
The Composition and Assembly of Soil Microbial Communities Differ across Vegetation Cover Types of Urban Green Spaces

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Location and Sample Collection
2.2. Soil Physicochemical Analysis
2.3. DNA Extraction and Sequencing
2.4. Sequence Processing and Putative Pathogen Identification
2.5. Co-Occurrence Network Construction and Community Stability Analysis
2.6. Neutral Community Model
2.7. Diversity, Correlation, and Multivariate Analyses
3. Results
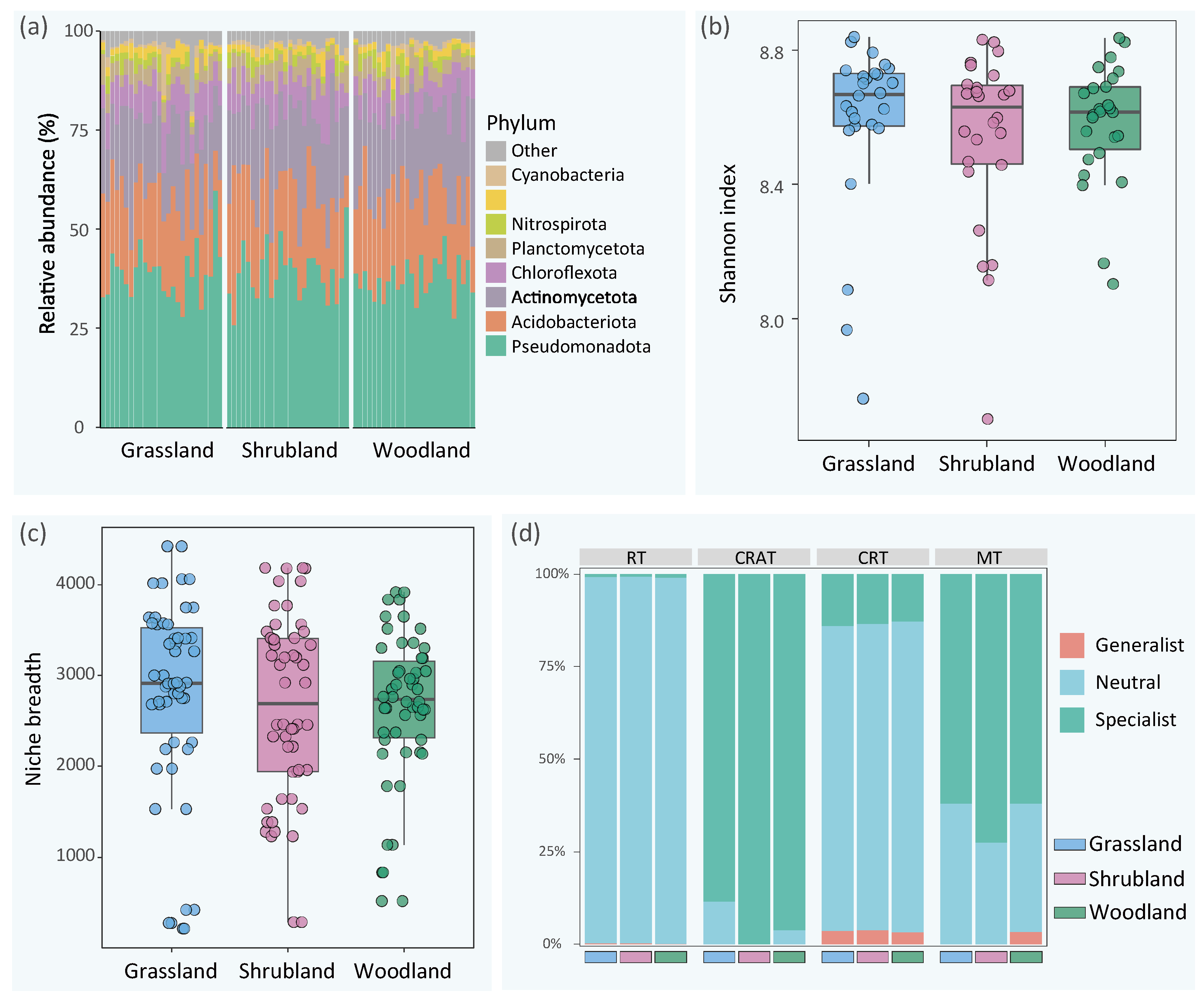
3.1. Composition, Diversity, and Ecological Niche of the Microbial Community in Urban Green Spaces
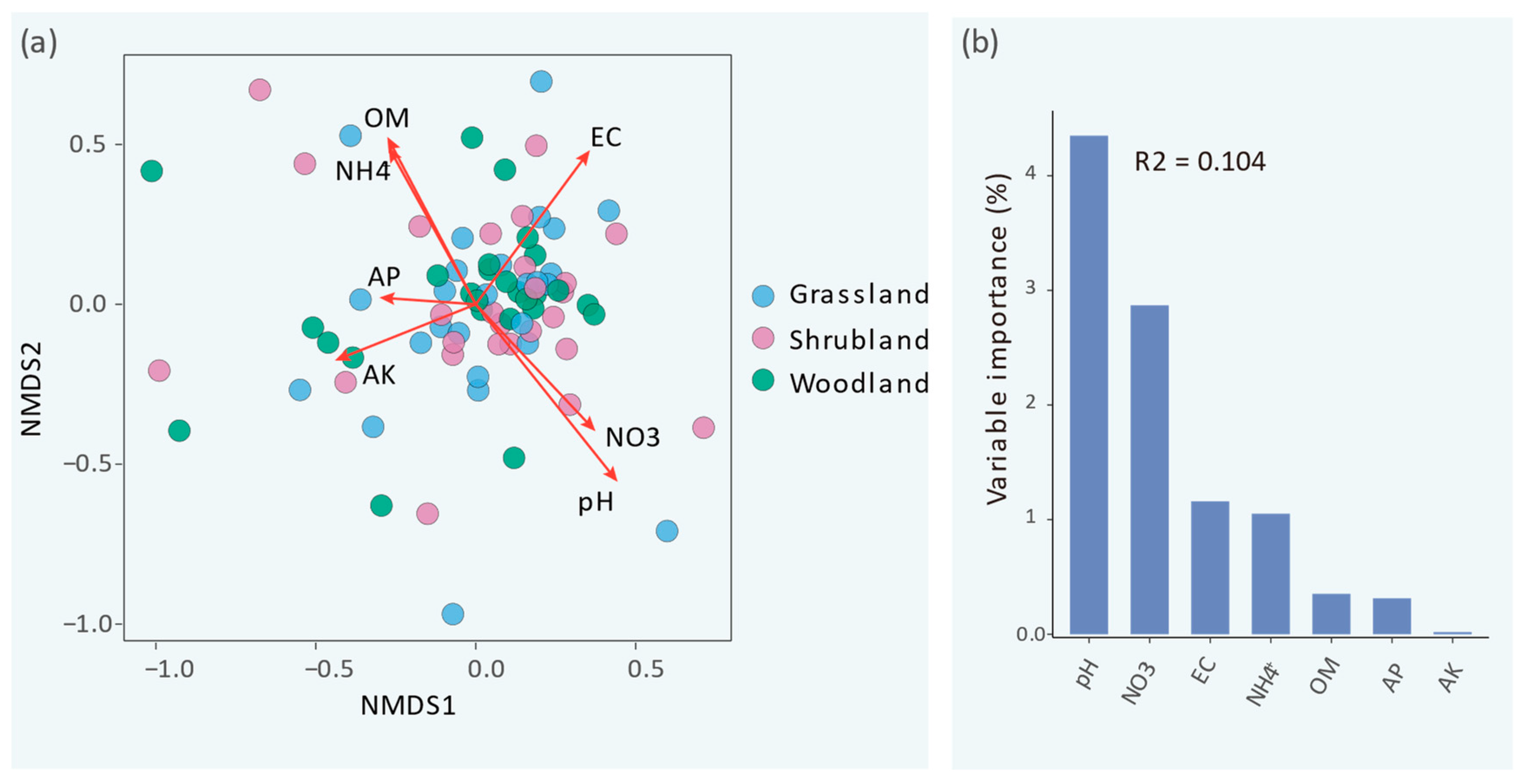
3.2. Factors Related to the Variation in Microbial Communities
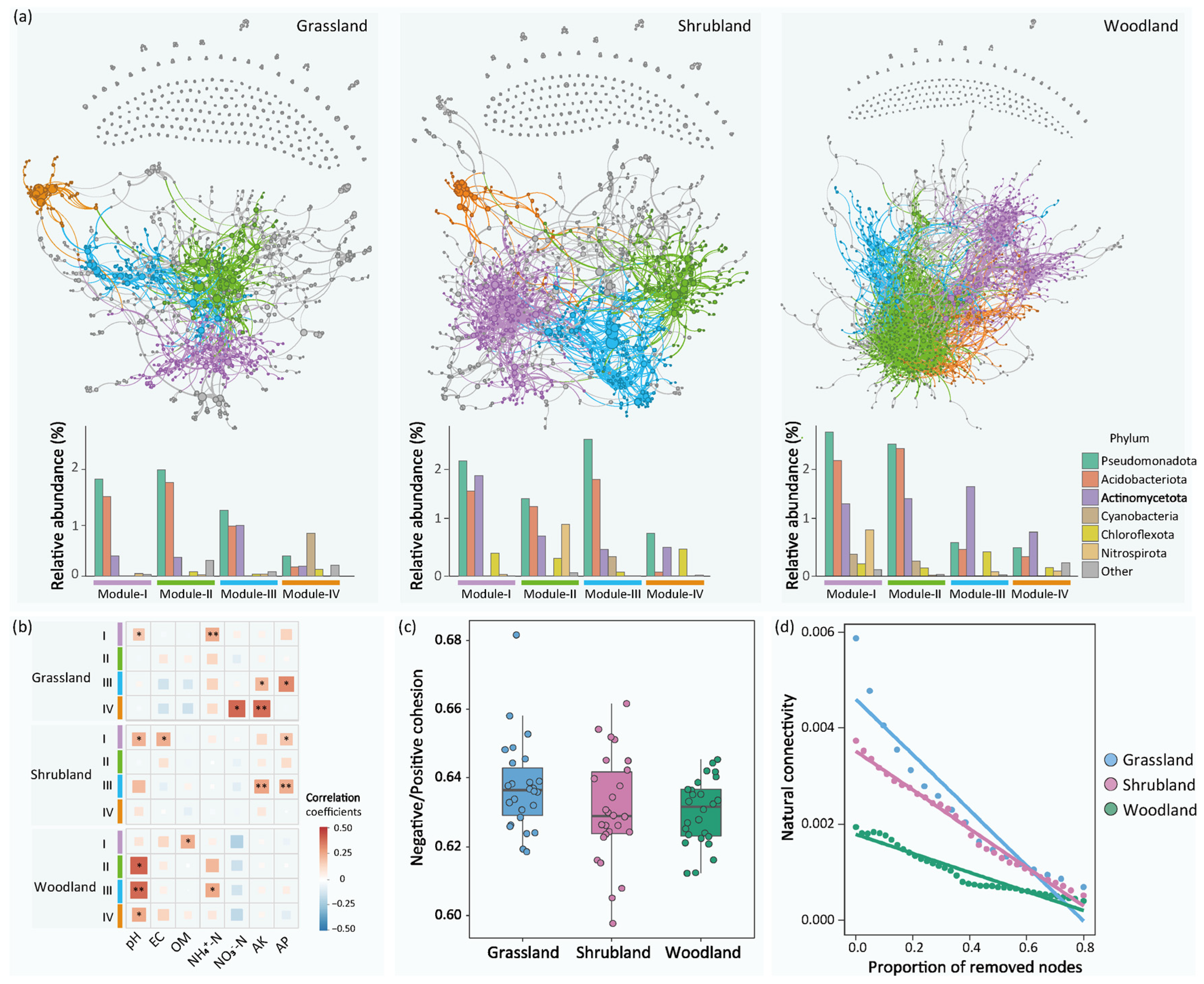
3.3. Microbial Networks and Community Assembly Processes
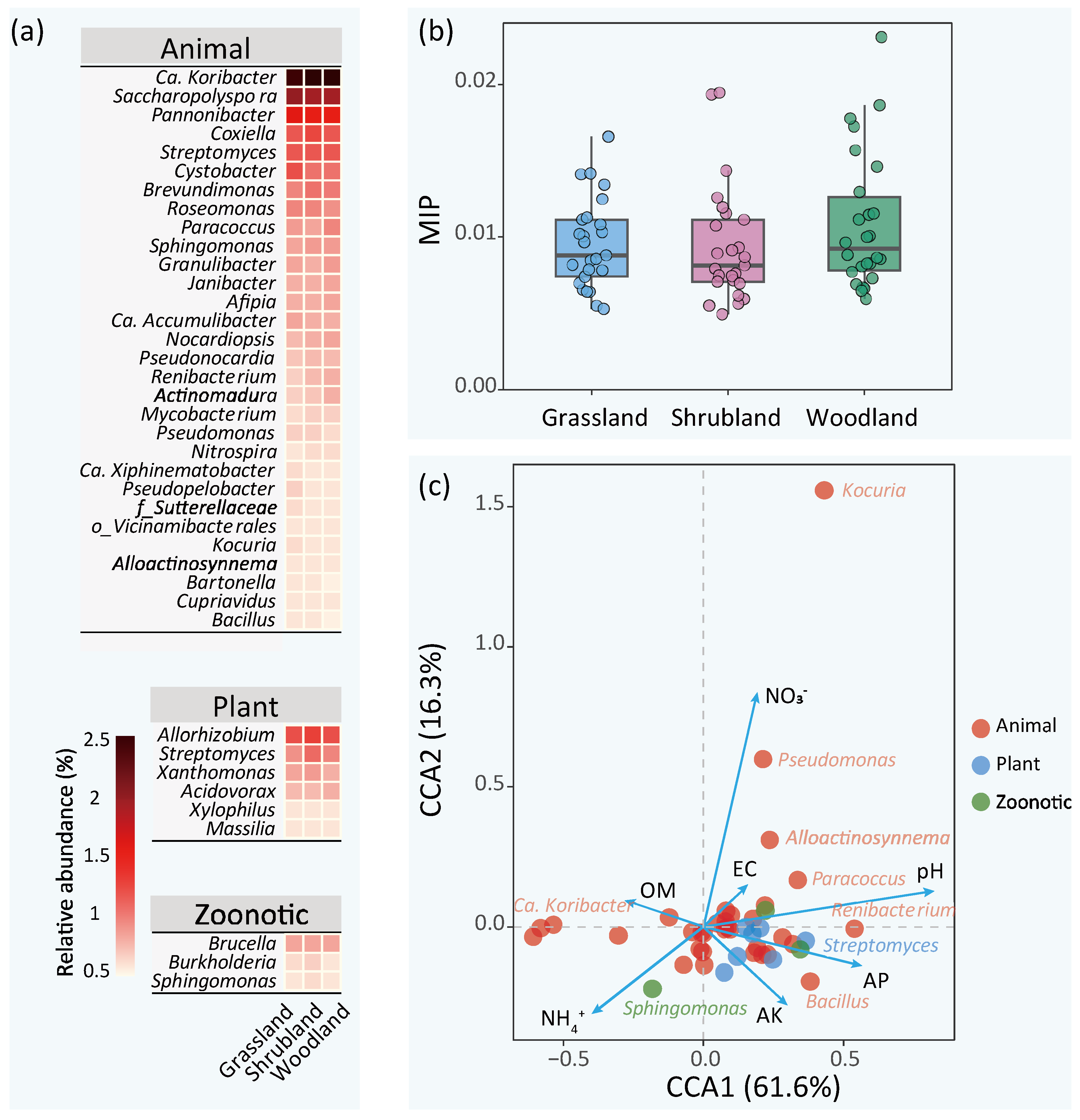
3.4. Putative Pathogens in Urban Green Land
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UN. United Nations Population Division World Urbanization Prospects: The 2018 Re-Vision. 2018. Available online: https://population.un.org/wup/ (accessed on 9 May 2020).
- Jabbar, M.; Yusoff, M.M.; Shafie, A. Assessing the role of urban green spaces for human well-being: A systematic review. GeoJournal 2021, 87, 4405–4423. [Google Scholar] [CrossRef]
- Haase, D.; Haase, A.; Rink, D. Conceptualizing the nexus between urban shrinkage and ecosystem services. Landsc. Urban Plan. 2014, 132, 159–169. [Google Scholar] [CrossRef]
- Gómez-Baggethun, E.; Gren, Å.; Barton, D.N.; Langemeyer, J.; McPhearson, T.; O’Farrell, P.; Andersson, E.; Hamstead, Z.; Kremer, P. Urban Ecosystem Services. In Urbanization, Biodiversity and Ecosystem Services: Challenges and Opportunities: A Global Assessment; Springer Nature: Berlin/Heidelberg, Germany, 2013; pp. 175–251. [Google Scholar]
- Ramirez, K.S.; Leff, J.W.; Barberán, A.; Bates, S.T.; Betley, J.; Crowther, T.W.; Kelly, E.F.; Oldfield, E.E.; Shaw, E.A.; Steenbock, C.; et al. Biogeographic patterns in below-ground diversity in New York City’s Central Park are similar to those observed globally. Proc. R. Soc. B Boil. Sci. 2014, 281, 20141988. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Baquerizo, M.; Eldridge, D.J.; Liu, Y.R.; Sokoya, B.; Wang, J.T.; Hu, H.W.; He, J.Z.; Bastida, F.; Moreno, J.L.; Bamigboye, A.R.; et al. Global homogenization of the structure and function in the soil microbiome of urban greenspaces. Sci. Adv. 2021, 7, eabg5809. [Google Scholar] [CrossRef]
- Cockell, C.S.; Jones, H.L. Advancing the case for microbial conservation. Oryx 2009, 43, 520–526. [Google Scholar] [CrossRef]
- Rousk, J.; Bengtson, P. Microbial regulation of global biogeochemical cycles. Front. Microbiol. 2014, 5, 103. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Li, J.; Xiao, N.; Qi, Y.; Fu, G.; Liu, G.; Qiao, M. Urban-development-induced Changes in the Diversity and Composition of the Soil Bacterial Community in Beijing. Sci. Rep. 2016, 6, 38811. [Google Scholar] [CrossRef]
- Tan, X.; Kan, L.; Su, Z.; Liu, X.; Zhang, L. The Composition and Diversity of Soil Bacterial and Fungal Communities Along an Urban-To-Rural Gradient in South China. Forests 2019, 10, 797. [Google Scholar] [CrossRef]
- Pereira, M.C.; O’riordan, R.; Stevens, C. Urban soil microbial community and microbial-related carbon storage are severely limited by sealing. J. Soils Sediments 2021, 21, 1455–1465. [Google Scholar] [CrossRef]
- Rai, P.K.; Rai, A.; Singh, S. Change in soil microbial biomass along a rural-urban gradient in Varanasi (U.P., India). Geol. Ecol. Landscapes 2018, 2, 15–21. [Google Scholar] [CrossRef]
- Zhu, R.; Liu, J.; Wang, J.; Han, W.; Shen, Z.; Muraina, T.O.; Chen, J.; Sun, D. Comparison of soil microbial community between reseeding grassland and natural grassland in Songnen Meadow. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.G.; Selway, C.A.; Weyrich, L.S.; Skelly, C.; Weinstein, P.; Thomas, T.; Young, J.M.; Marczylo, E.; Yadav, S.; Yadav, V.; et al. Rare genera differentiate urban green space soil bacterial communities in three cities across the world. Access Microbiol. 2022, 4, 000320. [Google Scholar] [CrossRef]
- Steinauer, K.; Tilman, D.; Wragg, P.D.; Cesarz, S.; Cowles, J.M.; Pritsch, K.; Reich, P.B.; Weisser, W.W.; Eisenhauer, N. Plant diversity effects on soil microbial functions and enzymes are stronger than warming in a grassland experiment. Ecology 2015, 96, 99–112. [Google Scholar] [CrossRef]
- Zhao, C.; Long, J.; Liao, H.K.; Zheng, C.L.; Li, J.; Liu, L.F.; Zhang, M.J. Dynamics of soil microbial communities following vegetation succession in a karst mountain ecosystem, Southwest China. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yang, X.; Jiang, L.; Guo, L.; Chen, Y.; Yang, S.; Liu, L. Partitioning of beta-diversity reveals distinct assembly mechanisms of plant and soil microbial communities in response to nitrogen enrichment. Ecol. Evol. 2022, 12, e9016. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef]
- Hubbell, S.P. Neutral theory in community ecology and the hypothesis of functional equivalence. Funct. Ecol. 2005, 19, 166–172. [Google Scholar] [CrossRef]
- Hirt, H. Healthy soils for healthy plants for healthy humans: How beneficial microbes in the soil, food and gut are interconnected and how agriculture can contribute to human health. EMBO Rep. 2020, 21, e51069. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; van der Heijden, M.G.A. Soil microbiomes and one health. Nat. Rev. Microbiol. 2023, 21, 6–20. [Google Scholar] [CrossRef]
- Silver, W.L.; Perez, T.; Mayer, A.; Jones, A.R. The role of soil in the contribution of food and feed. Philos. Trans. R. Soc. B 2021, 376, 20200181. [Google Scholar] [CrossRef] [PubMed]
- Baumgardner, D.J. Soil-Related Bacterial and Fungal Infections. J. Am. Board Fam. Med. 2012, 25, 734–744. [Google Scholar] [CrossRef]
- Khan, N.A. Acanthamoeba: Biology and increasing importance in human health. FEMS Microbiol. Rev. 2006, 30, 564–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, Z.-F.; Yang, X.-R.; An, X.-L.; Ren, Y.; Su, J.-Q. Urban greenness and plant species are key factors in shaping air microbiomes and reducing airborne pathogens. Environ. Int. 2021, 153, 106539. [Google Scholar] [CrossRef] [PubMed]
- Certini, G.; Forte, C.; D’acqui, L.P.; Santi, C.A. Spectroscopic properties of bulk and dichromate oxidation resistant soil organic matter from an anthroposequence in a Mediterranean environment. Plant Soil 2007, 291, 55–65. [Google Scholar] [CrossRef]
- Narihiro, T.; Nobu, M.K.; Kim, N.-K.; Kamagata, Y.; Liu, W.-T. The nexus of syntrophy-associated microbiota in anaerobic digestion revealed by long-term enrichment and community survey. Environ. Microbiol. 2014, 17, 1707–1720. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 15 March 2023).
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Dai, T.; Zhang, Y.; Tang, Y.; Bai, Y.; Tao, Y.; Huang, B.; Wen, D. Identifying the key taxonomic categories that characterize microbial community diversity using full-scale classification: A case study of microbial communities in the sediments of Hangzhou Bay. FEMS Microbiol. Ecol. 2016, 92, fiw150. [Google Scholar] [CrossRef]
- Yang, X.; Jiang, G.; Zhang, Y.; Wang, N.; Zhang, Y.; Wang, X.; Zhao, F.; Xu, Y.; Shen, Q.; Wei, Z. MBPD: A multiple bacterial pathogen detection pipeline for One Health practices. iMeta 2023, 2, e82. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, X.; Jing, G.; Chen, Y.; Jiang, S.; Zhang, M.; Liu, J.; Xu, J.; Su, X. Comprehensive understanding to the public health risk of environmental microbes via a microbiome-based index. J. Genet. Genom. 2022, 49, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Alm, E.J. Inferring Correlation Networks from Genomic Survey Data. PLOS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef]
- Herren, C.M.; McMahon, K.D. Cohesion: A method for quantifying the connectivity of microbial communities. ISME J. 2017, 11, 2426–2438. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.J.; David, A.S.; Menges, E.S.; Searcy, C.A.; Afkhami, M.E. Environmental stress destabilizes microbial networks. ISME J. 2021, 15, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-H.; Chen, S.-Y.; Chen, J.-W.; Xue, K.; Chen, S.-L.; Wang, X.-M.; Chen, T.; Kang, S.-C.; Rui, J.-P.; Thies, J.E.; et al. Reduced microbial stability in the active layer is associated with carbon loss under alpine permafrost degradation. Proc. Natl. Acad. Sci. USA 2021, 118, e2025321118. [Google Scholar] [CrossRef] [PubMed]
- Albert, R.; Jeong, H.; Barabasi, A. Error and attack tolerance of complex networks. Nature 2000, 406, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.-S.; Wu, J. Optimal network topology for structural robustness based on natural connectivity. Phys. A Stat. Mech. Its Appl. 2016, 443, 212–220. [Google Scholar] [CrossRef]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef]
- Burns, A.R.; Stephens, W.Z.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 2015, 10, 655–664. [Google Scholar] [CrossRef]
- Harrell, F.E.; Harrell, M., Jr. Package ‘hmisc’. CRAN 2018, 2019, 235–236. Available online: https://hbiostat.org/r/hmisc/ (accessed on 26 July 2023).
- Blanchet, F.G.; Legendre, P.; Borcard, D. Forward selection of explanatory variables. Ecology 2008, 89, 2623–2632. [Google Scholar] [CrossRef]
- Lichstein, J.W. Multiple regression on distance matrices: A multivariate spatial analysis tool. Plant Ecol. 2006, 188, 117–131. [Google Scholar] [CrossRef]
- Groemping, U. Relative Importance for Linear Regression in R: The Package relaimpo. J. Stat. Softw. 2006, 17, 1–27. [Google Scholar] [CrossRef]
- Kembel, S.W.; Cowan, P.D.; Helmus, M.R.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 2010, 26, 1463–1464. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Microbial community structure and its functional implications. Nature 2009, 459, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Cassman, N.; de Hollander, M.; Mendes, L.W.; Korevaar, H.; Geerts, R.H.; van Veen, J.A.; Kuramae, E.E. Impact of long-term N, P, K, and NPK fertilization on the composition and potential functions of the bacterial community in grassland soil. FEMS Microbiol. Ecol. 2014, 90, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-Based Assessment of Soil pH as a Predictor of Soil Bacterial Community Structure at the Continental Scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M.; Gupta, S.; Delgado-Baquerizo, M.; Morriën, E.; Garkoti, S.C.; Chaturvedi, R.; Ahmad, S. Successional trajectory of bacterial communities in soil are shaped by plant-driven changes during secondary succession. Sci. Rep. 2020, 10, 9864. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Royer, T.V.; Leff, L.G. Diversity of Fungi, Bacteria, and Actinomycetes on Leaves Decomposing in a Stream. Appl. Environ. Microbiol. 2007, 73, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Lewin, G.R.; Carlos, C.; Chevrette, M.G.; Horn, H.A.; McDonald, B.R.; Stankey, R.J.; Fox, B.G.; Currie, C.R. Evolution and Ecology of Actinobacteria and Their Bioenergy Applications. Annu. Rev. Microbiol. 2016, 70, 235–254. [Google Scholar] [CrossRef]
- Buckley, D.H.; Huangyutitham, V.; Nelson, T.A.; Rumberger, A.; Thies, J.E. Diversity of Planctomycetes in soil in relation to soil history and environmental heterogeneity. Appl. Environ. Microbiol. 2006, 72, 4522–4531. [Google Scholar] [CrossRef] [PubMed]
- Barnard, R.L.; Osborne, C.A.; Firestone, M.K. Responses of soil bacterial and fungal communities to extreme desiccation and rewetting. ISME J. 2013, 7, 2229–2241. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.E.L. Determining Microbial Niche Breadth in the Environment for Better Ecosystem Fate Predictions. mSystems 2019, 4, e00080-19. [Google Scholar] [CrossRef]
- Fan, K.; Weisenhorn, P.; Gilbert, J.A.; Shi, Y.; Bai, Y.; Chu, H. Soil pH correlates with the co-occurrence and assemblage process of diazotrophic communities in rhizosphere and bulk soils of wheat fields. Soil Biol. Biochem. 2018, 121, 185–192. [Google Scholar] [CrossRef]
- Chu, H.; Gao, G.-F.; Ma, Y.; Fan, K.; Delgado-Baquerizo, M. Soil Microbial Biogeography in a Changing World: Recent Advances and Future Perspectives. mSystems 2020, 5, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Hu, H.; Ma, W.; Deng, Y.; Wang, Q.; Luo, A.; Meng, J.; Feng, X.; Wang, Z. Relative Importance of Deterministic and Stochastic Processes on Soil Microbial Community Assembly in Temperate Grasslands. Microorganisms 2021, 9, 1929. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Wu, H.; Zhang, Y.; Wu, Q.; Guan, Q.; Lu, K.; Lin, Y. Differential distribution patterns and assembly processes of soil microbial communities under contrasting vegetation types at distinctive altitudes in the Changbai Mountain. Front. Microbiol. 2023, 14, 1152818. [Google Scholar] [CrossRef]
- Zhou, J.; Ning, D. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002–e00017. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Peng, Z.; Qi, J.; Gao, J.; Wei, G. Linking Bacterial-Fungal Relationships to Microbial Diversity and Soil Nutrient Cycling. mSystems 2021, 6, e01052-20. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.-Z.; Chen, Q.-L.; Zhang, Y.-J.; He, J.-Z.; Hu, H.-W. Antibiotic resistance in urban green spaces mirrors the pattern of industrial distribution. Environ. Int. 2019, 132, 105106. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Jiang, T.; Hu, S.; Wang, M.; Ming, D.; Chen, S. Genomic insights of Pannonibacter phragmitetus strain 31801 isolated from a patient with a liver abscess. Microbiologyopen 2017, 6, e00515. [Google Scholar] [CrossRef] [PubMed]
- Kuzmanović, N.; Biondi, E.; Overmann, J.; Puławska, J.; Verbarg, S.; Smalla, K.; Lassalle, F. Genomic analysis provides novel insights into diversification and taxonomy of Allorhizobium vitis (i.e., Agrobacterium vitis). BMC Genom. 2022, 23, 1–17. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Wang, J. The Composition and Assembly of Soil Microbial Communities Differ across Vegetation Cover Types of Urban Green Spaces. Sustainability 2023, 15, 13105. https://doi.org/10.3390/su151713105
Zhou Y, Wang J. The Composition and Assembly of Soil Microbial Communities Differ across Vegetation Cover Types of Urban Green Spaces. Sustainability. 2023; 15(17):13105. https://doi.org/10.3390/su151713105
Chicago/Turabian StyleZhou, Yangyi, and Jiangping Wang. 2023. "The Composition and Assembly of Soil Microbial Communities Differ across Vegetation Cover Types of Urban Green Spaces" Sustainability 15, no. 17: 13105. https://doi.org/10.3390/su151713105
APA StyleZhou, Y., & Wang, J. (2023). The Composition and Assembly of Soil Microbial Communities Differ across Vegetation Cover Types of Urban Green Spaces. Sustainability, 15(17), 13105. https://doi.org/10.3390/su151713105