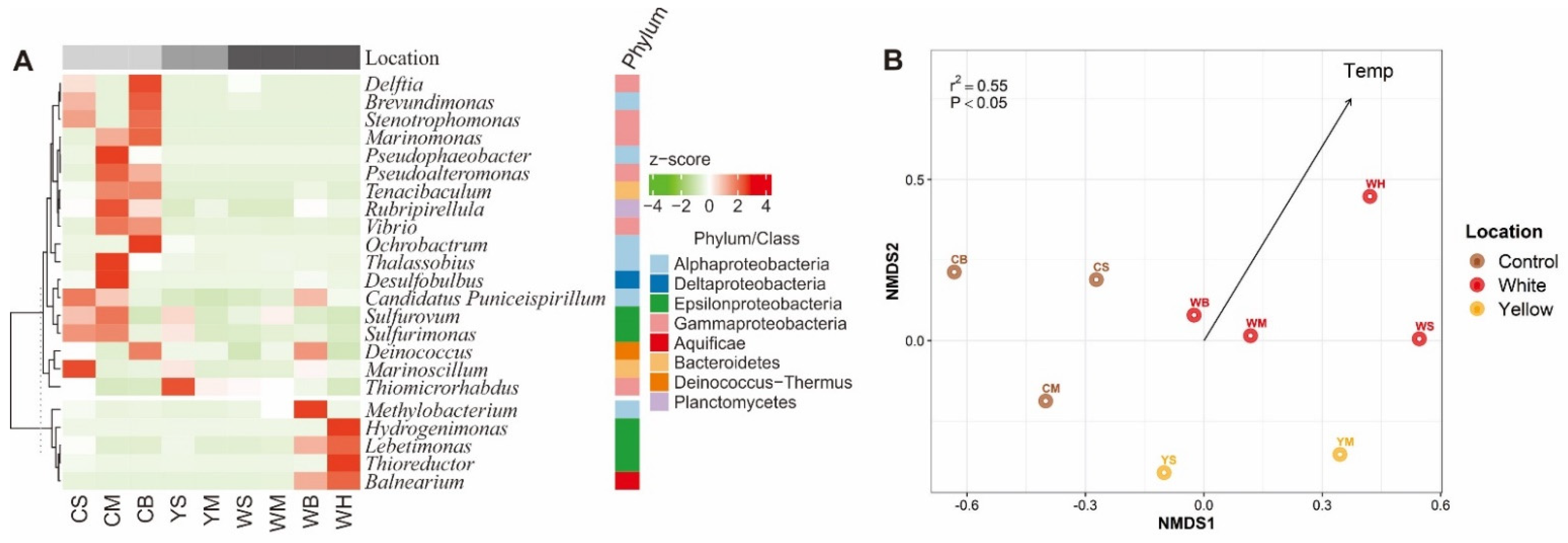
3.3. Bacterial Community Compositions of the Shallow-Sea Thermal Vent Ecosystem
3.4. Driving Forces of the ARG Profiles of the Shallow-Sea Hydrothermal Vent Ecosystem
3.5. Implications for the Baseline Levels of Environmental ARGs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, M.W.; Pierneef, R.; Bezuidt, O.K.I.; Van De Peer, Y.; Cowan, D.A.; Makhalanyane, T.P. A reservoir of ’historical’ antibiotic resistance genes in remote pristine Antarctic soils. Microbiome 2018, 6, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruden, A.; Pei, R.; Storteboom, H.; Carlson, K. Antibiotic resistance genes as emerging contaminants studies in northern Colorado. Environ. Sci. Technol. 2006, 40, 7445–7450. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 2015, 13, 116–123. [Google Scholar] [CrossRef]
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Zhu, Y.G.; Zhao, Y.; Li, B.; Huang, C.L.; Zhang, S.Y.; Yu, S.; Chen, Y.S.; Zhang, T.; Gillings, M.R.; Su, J.Q. Continental-scale pollution of estuaries with antibiotic resistance genes. Nat. Microbiol. 2017, 2, 16270. [Google Scholar] [CrossRef]
- Yang, Y.; Li, B.; Ju, F.; Zhang, T. Exploring variation of antibiotic resistance genes in activated sludge over a four-year period through a metagenomic approach. Environ. Sci. Technol. 2013, 47, 10197–10205. [Google Scholar] [CrossRef]
- Hu, A.; Wang, H.; Li, J.; Mulla, S.I.; Qiu, Q.; Tang, L.; Rashid, A.; Wu, Y.; Sun, Q.; Yu, C.P. Homogeneous selection drives antibiotic resistome in two adjacent sub-watersheds, China. J. Hazard Mater. 2020, 398, 122820. [Google Scholar] [CrossRef]
- Pal, C.; Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G. The structure and diversity of human, animal and environmental resistomes. Microbiome 2016, 4, 54. [Google Scholar] [CrossRef] [Green Version]
- Hendriksen, R.S.; Munk, P.; Njage, P.; van Bunnik, B.; McNally, L.; Lukjancenko, O.; Roder, T.; Nieuwenhuijse, D.; Pedersen, S.K.; Kjeldgaard, J.; et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Commun. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. Environmental factors influencing the development and spread of antibiotic resistance. FEMS Microbiol. Rev. 2018, 42, fux053. [Google Scholar] [CrossRef]
- Zhu, Y.G.; Gillings, M.; Simonet, P.; Stekel, D.; Banwart, S.; Penuelas, J. Human dissemination of genes and microorganisms in Earth’s Critical Zone. Glob. Chang. Biol. 2018, 24, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Karkman, A.; Parnanen, K.; Larsson, D.G.J. Fecal pollution can explain antibiotic resistance gene abundances in anthropogenically impacted environments. Nat. Commun. 2019, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Wang, H.; Chen, Q.; Su, J.Q.; Gad, M.; Li, J.; Mulla, S.I.; Yu, C.P.; Hu, A. Fecal pollution mediates the dominance of stochastic assembly of antibiotic resistome in an urban lagoon (Yundang lagoon), China. J. Hazard. Mater. 2021, 417, 126083. [Google Scholar] [CrossRef] [PubMed]
- Su, J.Q.; An, X.L.; Li, B.; Chen, Q.L.; Gillings, M.R.; Chen, H.; Zhang, T.; Zhu, Y.G. Metagenomics of urban sewage identifies an extensively shared antibiotic resistome in China. Microbiome 2017, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Li, L.G.; Yin, X.L.; Zhang, T. Tracking antibiotic resistance gene pollution from different sources using machine-learning classification. Microbiome 2018, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.J.; Patel, S.; Gibson, M.K.; Lauber, C.L.; Knight, R.; Fierer, N.; Dantas, G. Bacterial phylogeny structures soil resistomes across habitats. Nature 2014, 509, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Li, P.; Huang, Y.; Yu, K.; Chen, H.; Cui, K.; Huang, Q.; Zhang, J.; Yew-Hoong Gin, K.; He, Y. Environmental media exert a bottleneck in driving the dynamics of antibiotic resistance genes in modern aquatic environment. Water Res. 2019, 162, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hou, L.; Liu, Y.; Liu, K.; Zhang, L.; Huang, F.; Wang, L.; Rashid, A.; Hu, A.; Yu, C.P. Horizontal and vertical gene transfer drive sediment antibiotic resistome in an urban lagoon system. J. Environ. Sci. 2021, 102, 11–23. [Google Scholar] [CrossRef]
- Huang, J.; Zhu, J.; Liu, S.; Luo, Y.; Zhao, R.; Guo, F.; Li, B. Estuarine salinity gradient governs sedimentary bacterial community but not antibiotic resistance gene profile. Sci. Total Environ. 2022, 806, 151390. [Google Scholar] [CrossRef]
- Li, X.; Rensing, C.; Vestergaard, G.; Arumugam, M.; Nesme, J.; Gupta, S.; Brejnrod, A.D.; Sorensen, S.J. Metagenomic evidence for co-occurrence of antibiotic, biocide and metal resistance genes in pigs. Environ. Int. 2022, 158, 106899. [Google Scholar] [CrossRef]
- Popi, K.; Sotirios, V.; Stella, M.G.; Dimitrios, K.G.; Despo, F.K. Shotgun metagenomics assessment of the resistome, mobilome, pathogen dynamics and their ecological control modes in full-scale urban wastewater treatment plants. J. Hazard Mater. 2021, 418, 126387. [Google Scholar] [CrossRef]
- Lee, J.; Beck, K.; Burgmann, H. Wastewater bypass is a major temporary point-source of antibiotic resistance genes and multi-resistance risk factors in a Swiss river. Water Res. 2022, 208, 117827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, Z.; Chen, C.-T.A.; Tang, K.; Su, J.; Jiao, N. Sulfur Metabolizing Microbes Dominate Microbial Communities in Andesite-Hosted Shallow-Sea Hydrothermal Systems. PLoS ONE 2012, 7, e44593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop Ristova, P.; Pichler, T.; Friedrich, M.W.; Buhring, S.I. Bacterial Diversity and Biogeochemistry of Two Marine Shallow-Water Hydrothermal Systems off Dominica (Lesser Antilles). Front. Microbiol. 2017, 8, 2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasov, V.G.; Gebruk, A.V.; Mironov, A.N.; Moskalev, L.I. Deep-sea and shallow-water hydrothermal vent communities: Two different phenomena? Chem. Geol. 2005, 224, 5–39. [Google Scholar] [CrossRef]
- Tang, K.; Liu, K.; Jiao, N.; Zhang, Y.; Chen, C.T. Functional metagenomic investigations of microbial communities in a shallow-sea hydrothermal system. PLoS ONE 2013, 8, e72958. [Google Scholar] [CrossRef] [Green Version]
- Rubelmann, H.; Karlen, D.J.; Garey, J.R. Changes in Eukaryotic and Bacterial Communities along a 120 m Transect Associated with a Shallow Marine Hydrothermal Vent. Front. Mar. Sci. 2017, 4, 177. [Google Scholar] [CrossRef]
- Bellec, L.; Cambon-Bonavita, M.A.; Durand, L.; Aube, J.; Gayet, N.; Sandulli, R.; Brandily, C.; Zeppilli, D. Microbial Communities of the Shallow-Water Hydrothermal Vent Near Naples, Italy, and Chemosynthetic Symbionts Associated With a Free-Living Marine Nematode. Front. Microbiol. 2020, 11, 2023. [Google Scholar] [CrossRef]
- Li, Y.; Tang, K.; Zhang, L.; Zhao, Z.; Xie, X.; Chen, C.A.; Wang, D.; Jiao, N.; Zhang, Y. Coupled Carbon, Sulfur, and Nitrogen Cycles Mediated by Microorganisms in the Water Column of a Shallow-Water Hydrothermal Ecosystem. Front. Microbiol. 2018, 9, 2718. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.; Zhang, Y.; Lin, D.; Han, Y.; Chen, C.A.; Wang, D.; Lin, Y.S.; Sun, J.; Zheng, Q.; Jiao, N. Cultivation-Independent and Cultivation-Dependent Analysis of Microbes in the Shallow-Sea Hydrothermal System Off Kueishantao Island, Taiwan: Unmasking Heterotrophic Bacterial Diversity and Functional Capacity. Front. Microbiol. 2018, 9, 279. [Google Scholar] [CrossRef]
- Lin, Y.S.; Lin, H.T.; Wang, B.S.; Huang, W.J.; Lin, L.H.; Tsai, A.Y. Intense but variable autotrophic activity in a rapidly flushed shallow-water hydrothermal plume (Kueishantao Islet, Taiwan). Geobiol. 2021, 19, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Elsaied, H.; Stokes, H.W.; Nakamura, T.; Kitamura, K.; Fuse, H.; Maruyama, A. Novel and diverse integron integrase genes and integron-like gene cassettes are prevalent in deep-sea hydrothermal vents. Environ. Microbiol. 2007, 9, 2298–2312. [Google Scholar] [CrossRef] [PubMed]
- Bravakos, P.; Mandalakis, M.; Nomikou, P.; Anastasiou, T.I.; Kristoffersen, J.B.; Stavroulaki, M.; Kilias, S.; Kotoulas, G.; Magoulas, A.; Polymenakou, P.N. Genomic adaptation of Pseudomonas strains to acidity and antibiotics in hydrothermal vents at Kolumbo submarine volcano, Greece. Sci. Rep. 2021, 11, 1336. [Google Scholar] [CrossRef] [PubMed]
- Farias, P.; Espirito Santo, C.; Branco, R.; Francisco, R.; Santos, S.; Hansen, L.; Sorensen, S.; Morais, P.V. Natural hot spots for gain of multiple resistances: Arsenic and antibiotic resistances in heterotrophic, aerobic bacteria from marine hydrothermal vent fields. Appl. Environ. Microbiol. 2015, 81, 2534–2543. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Yuan, K.; Chen, X.; Yang, Y.; Zhang, T.; Wang, Y.; Luan, T.; Zou, S.; Li, X. Metagenomic Analysis Revealing Antibiotic Resistance Genes (ARGs) and Their Genetic Compartments in the Tibetan Environment. Environ. Sci. Technol. 2016, 50, 6670–6679. [Google Scholar] [CrossRef]
- McCann, C.M.; Christgen, B.; Roberts, J.A.; Su, J.Q.; Arnold, K.E.; Gray, N.D.; Zhu, Y.G.; Graham, D.W. Understanding drivers of antibiotic resistance genes in High Arctic soil ecosystems. Environ. Int. 2019, 125, 497–504. [Google Scholar] [CrossRef]
- Jardine, J.; Mavumengwana, V.; Ubomba-Jaswa, E. Antibiotic resistance and heavy metal tolerance in cultured bacteria from hot springs as indicators of environmental intrinsic resistance and tolerance levels. Environ. Pollut. 2019, 249, 696–702. [Google Scholar] [CrossRef]
- Najar, I.N.; Sherpa, M.T.; Das, S.; Das, S.; Thakur, N. Diversity analysis and metagenomic insights into antibiotic and metal resistance among Himalayan hot spring bacteriobiome insinuating inherent environmental baseline levels of antibiotic and metal tolerance. J. Global Antimicrob. Resist. 2020, 21, 342–352. [Google Scholar] [CrossRef]
- Hu, A.; Li, S.; Zhang, L.; Wang, H.; Yang, J.; Luo, Z.; Rashid, A.; Chen, S.; Huang, W.; Yu, C.P. Prokaryotic footprints in urban water ecosystems: A case study of urban landscape ponds in a coastal city, China. Environ. Pollut. 2018, 242, 1729–1739. [Google Scholar] [CrossRef]
- Hou, L.Y.; Zhang, L.P.; Li, F.R.; Huang, S.J.; Yang, J.; Ma, C.; Zhang, D.X.; Yu, C.P.; Hu, A.Y. Urban ponds as hotspots of antibiotic resistome in the urban environment. J. Hazard. Mater. 2021, 403, 124008. [Google Scholar] [CrossRef]
- Hu, A.; Ju, F.; Hou, L.; Li, J.; Yang, X.; Wang, H.; Mulla, S.I.; Sun, Q.; Bürgmann, H.; Yu, C.-P. Strong impact of anthropogenic contamination on the co-occurrence patterns of a riverine microbial community. Environ. Microbiol. 2017, 19, 4993–5009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Meth. 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Adyari, B.; Shen, D.; Li, S.; Zhang, L.; Rashid, A.; Sun, Q.; Hu, A.; Chen, N.; Yu, C.-P. Strong impact of micropollutants on prokaryotic communities at the horizontal but not vertical scales in a subtropical reservoir, China. Sci. Total Environ. 2020, 721, 137767. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. R Package Version 1.17-1. Available online: http://CRAN.R-project.org/package=vegan (accessed on 10 March 2010).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Gu, Z.G.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Li, B.; Jiang, X.T.; Wang, Y.L.; Xia, Y.; Li, A.D.; Zhang, T. Catalogue of antibiotic resistome and host-tracking in drinking water deciphered by a large scale survey. Microbiome 2017, 5, 154. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.L. Ecology and Evolution of Chromosomal Gene Transfer between Environmental Microorganisms and Pathogens. Microbiol. Spectr. 2021, 6, 1. [Google Scholar]
- Murdock, S.A.; Tunnicliffe, V.; Boschen-Rose, R.E.; Juniper, S.K. Emergent “core communities” of microbes, meiofauna and macrofauna at hydrothermal vents. ISME J. 2021, 1, 27. [Google Scholar] [CrossRef]
- Lanzen, A.; Jorgensen, S.L.; Bengtsson, M.M.; Jonassen, I.; Ovreas, L.; Urich, T. Exploring the composition and diversity of microbial communities at the Jan Mayen hydrothermal vent field using RNA and DNA. FEMS Microbiol. Ecol. 2011, 77, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Wang, F.; Guo, L.; Chen, Z.; Sievert, S.M.; Meng, J.; Huang, G.; Li, Y.; Yan, Q.; Wu, S.; et al. Comparative metagenomics of microbial communities inhabiting deep-sea hydrothermal vent chimneys with contrasting chemistries. ISME J. 2011, 5, 414–426. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, L.; Hu, Q.; Lyu, J.; Shao, Z. Thiomicrorhabdus indica sp. nov., an obligately chemolithoautotrophic, sulfur-oxidizing bacterium isolated from a deep-sea hydrothermal vent environment. Int. J. Syst. Evol. Microbiol. 2020, 70, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Takai, K. Deep-sea vent chemoautotrophs: Diversity, biochemistry and ecological significance. FEMS Microbiol. Ecol. 2008, 65, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.Y.; Jing, L.J.; Yao, Z.P.; Meng, F.S.; Teng, Y.G. Prevalence, source and risk of antibiotic resistance genes in the sediments of Lake Tai (China) deciphered by metagenomic assembly: A comparison with other global lakes. Environ. Int. 2019, 127, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, W.; Lin, H.; Wang, W.; Du, L.; Xing, W. Antibiotics and antibiotic resistance genes in global lakes: A review and meta-analysis. Environ. Int. 2018, 116, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Amos, G.C.A.; Ploumakis, S.; Zhang, L.; Hawkey, P.M.; Gaze, W.H.; Wellington, E.M.H. The widespread dissemination of integrons throughout bacterial communities in a riverine system. ISME J. 2018, 12, 681–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.W.; Wang, J.T.; Li, J.; Shi, X.Z.; Ma, Y.B.; Chen, D.; He, J.Z. Long-Term Nickel Contamination Increases the Occurrence of Antibiotic Resistance Genes in Agricultural Soils. Environ. Sci. Technol. 2017, 51, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Su, J.Q.; Wei, B.; Ou-Yang, W.Y.; Huang, F.Y.; Zhao, Y.; Xu, H.J.; Zhu, Y.G. Antibiotic resistome and its association with bacterial communities during sewage sludge composting. Environ. Sci. Technol. 2015, 49, 7356–7363. [Google Scholar] [CrossRef]
- An, X.L.; Su, J.Q.; Li, B.; Ouyang, W.Y.; Zhao, Y.; Chen, Q.L.; Cui, L.; Chen, H.; Gillings, M.R.; Zhang, T.; et al. Tracking antibiotic resistome during wastewater treatment using high throughput quantitative PCR. Environ. Int. 2018, 117, 146–153. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Larsson, D.G. Antibiotic resistance genes in the environment: Prioritizing risks. Nat. Rev. Microbiol. 2015, 13, 396. [Google Scholar] [CrossRef]
- Lupo, A.; Coyne, S.; Berendonk, T.U. Origin and evolution of antibiotic resistance: The common mechanisms of emergence and spread in water bodies. Front. Microbiol. 2012, 3, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manaia, C.M.; Rocha, J.; Scaccia, N.; Marano, R.; Radu, E.; Biancullo, F.; Cerqueira, F.; Fortunato, G.; Iakovides, I.C.; Zammit, I.; et al. Antibiotic resistance in wastewater treatment plants: Tackling the black box. Environ. Int. 2018, 115, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, R.; Liu, H.; Wang, C.; Yin, X.; Zhang, M.; Fang, J.; Zhang, T.; Ma, L. Evidence for Long-Term Anthropogenic Pollution: The Hadal Trench as a Depository and Indicator for Dissemination of Antibiotic Resistance Genes. Environ. Sci. Technol. 2021, 55, 15136–115148. [Google Scholar] [CrossRef] [PubMed]
- Stedtfeld, R.D.; Guo, X.; Stedtfeld, T.M.; Sheng, H.; Williams, M.R.; Hauschild, K.; Gunturu, S.; Tift, L.; Wang, F.; Howe, A.; et al. Primer set 2.0 for highly parallel qPCR array targeting antibiotic resistance genes and mobile genetic elements. FEMS Microbiol. Ecol. 2018, 94, 1–8. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhang, L.; Li, Y.; Liu, K.; Liu, Y.; Huang, S.; Li, F.; Chen, C.-T.A.; Zhang, Y.; Hu, A. A Comprehensive Profile of Antibiotic Resistance Genes in the Water Column of a Shallow-Sea Hydrothermal Vent Ecosystem. Sustainability 2022, 14, 1776. https://doi.org/10.3390/su14031776
Li J, Zhang L, Li Y, Liu K, Liu Y, Huang S, Li F, Chen C-TA, Zhang Y, Hu A. A Comprehensive Profile of Antibiotic Resistance Genes in the Water Column of a Shallow-Sea Hydrothermal Vent Ecosystem. Sustainability. 2022; 14(3):1776. https://doi.org/10.3390/su14031776
Chicago/Turabian StyleLi, Jiangwei, Lanping Zhang, Yufang Li, Keshao Liu, Yongqin Liu, Sijun Huang, Furun Li, Chen-Tung A. Chen, Yao Zhang, and Anyi Hu. 2022. "A Comprehensive Profile of Antibiotic Resistance Genes in the Water Column of a Shallow-Sea Hydrothermal Vent Ecosystem" Sustainability 14, no. 3: 1776. https://doi.org/10.3390/su14031776
APA StyleLi, J., Zhang, L., Li, Y., Liu, K., Liu, Y., Huang, S., Li, F., Chen, C.-T. A., Zhang, Y., & Hu, A. (2022). A Comprehensive Profile of Antibiotic Resistance Genes in the Water Column of a Shallow-Sea Hydrothermal Vent Ecosystem. Sustainability, 14(3), 1776. https://doi.org/10.3390/su14031776