
Development and Application of EST-SSR Markers Related to Lead Stress Responses in Kenaf Based on Transcriptome Sequencing Data
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and SSR Marker Development
2.2. PCR Reaction System, Conditions, and Data Analysis
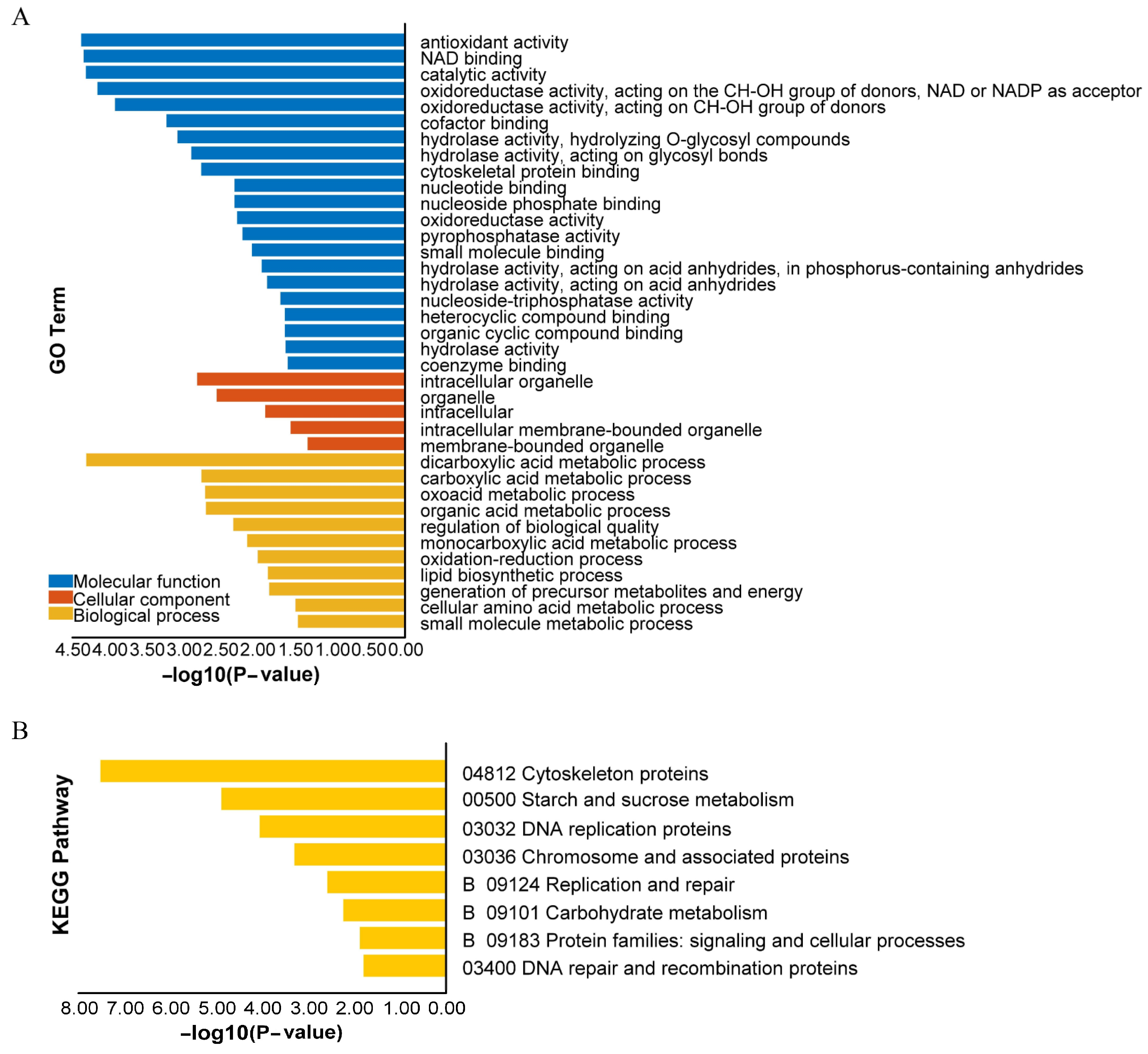
2.3. GO and KEGG Enrichment Analyses
2.4. Analysis of the DEGs in the PPI Network
2.5. Genetic Diversity and Structure Analysis
3. Results and Discussion
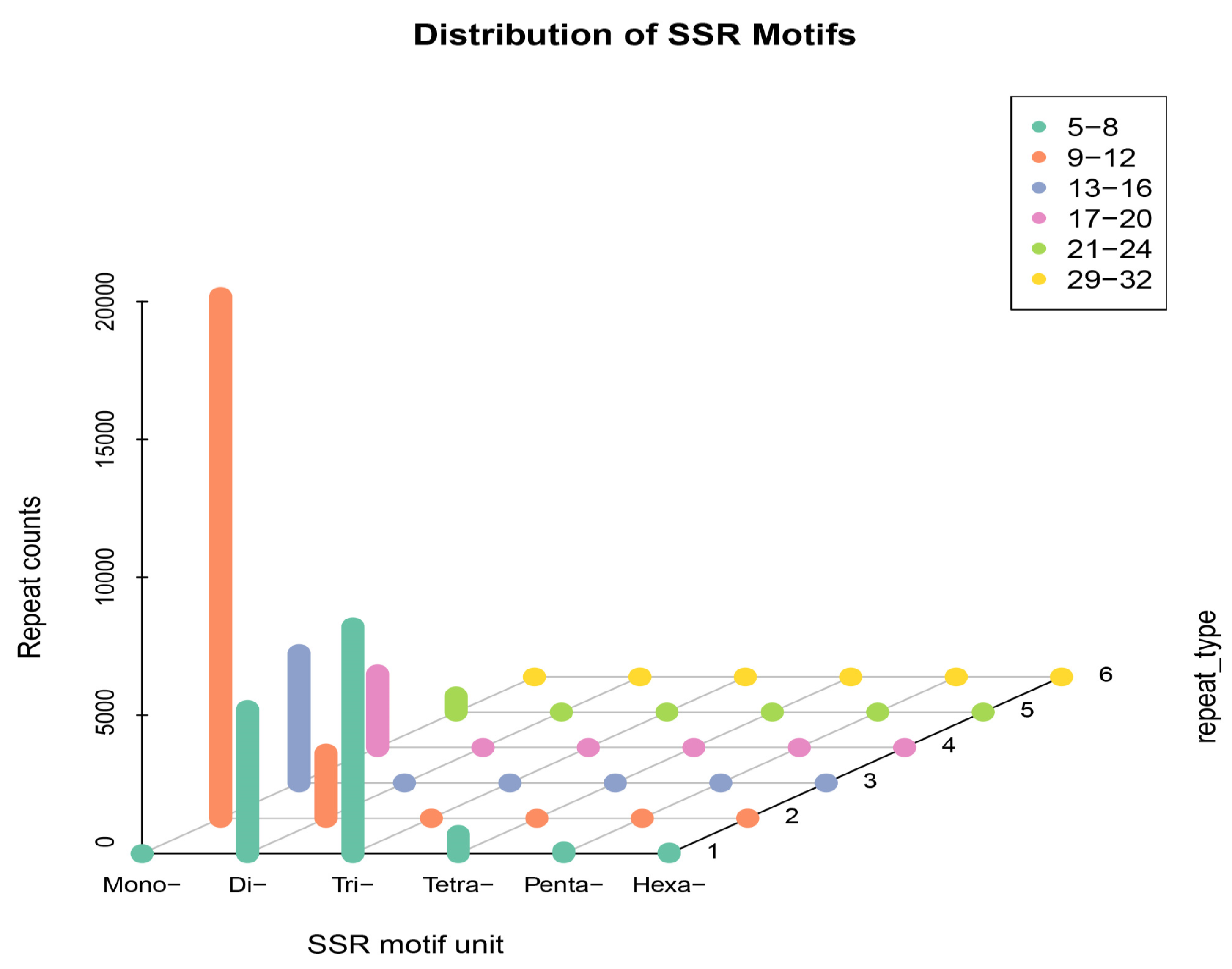
3.1. Analysis of SSR Characteristics
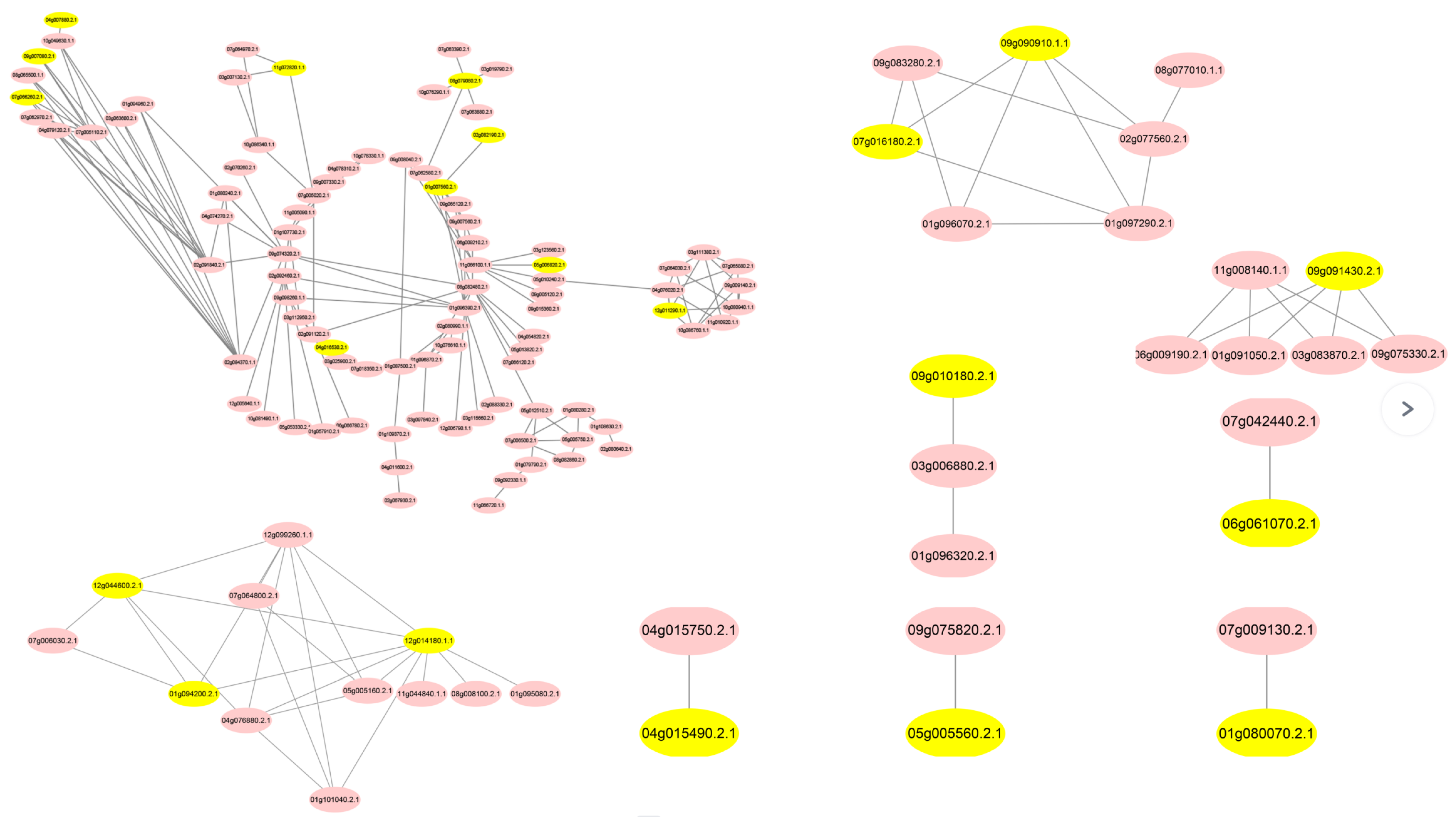
3.2. Analysis of DEGs in the PPI Network
3.3. Validation of EST-SSR Molecular Markers
3.4. Genetic Diversity of a Single Locus
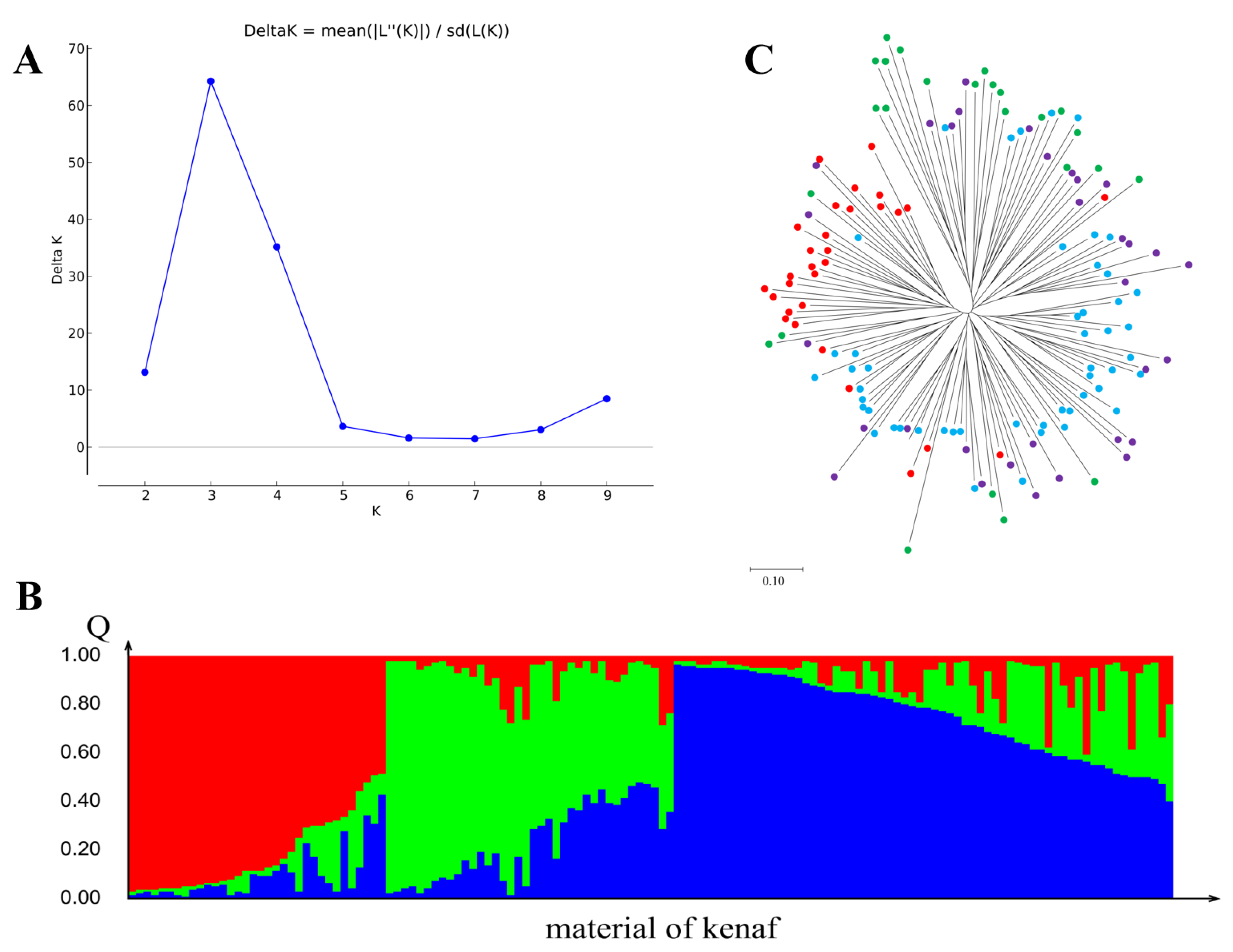
3.5. Genetic Structure Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- An, X.; Jin, G.; Zhang, J.; Luo, X.; Chen, C.; Li, W.; Ma, G.; Jin, L.; Dai, L.; Shi, X.; et al. Protein responses in kenaf plants exposed to drought conditions determined using iTRAQ technology. FEBS Open Bio 2018, 8, 1572–1583. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Chen, T.; Li, Z.; Jia, R.; Luo, D.; Tang, M.; Lu, H.; Hu, Y.; Yue, J.; Huang, Z. Transcriptome Analysis Revealed Key Genes and Pathways Related to Cadmium-Stress Tolerance in Kenaf (Hibiscus cannabinus L.). Ind. Crops Prod. 2020, 158, 112970. [Google Scholar] [CrossRef]
- An, X.; Chen, J.; Jin, G. Transcriptome profiling of kenaf (Hibiscus cannabinus L.) under plumbic stress conditions implies the involvement of NAC transcription factors regulating reactive oxygen species-dependent programmed cell death. PeerJ 2020, 8, e8733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Guo, X.; Zhao, P.; Ruan, M.; Yu, X.; Zou, L.; Yang, Y.; Li, X.; Deng, D.; Xiao, J.; et al. Molecular Diversity Analysis, Drought Related Marker-Traits Association Mapping and Discovery of Excellent Alleles for 100-Day Old Plants by Est-Ssrs in Cassava Germplasms (Manihot esculenta Cranz). PLoS ONE 2017, 12, e0177456. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Lu, B.R.; Baldwin, B.S.; Sameshima, K.; Chen, J.K. Comparative Studies of Genetic Diversity in Kenaf (Hibiscus cannabinus L.) Varieties Based on Analysis of Agronomic and Rapd Data. Hereditas 2002, 136, 231–239. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Y.; Zhang, X.; Ma, X.; Zhang, L.; Liao, Z.; Zhang, Q.; Wan, X.; Cheng, Y.; Zhang, J.; et al. The Genome of Kenaf (Hibiscus cannabinus L.) Provides Insights into Bast Fibre and Leaf Shape Biogenesis. Plant Biotechnol. J. 2020, 18, 1796–1809. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, D.; Chen, A.; Tang, H.; Li, J.; Huang, S. Characterization of the Kenaf (Hibiscus cannabinus) Global Transcriptome Using Illumina Paired-End Sequencing and Development of EST-SSR Markers. PLoS ONE 2016, 11, e0150548. [Google Scholar] [CrossRef]
- Chowdhury, T.; Mandal, A.; Roy, S.C.; De Sarker, D. Diversity of the Genus Ocimum (Lamiaceae) through Morpho-Molecular (Rapd) and Chemical (Gc-Ms) Analysis. J. Genet. Eng. Biotechnol. 2017, 15, 275–286. [Google Scholar] [CrossRef]
- Frascaroli, E.; Schrag, T.A.; Melchinger, A.E. Genetic diversity analysis of elite European maize (Zea mays L.) inbred lines using AFLP, SSR, and SNP markers reveals ascertainment bias for a subset of SNPs. Theor. Appl. Genet. 2013, 126, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Aitken, K.S.; Jackson, P.A.; McIntyre, C.L. A combination of AFLP and SSR markers provides extensive map coverage and identification of homo(eo)logous linkage groups in a sugarcane cultivar. Theor. Appl. Genet. 2005, 110, 789–801. [Google Scholar] [CrossRef]
- Choudhary, S.B.; Sharma, H.K.; Kumar, A.A.; Maruthi, R.T.; Mitra, J.; Chowdhury, I.; Singh, B.K.; Karmakar, P.G. Ssr and Morphological Trait Based Population Structure Analysis of 130 Diverse Flax (Linum usitatissimum L.) Accessions. C. R. Biol. 2017, 340, 65–75. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, Q.; Wu, G.; Zhang, S.; Jiang, T. Development of Novel Ssr Markers for Flax (Linum usitatissimum L.) Using Reduced-Representation Genome Sequencing. Front. Plant Sci. 2016, 7, 2018. [Google Scholar] [CrossRef] [Green Version]
- Luan, M.B.; Zou, Z.Z.; Zhu, J.J.; Wang, X.F.; Xu, Y.; Ma, Q.H.; Sun, Z.M.; Chen, J.H. Development of a core collection for ramie by heuristic search based on SSR markers. Biotechnol. Biotechnol. Equip. 2014, 28, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.Y.; Zhang, C.; Wang, P.; Hu, S.; Chang, H.P.; Xiao, W.J.; Lu, X.T.; Jiang, S.B.; Ye, J.Z.; Guo, X.H. Genetic diversity analysis of okra (Abelmoschus esculentus L.) by inter-simple sequence repeat (ISSR) markers. Genet. Mol. Res. 2014, 13, 3165–3175. [Google Scholar] [CrossRef]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, X.; Luo, X.; Liu, T.; Li, W.; Zou, L. Development and Application of Fruit Color-Related Expressed Sequence Tag-Simple Sequence Repeat Markers in Abelmoschus esculentus on the Basis of Transcriptome Sequencing. Front. Plant Sci. 2022, 13, 907895. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. Tbtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.C.; Rohlf, F.J. Ecological character displacement in Plethodon: Biomechanical differences found from a geometric morphometric study. Proc. Natl. Acad. Sci. USA 2000, 97, 4106–4111. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Zhou, P.Y.; Zou, W.T.; Li, Y.G. Est-Ssr Marker Development Based on Transcriptome Sequencing and Genetic Analyses of Phoebe Bournei (Lauraceae). Mol. Biol. Rep. 2021, 48, 2201–2208. [Google Scholar] [CrossRef]
- Wei, Z.; Sun, Z.; Cui, B.; Zhang, Q.; Xiong, M.; Wang, X.; Zhou, D. Transcriptome Analysis of Colored Calla Lily (Zantedeschia rehmannii Engl.) by Illumina Sequencing: De Novo Assembly, Annotation and Est-Ssr Marker Development. PeerJ. 2016, 4, e2378. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, X.; Wei, J.; Li, Y.; Tigabu, M.; Zhao, X. Development and Transferability of Est-Ssr Markers for Pinus koraiensis from Cold-Stressed Transcriptome through Illumina Sequencing. Genes 2020, 11, 500. [Google Scholar] [CrossRef]
- Yang, Z.J.; Peng, Z.S.; Yang, H. Identification of Novel and Useful Est-Ssr Markers from De Novo Transcriptome Sequence of Wheat (Triticum aestivum L.). Genet. Mol. Res. 2016, 15, 15017509. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Cheng, Y.S.; Yu, L.; Xu, Y.Y.; Wang, Y. Bioinformatics Analysis of Differentially Expressed Genes and Protein-Protein Interaction Networks Associated with Functional Pathways in Ulcerative Colitis. Med. Sci. Monit. 2021, 27, e927917. [Google Scholar] [CrossRef]
- Liu, C.; Yuan, D.; Lin, Z. Construction of an EST-SSR-based interspecific transcriptome linkage map of fibre development in cotton. J. Genet. 2014, 93, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Gailing, O.; Bodénès, C.; Finkeldey, R.; Kremer, A.; Plomion, C. Genetic Mapping of Est-Derived Simple Sequence Repeats (Est-Ssrs) to Identify Qtl for Leaf Morphological Characters in a Quercus Robur Full-Sib Family. Tree Genet. Genomes 2013, 9, 1361–1367. [Google Scholar] [CrossRef]
- Xiang, C.; Duan, Y.; Li, H.; Ma, W.; Huang, S.; Sui, X.; Zhang, Z.; Wang, C. A High-Density Est-Ssr-Based Genetic Map and Qtl Analysis of Dwarf Trait in Cucurbita pepo L. Int. J. Mol. Sci. 2018, 19, 3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, D.; Taylor, P.W.J.; Inder, P.; Phan, H.T.T.; Ellwood, S.R.; Mathur, P.N.; Sarker, A.; Ford, R. Integration of Est-Ssr Markers of Medicago Truncatula into Intraspecific Linkage Map of Lentil and Identification of Qtl Conferring Resistance to Ascochyta Blight at Seedling and Pod Stages. Mol. Breed. 2011, 30, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Hadi, F.; Bano, A.; Fuller, M.P. Augmented Phytoextraction of Lead (Pb2+)-Polluted Soils: A Comparative Study of the Effectiveness of Plant Growth Regulators, EDTA, and Plant Growth–Promoting Rhizobacteria. Bioremediat. J. 2013, 17, 124–130. [Google Scholar] [CrossRef]
- Ji, P.; Tang, X.; Jiang, Y.; Tong, Y.; Gao, P.; Han, W. Potential of Gibberellic Acid 3 (Ga3) for Enhancing the Phytoremediation Efficiency of Solanum nigrum L. Bull. Environ. Contam. Toxicol. 2015, 95, 810–814. [Google Scholar] [CrossRef]
- Hadi, F.; Bano, A.; Fuller, M.P. The improved phytoextraction of lead (Pb) and the growth of maize (Zea mays L.): The role of plant growth regulators (GA3 and IAA) and EDTA alone and in combinations. Chemosphere 2010, 80, 457–462. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Content | Number |
|---|---|
| Total number of sequences examined | 136,854 |
| Total size of examined sequences (bp) | 151,754,502 |
| Total number of identified SSRs | 43,457 |
| Number of SSR-containing sequences | 34,180 |
| Number of sequences containing more than 1 SSR | 7421 |
| Number of SSRs present in compound formation | 2321 |
| Marker | Major.Allele.Frquency | Na | Ne | Heterozygosity | Exp_Het | PIC | I |
|---|---|---|---|---|---|---|---|
| KSSR01 | 0.38 | 17 | 5.08 | 0.57 | 0.81 | 0.79 | 2.05 |
| KSSR03 | 0.66 | 15 | 2.19 | 0.20 | 0.55 | 0.52 | 1.35 |
| KSSR05 | 0.60 | 6 | 2.45 | 0.30 | 0.60 | 0.55 | 1.19 |
| KSSR11 | 0.38 | 14 | 3.67 | 0.49 | 0.73 | 0.68 | 1.61 |
| KSSR12 | 0.33 | 15 | 5.24 | 0.50 | 0.81 | 0.79 | 2.00 |
| KSSR15 | 0.41 | 28 | 4.83 | 0.37 | 0.80 | 0.78 | 2.22 |
| KSSR16 | 0.40 | 20 | 3.42 | 0.20 | 0.71 | 0.66 | 1.64 |
| KSSR17 | 0.53 | 15 | 3.04 | 0.08 | 0.67 | 0.64 | 1.58 |
| KSSR18 | 0.74 | 9 | 1.77 | 0.21 | 0.44 | 0.42 | 1.02 |
| KSSR22 | 0.39 | 21 | 4.57 | 0.50 | 0.78 | 0.76 | 1.98 |
| KSSR24 | 0.46 | 13 | 2.88 | 0.46 | 0.65 | 0.59 | 1.40 |
| KSSR25 | 0.68 | 6 | 1.85 | 0.07 | 0.46 | 0.39 | 0.81 |
| KSSR26 | 0.83 | 16 | 1.43 | 0.16 | 0.30 | 0.30 | 0.85 |
| KSSR28 | 0.47 | 18 | 3.71 | 0.33 | 0.73 | 0.71 | 1.86 |
| KSSR31 | 0.25 | 18 | 5.88 | 0.41 | 0.83 | 0.81 | 2.08 |
| KSSR37 | 0.45 | 19 | 3.93 | 0.41 | 0.75 | 0.72 | 1.88 |
| KSSR38 | 0.49 | 17 | 3.45 | 0.16 | 0.71 | 0.69 | 1.80 |
| KSSR40 | 0.64 | 18 | 2.31 | 0.10 | 0.57 | 0.54 | 1.42 |
| KSSR41 | 0.78 | 14 | 1.61 | 0.09 | 0.38 | 0.37 | 0.99 |
| KSSR43 | 0.26 | 8 | 5.31 | 0.00 | 0.81 | 0.79 | 1.79 |
| KSSR44 | 0.46 | 17 | 3.22 | 0.14 | 0.69 | 0.65 | 1.59 |
| KSSR49 | 0.39 | 20 | 3.40 | 0.16 | 0.71 | 0.66 | 1.72 |
| Mean | 0.50 | 15.64 | 3.42 | 0.27 | 0.66 | 0.63 | 1.58 |
| SSR ID | Gene ID | SSR Motif | Forward Primer (5′–3′) | Tm (°C) | Size (bp) | Reverse Primer (5′–3′) | Tm (°C) | Size (bp) | Product Size (bp) |
|---|---|---|---|---|---|---|---|---|---|
| KSSR01 | Cluster-5711.2645 | (A)10 | GTCGACCGGGTCTACGAATC | 59.971 | 20 | TCAACTACGGTAGGGCAGGA | 59.96 | 20 | 113–133 |
| KSSR03 | Cluster-5711.55898 | (GCA)7 | CCTCCGTTCTTACCACCACC | 60.037 | 20 | TCCCAAAGGAATGCCCAACA | 59.81 | 20 | 144–161 |
| KSSR05 | Cluster-5711.54607 | (A)10 | AGAAAAGGCCATTCCTGATCCA | 59.687 | 22 | ATTGGTGGACTGCAGTACGG | 60.036 | 20 | 158–163 |
| KSSR11 | Cluster-5711.64891 | (A)10 | TGTAACCCACGGTGGCTTTT | 60.107 | 20 | CACATTACAAATCTAAACCTTCCCTGA | 59.607 | 27 | 174–187 |
| KSSR12 | Cluster-5711.75272 | (T)10 | CGTCGAATCAGTCATGCTGC | 59.7 | 20 | TGCTCATTGCTCAATAGATCAAGA | 58.322 | 24 | 171–189 |
| KSSR15 | Cluster-5711.57149 | (TCT)5 | CTCTTCCAACGCAGCCAAAC | 60.04 | 20 | AATGGGTTTTCCGACACCGA | 59.891 | 20 | 174–228 |
| KSSR16 | Cluster-5711.67236 | (A)11 | ACGGCCGACTTTCAGAATGT | 59.965 | 20 | TCCAGGCTCCAGCTATCTCA | 59.737 | 20 | 193–219 |
| KSSR17 | Cluster-5711.53839 | (ACC)5 | AGGCTTTCGTTGCTCACCAT | 60.251 | 20 | TTTGGAGGCACGGGAGATTC | 60.035 | 20 | 198–214 |
| KSSR18 | Cluster-5711.66135 | (A)11 | TTCCGGTGCAGATAGGGAGA | 60.033 | 20 | ACAAGGAAACAGAGGCAGCA | 59.817 | 20 | 201–209 |
| KSSR22 | Cluster-5711.46957 | (TGG)5 | CCACCGTATTCTCATCGGCA | 59.896 | 20 | CTCCAATCCATCGGAGCCTC | 59.965 | 20 | 188–222 |
| KSSR24 | Cluster-5711.73527 | (CCA)5 | CACATCCCATTTGACCCCCA | 59.959 | 20 | TCTGTGCCAATGGAAGAGCA | 59.599 | 20 | 207–222 |
| KSSR25 | Cluster-5711.63058 | (A)10 | CCCGTCTCAAATTCTCAGCCA | 60.339 | 21 | CGAAATGCCAGCGTTGTTCA | 60.041 | 20 | 213–224 |
| KSSR26 | Cluster-5711.44096 | (CAGGCA)6 | ACCTCTCTCTGTGTTTCCGC | 59.681 | 20 | CCAGTCACCAGCACGTACTT | 59.966 | 20 | 209–234 |
| KSSR28 | Cluster-5711.58049 | (A)14 | CATTGCCTGCCAGACCAAAC | 60.038 | 20 | GGTGTCCACATGGTATTCGGT | 60.066 | 21 | 216–235 |
| KSSR31 | Cluster-5711.40164 | (A)11 | TGAGGCCCTCTGAGGCTAAT | 60.031 | 20 | CGACGACTCTAACAAGCGGT | 60.11 | 20 | 203–245 |
| KSSR37 | Cluster-5711.78093 | (T)12 | GCAGACTGCAGAAGACCCTT | 59.964 | 20 | TCTTTCTCACCACAGTTGACA | 57.095 | 21 | 230–249 |
| KSSR38 | Cluster-5711.5810 | (CT)6 | AAACGATGTCAGGCGTCAGT | 59.966 | 20 | CACTGTGTGGCGTGTTTCAG | 59.972 | 20 | 229–250 |
| KSSR40 | Cluster-3733.0 | (C)11 | TGTGCTCTGTACTGGCAAGT | 59.241 | 20 | GCATGAACTCAACTCCCCGA | 60.036 | 20 | 242–262 |
| KSSR41 | Cluster-5711.54732 | (A)14 | GCAGGGGTGGGGTTGTTATT | 60.252 | 20 | ACAGGAGAGTGTGGGGAGAA | 59.809 | 20 | 246–261 |
| KSSR43 | Cluster-5711.50385 | (A)10 | TGTTGGCAGCCTATGAAGCA | 59.962 | 20 | CGGTTTGGGGGAGCAAAGTA | 60.251 | 20 | 120–127 |
| KSSR44 | Cluster-5711.47108 | (T)10 | CAACCAGTACTCTACCGGCC | 59.825 | 20 | GAACAGTTGGGAGGACTGCA | 59.891 | 20 | 247–267 |
| KSSR49 | Cluster-5711.50393 | (A)10 | TGTTGGCAGCCTATGAAGCA | 59.962 | 20 | CGGTTTGGGGGAGCAAAGTA | 60.251 | 20 | 252–276 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, X.; Luo, X.; Li, W.; Liu, T.; Zou, L. Development and Application of EST-SSR Markers Related to Lead Stress Responses in Kenaf Based on Transcriptome Sequencing Data. Sustainability 2023, 15, 1514. https://doi.org/10.3390/su15021514
An X, Luo X, Li W, Liu T, Zou L. Development and Application of EST-SSR Markers Related to Lead Stress Responses in Kenaf Based on Transcriptome Sequencing Data. Sustainability. 2023; 15(2):1514. https://doi.org/10.3390/su15021514
Chicago/Turabian StyleAn, Xia, Xiahong Luo, Wenlue Li, Tingting Liu, and Lina Zou. 2023. "Development and Application of EST-SSR Markers Related to Lead Stress Responses in Kenaf Based on Transcriptome Sequencing Data" Sustainability 15, no. 2: 1514. https://doi.org/10.3390/su15021514