Diet-Modulated Lipoprotein Metabolism and Vascular Inflammation Evaluated by 18F-fluorodeoxyglucose Positron Emission Tomography


{kind=link}
Abstract
:1. Introduction
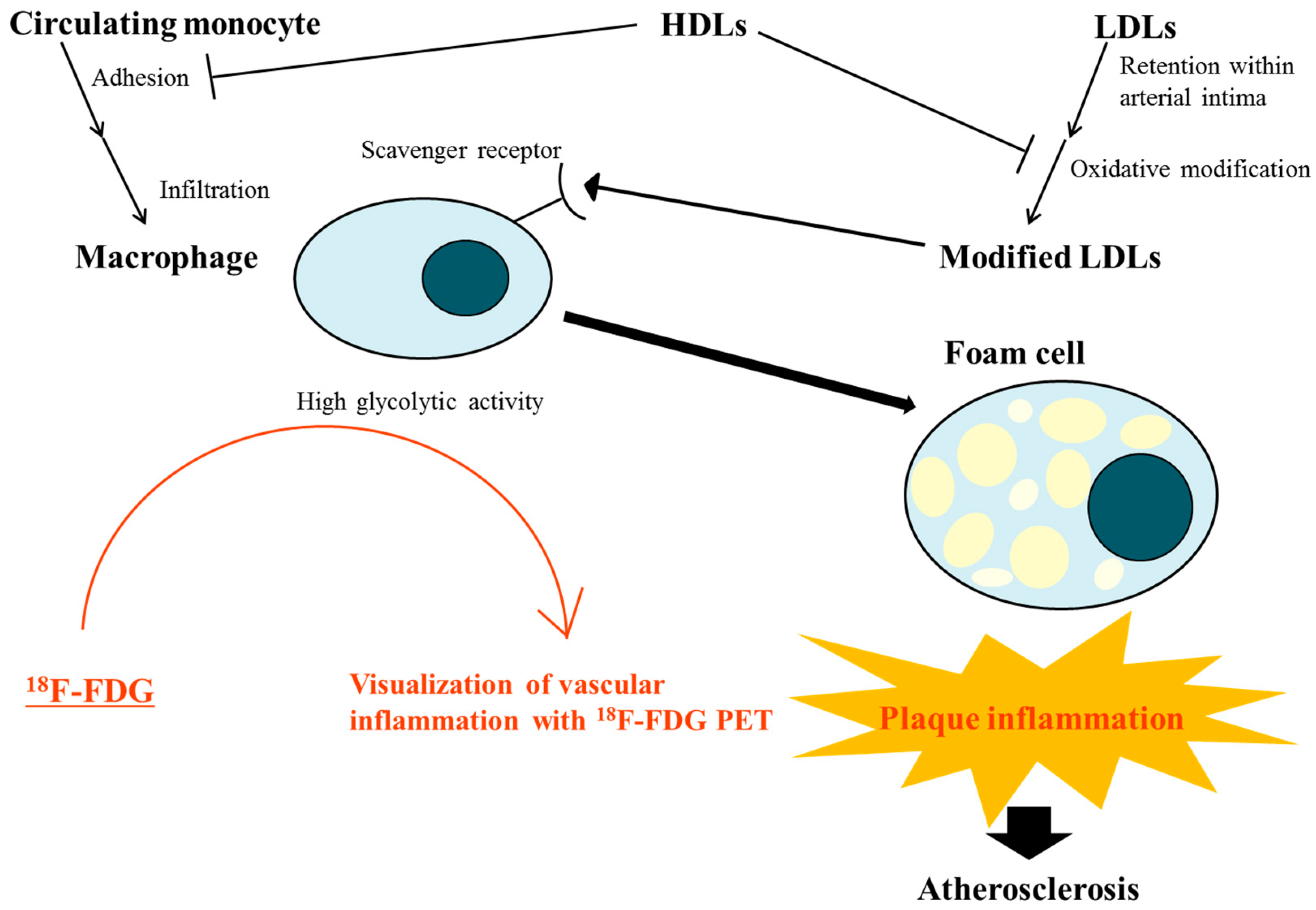
2. Atherosclerosis, an Inflammatory Disease
3. Effects of Lipoproteins on Vascular Inflammation
4. Impact of Diets on Inflammation and Lipoprotein Oxidation Modulating Atherosclerotic Process
5. Utility of 18F-fluorodeoxyglucose (FDG) Positron Emission Tomography (PET) for Identifying Vascular Inflammation
6. Association of Vascular Inflammation Assessed by Positron Emission Tomography (PET) with Markers of Lipoprotein Metabolism and other Risk Factors for Atherosclerotic Cardiovascular Disease (ASCVD)
7. Improvement in Vascular Inflammation Assessed by Positron Emission Tomography (PET) by the Medical Manage and Lifestyle Intervention Including Diet Control
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics--2015 update: A report from the american heart association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Rached, F.; Kontush, A.; Chapman, M.J. Impact of lipoproteins on atherobiology: Emerging insights. Cardiol. Clin. 2018, 36, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Mata, P.; Alonso, R.; Lopez-Farre, A.; Ordovas, J.M.; Lahoz, C.; Garces, C.; Caramelo, C.; Codoceo, R.; Blazquez, E.; de Oya, M. Effect of dietary fat saturation on ldl oxidation and monocyte adhesion to human endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Giugliano, D. Diet and inflammation: A link to metabolic and cardiovascular diseases. Eur. Heart J. 2006, 27, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Guo, L.; Zhang, L.; Li, Y.; He, R.; Cheng, G. Inflammatory potential of diet and risk of cardiovascular disease or mortality: A meta-analysis. Sci. Rep. 2017, 7, 6367. [Google Scholar] [CrossRef] [PubMed]
- Van Lennep, J.E.; Westerveld, H.T.; van Lennep, H.W.; Zwinderman, A.H.; Erkelens, D.W.; van der Wall, E.E. Apolipoprotein concentrations during treatment and recurrent coronary artery disease events. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2408–2413. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. High-sensitivity C-reactive protein: Potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 2001, 103, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Bui, A.V.; Diesch, J.; Manasseh, R.; Hausding, C.; Rivera, J.; Haviv, I.; Agrotis, A.; Htun, N.M.; Jowett, J.; et al. A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microrna expression profiling. Circ. Res. 2013, 113, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.K.; Sukhova, G.K.; Herrington, D.M.; Libby, P. Pravastatin has cholesterol-lowering independent effects on the artery wall of atherosclerotic monkeys. J. Am. Coll. Cardiol. 1998, 31, 684–691. [Google Scholar] [CrossRef]
- Fukumoto, Y.; Libby, P.; Rabkin, E.; Hill, C.C.; Enomoto, M.; Hirouchi, Y.; Shiomi, M.; Aikawa, M. Statins alter smooth muscle cell accumulation and collagen content in established atheroma of watanabe heritable hyperlipidemic rabbits. Circulation 2001, 103, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhao, S.; Zhou, H.; Ye, H.; Li, J. Atorvastatin lowers plasma matrix metalloproteinase-9 in patients with acute coronary syndrome. Clin. Chem. 2004, 50, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Bellosta, S.; Via, D.; Canavesi, M.; Pfister, P.; Fumagalli, R.; Paoletti, R.; Bernini, F. Hmg-coa reductase inhibitors reduce mmp-9 secretion by macrophages. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Bustos, C.; Hernandez-Presa, M.A.; Ortego, M.; Tunon, J.; Ortega, L.; Perez, F.; Diaz, C.; Hernandez, G.; Egido, J. HMG-CoA reductase inhibition by atorvastatin reduces neointimal inflammation in a rabbit model of atherosclerosis. J. Am. Coll. Cardiol. 1998, 32, 2057–2064. [Google Scholar] [CrossRef] [Green Version]
- Niwa, S.; Totsuka, T.; Hayashi, S. Inhibitory effect of fluvastatin, an HMG-CoA reductase inhibitor, on the expression of adhesion molecules on human monocyte cell line. Int. J. Immunopharmacol. 1996, 18, 669–675. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Pfeffer, M.A.; Sacks, F.; Braunwald, E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The cholesterol and recurrent events (care) investigators. Circulation 1999, 100, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.A.; Danielson, E.; Rifai, N.; Ridker, P.M. Effect of statin therapy on c-reactive protein levels: The pravastatin inflammation/CRP evaluation (PRINCE): A randomized trial and cohort study. JAMA 2001, 286, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Jialal, I.; Stein, D.; Balis, D.; Grundy, S.M.; Adams-Huet, B.; Devaraj, S. Effect of hydroxymethyl glutaryl coenzyme a reductase inhibitor therapy on high sensitive C-reactive protein levels. Circulation 2001, 103, 1933–1935. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rifai, N.; Clearfield, M.; Downs, J.R.; Weis, S.E.; Miles, J.S.; Gotto, A.M., Jr. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N. Eng. J. Med. 2001, 344, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated c-reactive protein. N. Eng. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Rifai, N.; Pfeffer, M.A.; Sacks, F.M.; Moye, L.A.; Goldman, S.; Flaker, G.C.; Braunwald, E. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events (CARE) investigators. Circulation 1998, 98, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Williams, K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Eng. J. Med. 1989, 320, 915–924. [Google Scholar]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Witztum, J.L. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2311–2316. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.J.; Sposito, A.C. Hypertension and dyslipidaemia in obesity and insulin resistance: Pathophysiology, impact on atherosclerotic disease and pharmacotherapy. Pharmacol. Ther. 2008, 117, 354–373. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Van Linthout, S.; Spillmann, F.; Lorenz, M.; Meloni, M.; Jacobs, F.; Egorova, M.; Stangl, V.; De Geest, B.; Schultheiss, H.P.; Tschope, C. Vascular-protective effects of high-density lipoprotein include the downregulation of the angiotensin II type 1 receptor. Hypertension 2009, 53, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.A.; Bobik, A.; Murphy, A.; Kanellakis, P.; Blombery, P.; Mukhamedova, N.; Woollard, K.; Lyon, S.; Sviridov, D.; Dart, A.M. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ. Res. 2008, 103, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Van Leuven, S.I.; Birjmohun, R.S.; Franssen, R.; Bisoendial, R.J.; de Kort, H.; Levels, J.H.; Basser, R.L.; Meijers, J.C.; Kuivenhoven, J.A.; Kastelein, J.J.; et al. ApoAI-phosphatidylcholine infusion neutralizes the atherothrombotic effects of C-reactive protein in humans. J. Thromb. Haemost. 2009, 7, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockerill, G.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1987–1994. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, L.; Franceschini, G.; Sirtori, C.R.; De Palma, A.; Saresella, M.; Ferrante, P.; Taramelli, D. Inhibition of VCAM-1 expression in endothelial cells by reconstituted high density lipoproteins. Biochem. Biophys. Res. Commun. 1997, 238, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. Ability of reconstituted high density lipoproteins to inhibit cytokine-induced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. J. Lipid Res. 1999, 40, 345–353. [Google Scholar] [PubMed]
- Bursill, C.A.; Castro, M.L.; Beattie, D.T.; Nakhla, S.; van der Vorst, E.; Heather, A.K.; Barter, P.J.; Rye, K.A. High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1773–1778. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.G.; Duffy, S.J.; Formosa, M.F.; Natoli, A.K.; Henstridge, D.C.; Penfold, S.A.; Thomas, W.G.; Mukhamedova, N.; de Courten, B.; Forbes, J.M.; et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 2009, 119, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Dousset, N.; Ferretti, G.; Bacchetti, T.; Curatola, G.; Salvayre, R. Antioxidant and cytoprotective properties of high-density lipoproteins in vascular cells. Free Radic. Biol. Med. 2006, 41, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Speidel, M.T.; Booyse, F.M.; Abrams, A.; Moore, M.A.; Chung, B.H. Lipolyzed hypertriglyceridemic serum and triglyceride-rich lipoprotein cause lipid accumulation in and are cytotoxic to cultured human endothelial cells. High density lipoproteins inhibit this cytotoxicity. Thromb. Res. 1990, 58, 251–264. [Google Scholar] [CrossRef]
- Hamilton, K.K.; Zhao, J.; Sims, P.J. Interaction between apolipoproteins A-Iand A-II and the membrane attack complex of complement. Affinity of the apoproteins for polymeric C9. J. Biol. Chem. 1993, 268, 3632–3638. [Google Scholar] [PubMed]
- Suc, I.; Escargueil-Blanc, I.; Troly, M.; Salvayre, R.; Negre-Salvayre, A. HDL and ApoA prevent cell death of endothelial cells induced by oxidized HDL. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2158–2166. [Google Scholar] [CrossRef] [PubMed]
- Robbesyn, F.; Garcia, V.; Auge, N.; Vieira, O.; Frisach, M.F.; Salvayre, R.; Negre-Salvayre, A. HDL counterbalance the proinflammatory effect of oxidized HDL by inhibiting intracellular reactive oxygen species rise, proteasome activation, and subsequent NF-kappaB activation in smooth muscle cells. FASEB J. 2003, 17, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Pagler, T.A.; Seimon, T.A.; Thorp, E.; Welch, C.L.; Witztum, J.L.; Tabas, I.; Tall, A.R. ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ. Res. 2010, 106, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Ansell, B.J.; Navab, M.; Hama, S.; Kamranpour, N.; Fonarow, G.; Hough, G.; Rahmani, S.; Mottahedeh, R.; Dave, R.; Reddy, S.T.; et al. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation 2003, 108, 2751–2756. [Google Scholar] [CrossRef] [PubMed]
- Nappo, F.; Esposito, K.; Cioffi, M.; Giugliano, G.; Molinari, A.M.; Paolisso, G.; Marfella, R.; Giugliano, D. Postprandial endothelial activation in healthy subjects and in type 2 diabetic patients: Role of fat and carbohydrate meals. J. Am. Coll. Cardiol. 2002, 39, 1145–1150. [Google Scholar] [CrossRef] [Green Version]
- Bowen, P.E.; Borthakur, G. Postprandial lipid oxidation and cardiovascular disease risk. Curr. Atheroscler. Rep. 2004, 6, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Nappo, F.; Giugliano, F.; Di Palo, C.; Ciotola, M.; Barbieri, M.; Paolisso, G.; Giugliano, D. Meal modulation of circulating interleukin 18 and adiponectin concentrations in healthy subjects and in patients with type 2 diabetes mellitus. Am. J. Clin. Nutr. 2003, 78, 1135–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.B.; Willett, W.C. Optimal diets for prevention of coronary heart disease. JAMA 2002, 288, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, E.; Schulze, M.B.; Meigs, J.B.; Manson, J.E.; Rifai, N.; Stampfer, M.J.; Willett, W.C.; Hu, F.B. Consumption of trans fatty acids is related to plasma biomarkers of inflammation and endothelial dysfunction. J. Nutr. 2005, 135, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Pirro, M.; Schillaci, G.; Savarese, G.; Gemelli, F.; Mannarino, M.R.; Siepi, D.; Bagaglia, F.; Mannarino, E. Attenuation of inflammation with short-term dietary intervention is associated with a reduction of arterial stiffness in subjects with hypercholesterolaemia. Eur. J. Cardiovasc. Prev. Rehabil. 2004, 11, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, E.; Schulze, M.B.; Manson, J.E.; Meigs, J.B.; Albert, C.M.; Rifai, N.; Willett, W.C.; Hu, F.B. Consumption of (n-3) fatty acids is related to plasma biomarkers of inflammation and endothelial activation in women. J. Nutr. 2004, 134, 1806–1811. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Marfella, R.; Ciotola, M.; Di Palo, C.; Giugliano, F.; Giugliano, G.; D’Armiento, M.; D’Andrea, F.; Giugliano, D. Effect of a mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: A randomized trial. JAMA 2004, 292, 1440–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhamme, P.; Quarck, R.; Hao, H.; Knaapen, M.; Dymarkowski, S.; Bernar, H.; Van Cleemput, J.; Janssens, S.; Vermylen, J.; Gabbiani, G.; et al. Dietary cholesterol withdrawal reduces vascular inflammation and induces coronary plaque stabilization in miniature pigs. Cardiovasc. Res. 2002, 56, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Hartung, D.; Sarai, M.; Petrov, A.; Kolodgie, F.; Narula, N.; Verjans, J.; Virmani, R.; Reutelingsperger, C.; Hofstra, L.; Narula, J. Resolution of apoptosis in atherosclerotic plaque by dietary modification and statin therapy. J. Nucl. Med. 2005, 46, 2051–2056. [Google Scholar] [PubMed]
- Casas, R.; Sacanella, E.; Urpi-Sarda, M.; Chiva-Blanch, G.; Ros, E.; Martinez-Gonzalez, M.A.; Covas, M.I.; Salas-Salvado, J.; Fiol, M.; Aros, F.; et al. The effects of the mediterranean diet on biomarkers of vascular wall inflammation and plaque vulnerability in subjects with high risk for cardiovascular disease. A randomized trial. PLoS ONE 2014, 9, e100084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas, R.; Sacanella, E.; Urpi-Sarda, M.; Corella, D.; Castaner, O.; Lamuela-Raventos, R.M.; Salas-Salvado, J.; Martinez-Gonzalez, M.A.; Ros, E.; Estruch, R. Long-term immunomodulatory effects of a mediterranean diet in adults at high risk of cardiovascular disease in the prevencion con Dieta mediterranea (PREDIMED) randomized controlled trial. J. Nutr. 2016, 146, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Ben-Haim, S.; Kupzov, E.; Tamir, A.; Israel, O. Evaluation of 18F-FDG uptake and arterial wall calcifications using 18F-FDG PET/CT. J. Nucl. Med. 2004, 45, 1816–1821. [Google Scholar] [PubMed]
- Lee, S.J.; On, Y.K.; Lee, E.J.; Choi, J.Y.; Kim, B.T.; Lee, K.H. Reversal of vascular 18F-FDG uptake with plasma high-density lipoprotein elevation by atherogenic risk reduction. J. Nucl. Med. 2008, 49, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Delbeke, D. Oncological applications of FDG pet imaging. J. Nucl. Med. 1999, 40, 1706–1715. [Google Scholar] [PubMed]
- Tatsumi, M.; Cohade, C.; Nakamoto, Y.; Wahl, R.L. Fluorodeoxyglucose uptake in the aortic wall at PET/CT: Possible finding for active atherosclerosis. Radiology 2003, 229, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, M.P.; Freiman, A.; Larson, S.M.; Strauss, H.W. Association of vascular 18F-FDG uptake with vascular calcification. J. Nucl. Med. 2005, 46, 1278–1284. [Google Scholar] [PubMed]
- Tahara, N.; Kai, H.; Ishibashi, M.; Nakaura, H.; Kaida, H.; Baba, K.; Hayabuchi, N.; Imaizumi, T. Simvastatin attenuates plaque inflammation: Evaluation by fluorodeoxyglucose positron emission tomography. J. Am. Coll. Cardiol. 2006, 48, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.; Jang, S.; Cucchiara, A.; Newberg, A.B.; Alavi, A. 18F FDG uptake in the large arteries: A correlation study with the atherogenic risk factors. Semin. Nucl. Med. 2002, 32, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Kubota, K.; Kubota, R.; Ido, T.; Tamahashi, N. High accumulation of fluorine-18-fluorodeoxyglucose in turpentine-induced inflammatory tissue. J. Nucl. Med. 1995, 36, 1301–1306. [Google Scholar] [PubMed]
- Rudd, J.H.; Warburton, E.A.; Fryer, T.D.; Jones, H.A.; Clark, J.C.; Antoun, N.; Johnstrom, P.; Davenport, A.P.; Kirkpatrick, P.J.; Arch, B.N.; et al. Imaging atherosclerotic plaque inflammation with [18F]-fluorodeoxyglucose positron emission tomography. Circulation 2002, 105, 2708–2711. [Google Scholar] [CrossRef] [PubMed]
- Rogers, I.S.; Nasir, K.; Figueroa, A.L.; Cury, R.C.; Hoffmann, U.; Vermylen, D.A.; Brady, T.J.; Tawakol, A. Feasibility of FDG imaging of the coronary arteries: Comparison between acute coronary syndrome and stable angina. JACC Cardiovasc. Imaging 2010, 3, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Ishino, S.; Mukai, T.; Asano, D.; Teramoto, N.; Watabe, H.; Kudomi, N.; Shiomi, M.; Magata, Y.; Iida, H.; et al. (18)F-FDG accumulation in atherosclerotic plaques: Immunohistochemical and pet imaging study. J. Nucl. Med. 2004, 45, 1245–1250. [Google Scholar] [PubMed]
- Tawakol, A.; Migrino, R.Q.; Bashian, G.G.; Bedri, S.; Vermylen, D.; Cury, R.C.; Yates, D.; LaMuraglia, G.M.; Furie, K.; Houser, S.; et al. In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a noninvasive measure of carotid plaque inflammation in patients. J. Am. Coll. Cardiol. 2006, 48, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Tahara, N.; Kai, H.; Nakaura, H.; Mizoguchi, M.; Ishibashi, M.; Kaida, H.; Baba, K.; Hayabuchi, N.; Imaizumi, T. The prevalence of inflammation in carotid atherosclerosis: Analysis with fluorodeoxyglucose-positron emission tomography. Eur. Heart J. 2007, 28, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Rudd, J.H.; Myers, K.S.; Bansilal, S.; Machac, J.; Rafique, A.; Farkouh, M.; Fuster, V.; Fayad, Z.A. (18)fluorodeoxyglucose positron emission tomography imaging of atherosclerotic plaque inflammation is highly reproducible: Implications for atherosclerosis therapy trials. J. Am. Coll. Cardiol. 2007, 50, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Rudd, J.H.; Myers, K.S.; Bansilal, S.; Machac, J.; Pinto, C.A.; Tong, C.; Rafique, A.; Hargeaves, R.; Farkouh, M.; Fuster, V.; et al. Atherosclerosis inflammation imaging with 18F-FDG pet: Carotid, iliac, and femoral uptake reproducibility, quantification methods, and recommendations. J. Nucl. Med. 2008, 49, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Tahara, N.; Kai, H.; Yamagishi, S.; Mizoguchi, M.; Nakaura, H.; Ishibashi, M.; Kaida, H.; Baba, K.; Hayabuchi, N.; Imaizumi, T. Vascular inflammation evaluated by [18F]-fluorodeoxyglucose positron emission tomography is associated with the metabolic syndrome. J. Am. Coll. Cardiol. 2007, 49, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Yong, H.S.; Hwang, S.Y.; Eo, J.S.; Hong, H.C.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Choi, D.S.; Baik, S.H.; et al. Association of pooled cohort risk scores with vascular inflammation and coronary artery calcification in Korean adults. Metabolism 2016, 65, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Kim, S.; Park, M.S.; Yang, S.J.; Kim, T.N.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Seo, H.S.; Baik, S.H.; et al. Vascular inflammation stratified by C-reactive protein and low-density lipoprotein cholesterol levels: Analysis with 18F-FDG pet. J. Nucl. Med. 2011, 52, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Kim, S.; Yang, S.J.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; Choi, D.S.; Choi, K.M. Association of adiponectin, resistin, and vascular inflammation: Analysis with 18F-fluorodeoxyglucose positron emission tomography. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Tahara, N.; Nitta, Y.; Tahara, A.; Igata, S.; Bekki, M.; Nakamura, T.; Sugiyama, Y.; Kaida, H.; Kurata, S.; et al. Vascular inflammation evaluated by [18F]-fluorodeoxyglucose-positron emission tomography/computed tomography is associated with endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y. Adiponectin: Identification, physiology and clinical relevance in metabolic and vascular disease. Atheroscler. Suppl. 2005, 6, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Maemura, K.; Takeda, N.; Harada, T.; Nojiri, T.; Imai, Y.; Manabe, I.; Utsunomiya, K.; Nagai, R. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: A new insight into adipocytokine-endothelial cell interactions. Biochem. Biophys. Res. Commun. 2004, 314, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Lin, C.Y.; Tsai, J.Y.; Wu, Y.L.; Su, K.H.; Lu, K.Y.; Hsiao, S.H.; Pan, C.C.; Kou, Y.R.; Hsu, Y.P.; et al. Resistin increases lipid accumulation by affecting class A scavenger receptor, CD36 and ATP-binding cassette transporter-A1 in macrophages. Life Sci. 2009, 84, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Filkova, M.; Haluzik, M.; Gay, S.; Senolt, L. The role of resistin as a regulator of inflammation: Implications for various human pathologies. Clin. Immunol. 2009, 133, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Buckels, A.C.; Kinghorn, I.J.; Murdock, P.R.; Holbrook, J.D.; Plumpton, C.; Macphee, C.H.; Smith, S.A. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem. Biophys. Res. Commun. 2003, 300, 472–476. [Google Scholar] [CrossRef]
- Hong, H.C.; Hwang, S.Y.; Park, S.; Ryu, J.Y.; Choi, H.Y.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; et al. Implications of pericardial, visceral and subcutaneous adipose tissue on vascular inflammation measured using 18FDG-PET/CT. PLoS ONE 2015, 10, e0135294. [Google Scholar] [CrossRef] [PubMed]
- Cocker, M.S.; Mc Ardle, B.; Spence, J.D.; Lum, C.; Hammond, R.R.; Ongaro, D.C.; McDonald, M.A.; Dekemp, R.A.; Tardif, J.C.; Beanlands, R.S. Imaging atherosclerosis with hybrid [18F]fluorodeoxyglucose positron emission tomography/computed tomography imaging: What leonardo da vinci could not see. J. Nucl. Med. 2012, 19, 1211–1225. [Google Scholar] [CrossRef] [PubMed]
- Vucic, E.; Dickson, S.D.; Calcagno, C.; Rudd, J.H.; Moshier, E.; Hayashi, K.; Mounessa, J.S.; Roytman, M.; Moon, M.J.; Lin, J.; et al. Pioglitazone modulates vascular inflammation in atherosclerotic rabbits noninvasive assessment with FDG-PET-CT and dynamic contrast-enhanced mr imaging. JACC Cardiovasc. Imaging 2011, 4, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Magata, Y.; Kato, T.; Hatano, K.; Ishino, S.; Mukai, T.; Shiomi, M.; Ito, K.; Saji, H. Application of 18F-FDG pet for monitoring the therapeutic effect of antiinflammatory drugs on stabilization of vulnerable atherosclerotic plaques. J. Nucl. Med. 2006, 47, 1845–1850. [Google Scholar] [PubMed]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E. In vivo inhibition of foam cell development by probucol in watanabe rabbits. Am. J. Cardiol. 1988, 62, 6B–12B. [Google Scholar] [CrossRef]
- Marx, N.; Mach, F.; Sauty, A.; Leung, J.H.; Sarafi, M.N.; Ransohoff, R.M.; Libby, P.; Plutzky, J.; Luster, A.D. Peroxisome proliferator-activated receptor-gamma activators inhibit IFN-gamma-induced expression of the T cell-active CXC chemokines IP-10, Mig, and I-TAC in human endothelial cells. J. Immunol. 2000, 164, 6503–6508. [Google Scholar] [CrossRef] [PubMed]
- Takata, Y.; Kitami, Y.; Yang, Z.H.; Nakamura, M.; Okura, T.; Hiwada, K. Vascular inflammation is negatively autoregulated by interaction between CCAAT/enhancer-binding protein-delta and peroxisome proliferator-activated receptor-gamma. Circ. Res. 2002, 91, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Meehan, W.P.; Kintscher, U.; Jackson, S.; Wakino, S.; Noh, G.; Palinski, W.; Hsueh, W.A.; Law, R.E. Troglitazone inhibits formation of early atherosclerotic lesions in diabetic and nondiabetic low density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Li, A.C.; Brown, K.K.; Silvestre, M.J.; Willson, T.M.; Palinski, W.; Glass, C.K. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J. Clin. Investig. 2000, 106, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, T.; Meyer, P.M.; Feinstein, S.B.; Davidson, M.H.; Kondos, G.T.; D’Agostino, R.B., Sr.; Perez, A.; Provost, J.C.; Haffner, S.M. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: A randomized trial. JAMA 2006, 296, 2572–2581. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Nicholls, S.J.; Wolski, K.; Nesto, R.; Kupfer, S.; Perez, A.; Jure, H.; De Larochelliere, R.; Staniloae, C.S.; Mavromatis, K.; et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: The PERISCOPE randomized controlled trial. JAMA 2008, 299, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Dormandy, J.A.; Charbonnel, B.; Eckland, D.J.; Erdmann, E.; Massi-Benedetti, M.; Moules, I.K.; Skene, A.M.; Tan, M.H.; Lefebvre, P.J.; Murray, G.D.; et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the proactive study (PROspective pioglitazone clinical trial in macrovascular events): A randomised controlled trial. Lancet 2005, 366, 1279–1289. [Google Scholar] [CrossRef]
- Hellberg, S.; Sippola, S.; Liljenback, H.; Virta, J.; Silvola, J.M.U.; Stahle, M.; Savisto, N.; Metso, J.; Jauhiainen, M.; Saukko, P.; et al. Effects of atorvastatin and diet interventions on atherosclerotic plaque inflammation and [18F]FDG uptake in Ldlr-/-Apob100/100mice. Atherosclerosis 2017, 263, 369–376. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-B.; Choi, K.M. Diet-Modulated Lipoprotein Metabolism and Vascular Inflammation Evaluated by 18F-fluorodeoxyglucose Positron Emission Tomography. Nutrients 2018, 10, 1382. https://doi.org/10.3390/nu10101382
Lee Y-B, Choi KM. Diet-Modulated Lipoprotein Metabolism and Vascular Inflammation Evaluated by 18F-fluorodeoxyglucose Positron Emission Tomography. Nutrients. 2018; 10(10):1382. https://doi.org/10.3390/nu10101382
Chicago/Turabian StyleLee, You-Bin, and Kyung Mook Choi. 2018. "Diet-Modulated Lipoprotein Metabolism and Vascular Inflammation Evaluated by 18F-fluorodeoxyglucose Positron Emission Tomography" Nutrients 10, no. 10: 1382. https://doi.org/10.3390/nu10101382
APA StyleLee, Y.-B., & Choi, K. M. (2018). Diet-Modulated Lipoprotein Metabolism and Vascular Inflammation Evaluated by 18F-fluorodeoxyglucose Positron Emission Tomography. Nutrients, 10(10), 1382. https://doi.org/10.3390/nu10101382