Docoxahexaenoic Acid Induces Apoptosis of Pancreatic Cancer Cells by Suppressing Activation of STAT3 and NF-?B



Abstract
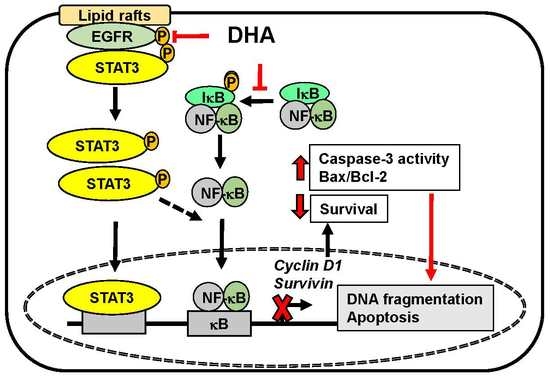
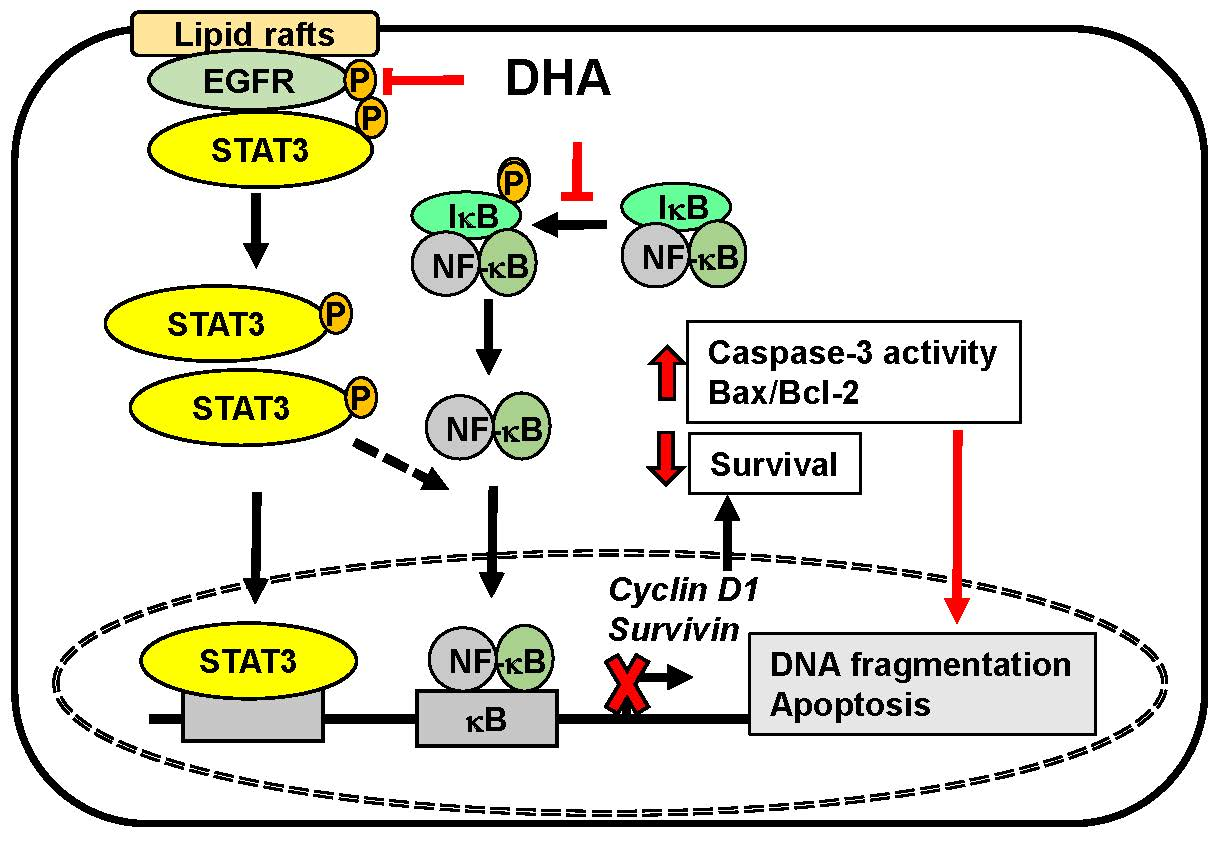
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Line and Culture Condition
2.2. Experimental Protocol
2.3. Determination of Cell Viability
2.4. DNA Fragmentation
2.5. Real-Time PCR Analysis for Survivin and Cyclin D1
2.6. Preparation of Cell Extracts
2.7. Western Blot Analysis for STAT3, p-STAT3, EGFR, p-EGFR, Bcl-2, Bax, IκBα, p-IκBα, Cyclin D1, Survivin, Caveolin-1, and Caspas-3
2.8. Immunoprecipitation of EGFR and STAT3
2.9. Luciferase Reporter Gene Assay for NF-κB Activity
2.10. Electrophoretic Mobility Shift Assay (EMSA) for NF-κB and STAT3
2.11. Immunofluorescence Staining for GM1 in Lipid Rafts
2.12. Isolation of Lipid Raft Fractions and Determination of Calveolin-1 and EGFR in Membrane Fractions
2.13. Statistical Analysis
3. Results
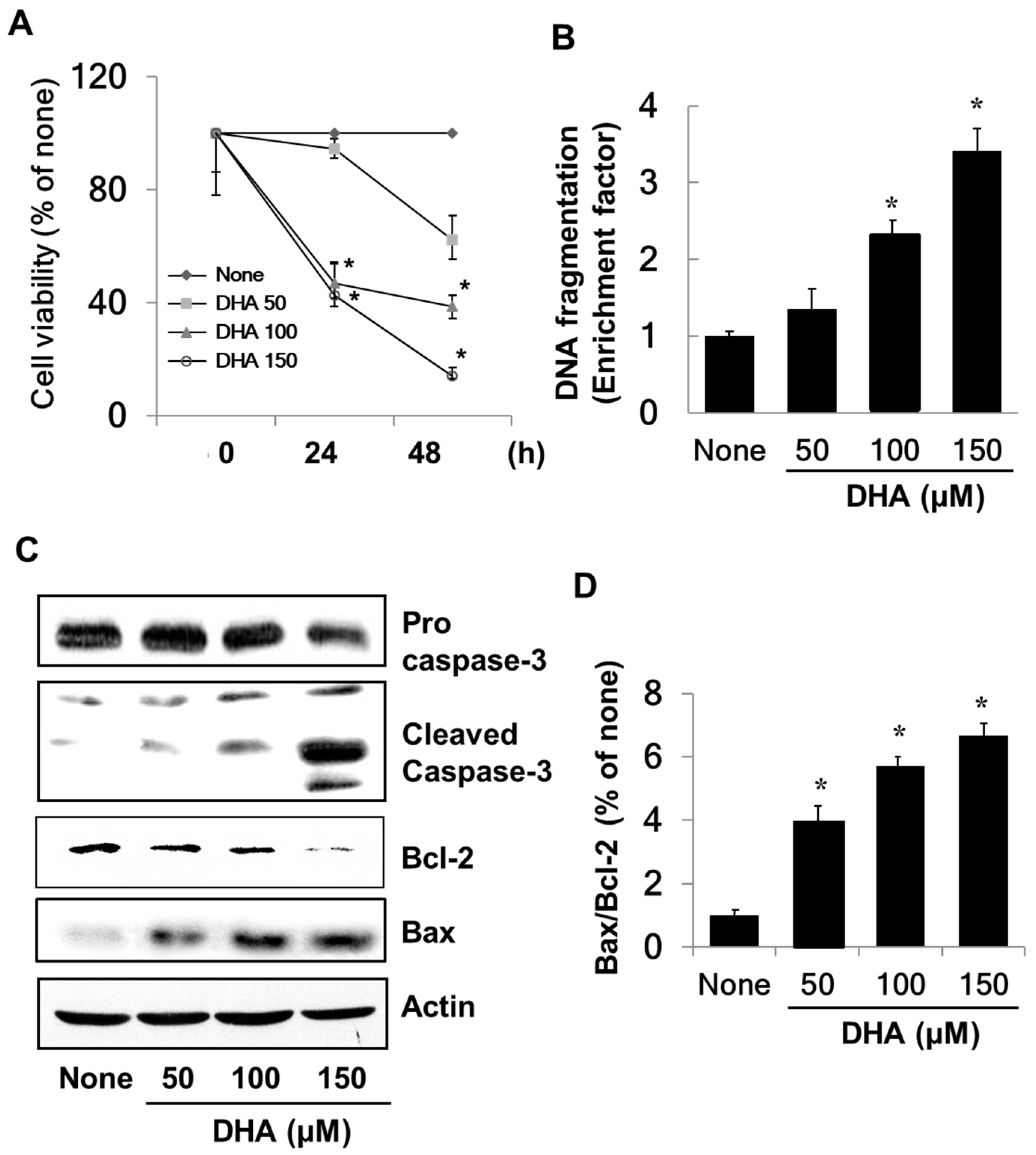
3.1. DHA Induces Apoptosis in PANC-1 Cells
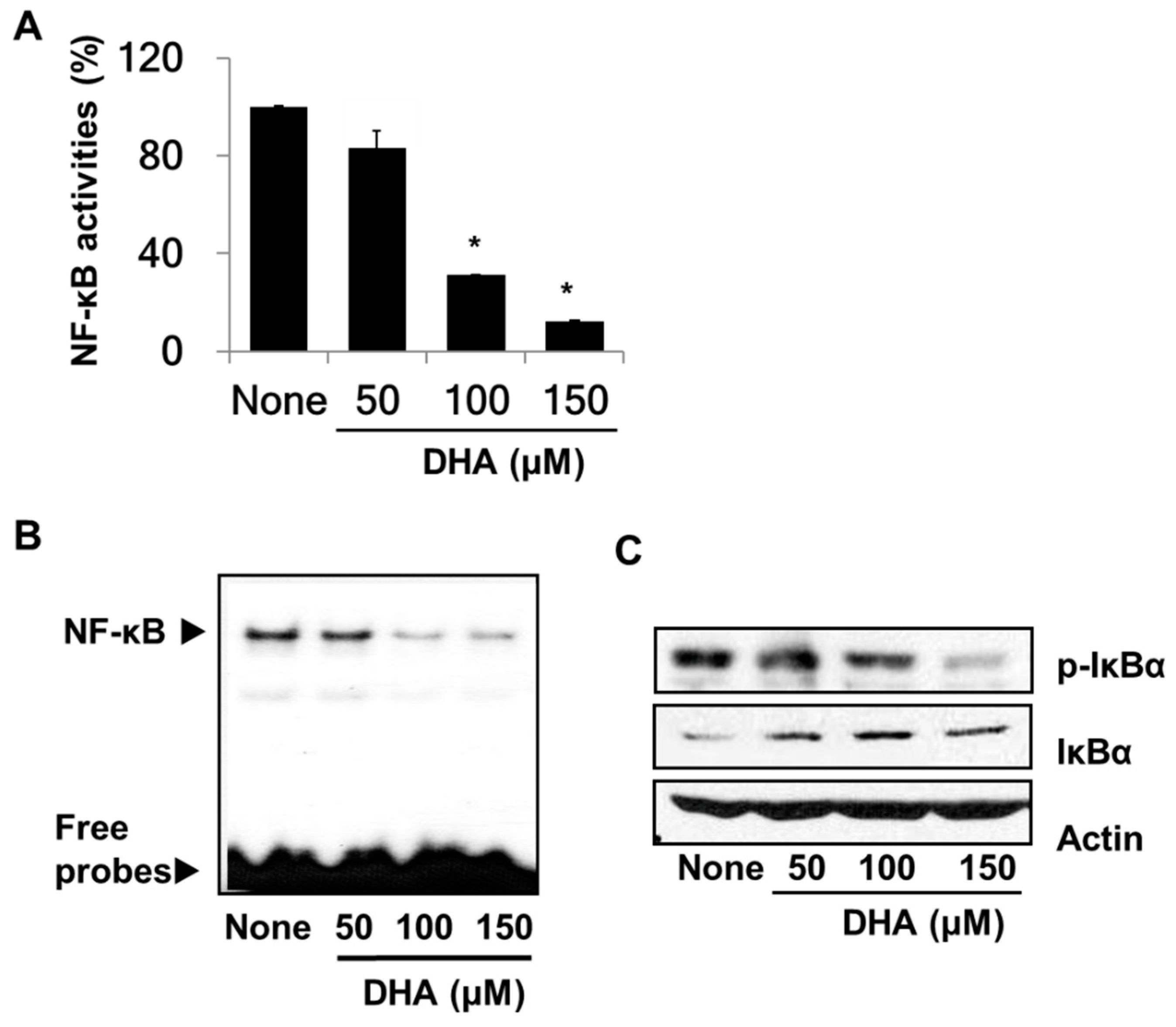
3.2. DHA Suppresses the Activity of NF-κB in PANC-1 Cells
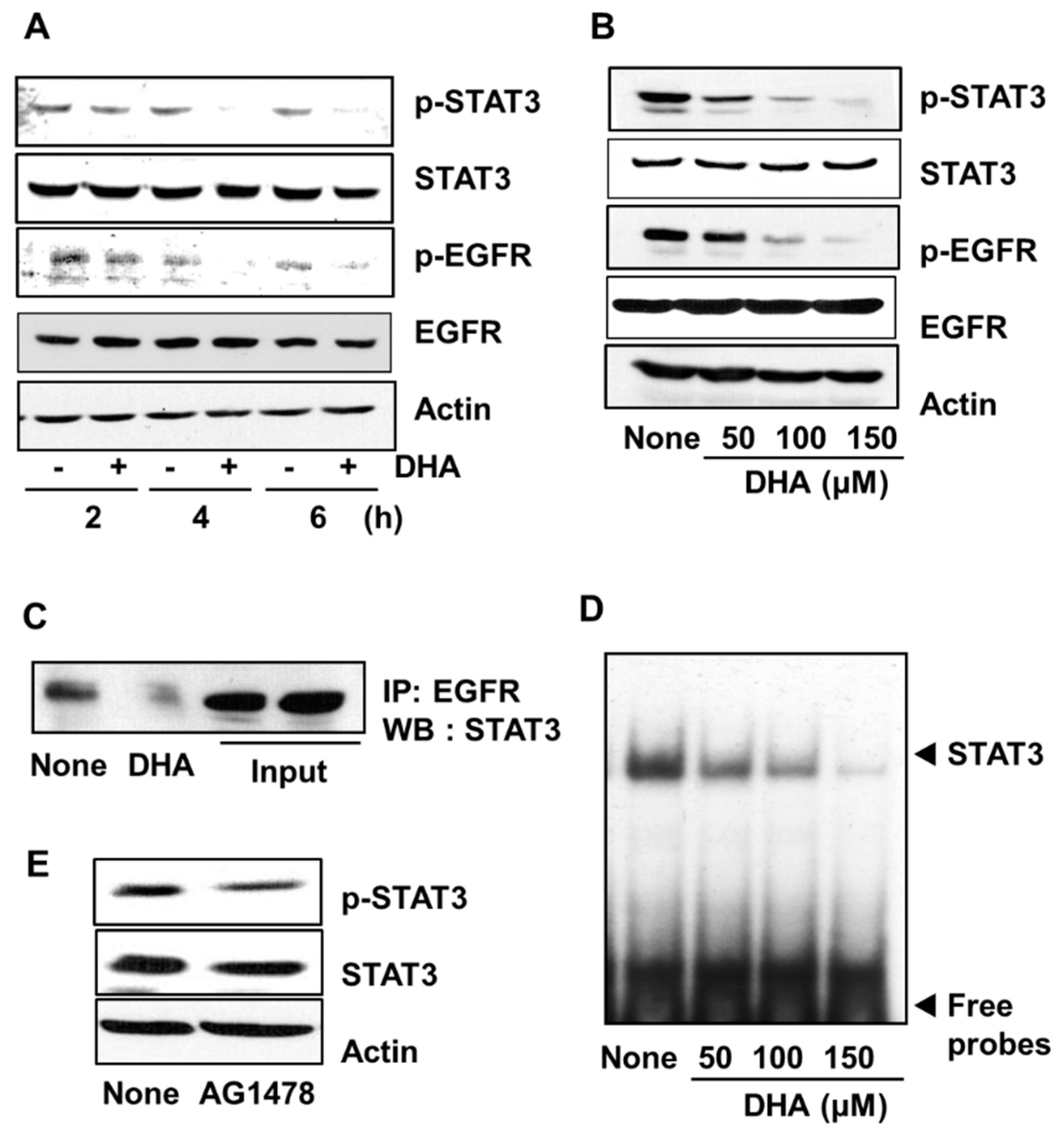
3.3. DHA Inhibits Activation of STAT3 and EGFR in PANC-1 Cells
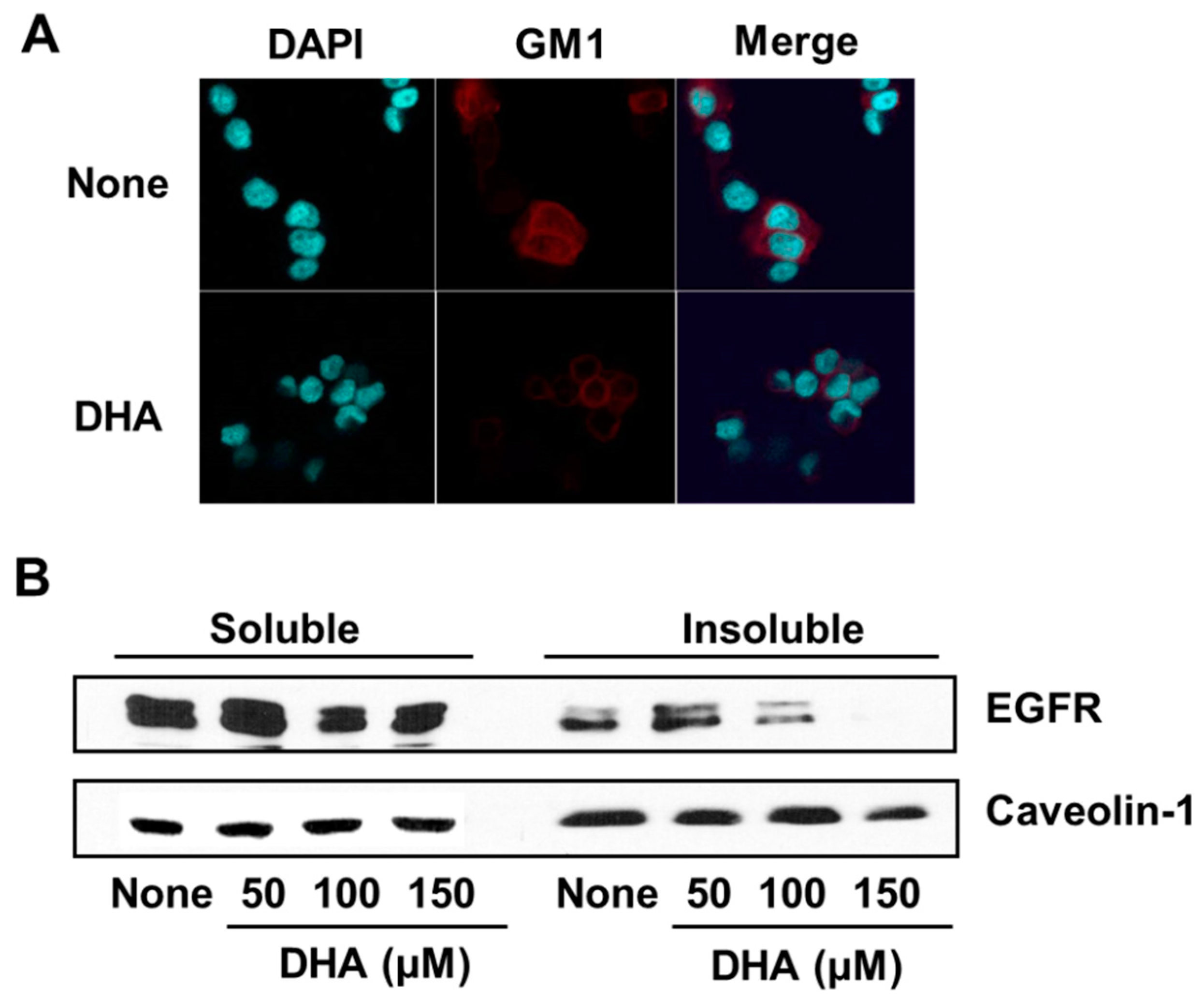
3.4. DHA Disintegrates Lipid Rafts, Resulting in Exclusion of EGFR from Lipid Rafts in PANC-1 Cells
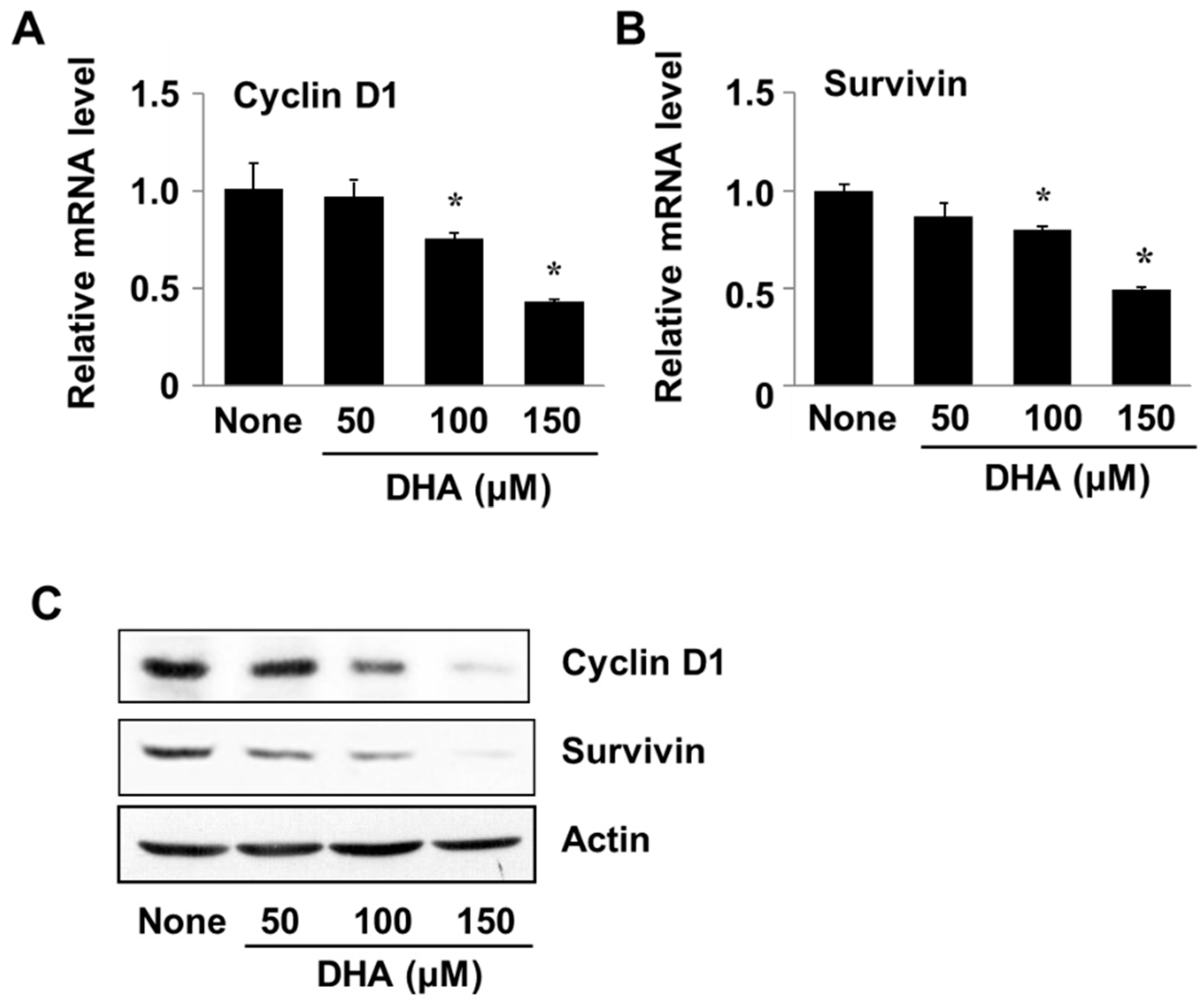
3.5. DHA Decreases Expression of Cyclin D1 and Survivin in PANC-1 Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ryu, K. The Early Detection of Pancreatic Cancer: Whom and How? Korean J. Pancreas Biliary Tract 2015, 20, 198–203. [Google Scholar] [CrossRef]
- Lennon, A.M.; Wolfgang, C.L.; Canto, M.I.; Klein, A.P.; Herman, J.M.; Goggins, M.; Fishman, E.K.; Kamel, I.; Weiss, M.J.; Diaz, L.A. The Early Detection of Pancreatic Cancer: What Will It Take to Diagnose and Treat Curable Pancreatic Neoplasia? Cancer Res. 2014, 74, 3381–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.K.; Shankar, S.; Srivastava, R.K. STAT3 as an emerging molecular target in pancreatic cancer. Gastrointest. Cancer Targets Ther. 2014, 4, 115–122. [Google Scholar]
- Xiong, A.; Yang, Z.; Shen, Y.; Zhou, J.; Shen, Q. Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers 2014, 6, 926–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Yan, R.; He, X.; He, J. Constitutive activation of STAT3 and cyclin D1 overexpression contribute to proliferation, migration and invasion in gastric cancer cells. Am. J. Transl. Res. 2017, 9, 5671–5677. [Google Scholar] [PubMed]
- Kanda, N.; Seno, H.; Konda1, Y.; Marusawa1, H.; Kanai, M.; Nakajima1, T.; Kawashima1, T.; Nanakin, A.; Sawabu, T.; Uenoyama, Y. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene 2004, 23, 4921–4929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganathan, S.; Yue, P.; Paladino, D.C.; Bogdanovic, J.; Huo, Q.; Turkson, J. A functional nuclear epidermal growth factor receptor, SRC and Stat3 heteromeric complex in pancreatic cancer cells. PLoS ONE 2011, 6, e19605. [Google Scholar] [CrossRef] [PubMed]
- Park, O.K.; Schaefer, T.S.; Nathans, D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Biochemistry 1996, 93, 13704–13708. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.J.; Derynck, R.; Korc, M. Production of transforming growth factor α in human pancreatic cancer cells: Evidence for a superagonist autocrine cycle. Proc. Natl. Acad. Sci. USA 1987, 84, 7567–7570. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, S.; Yue, P.; Turkson, J. Enhanced sensitivity of pancreatic cancer cells to concurrent inhibition of aberrant signal transducer and activator of transcription 3 and epidermal growth factor receptor or Src. J. Pharmacol. Exp. Ther. 2010, 333, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Gong, K.; Wohlfeld, B.; Hatanpaa, K.J.; Zhao, D.; Habib, A.A. Ligand-Independent EGFR Signaling. Cancer Res. 2015, 75, 3436–3441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.; Karin, M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Tomita, M.; Matsuda, T.; Ohta, T.; Tanaka, Y.; Fujii, M.; Hatano, M.; Tokuhisa, T.; Mori, N. Transcriptional activation of survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. Int. J. Cancer 2005, 115, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhang, W.; Kone, B.C. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem. J. 2002, 367, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Herrmnn, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed]
- Bollrath, J.; Greten, F.R. IKK/NF-kappaB and STAT3 pathways: Central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009, 10, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, G.P.; Nozell, S.E.; Benveniste, E.T. NF-kappaB and STAT3 signaling in glioma: Targets for future therapies. Expert Rev. Neurother. 2010, 10, 575–586. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Karin, M. NF-kappaB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Blanckaert, V.; Ulmann, L.; Minouni, V.; Antol, J.; Brancquart, L.; Chenais, B. Docosahexaenoic acid intake decreases proliferation, increases apoptosis and decreases the invasive potential of the human breast carcinoma cell line MDA-MB-231. Int. J. Oncol. 2010, 36, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Calviello, G.; Resci, F.; Serini, S.; Piccioni, E.; Toesca, A.; Boninsegna, A.; Monego, G.; Ranelletti, F.O.; Palozza, P. Docosahexaenoic acid induces proteasome-dependent degradation of beta-catenin, down-regulation of surviving and apoptosis in human colorectal cancer cells not expressing COX-2. Carcinogenesis 2007, 28, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.N.; Jia, W.D.; Chen, H.; Ma, J.L.; Ge, Y.S.; Yu, J.H.; Li, J.S. Docosahexaenoic acid (DHA) induces apoptosis in human hepatocellular carcinoma cells. Int. J. Clin. Exp. Pathol. 2013, 6, 281–289. [Google Scholar] [PubMed]
- Schley, P.D.; Jijon, H.B.; Robinson, L.E.; Field, C.J. Mechanisms of omega-3 fatty acid-induced growth inhibition in MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2005, 92, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Rogers, K.R.; Kikawa, K.D.; Mouradian, M.; Hernandez, K.; McKinnon, K.M.; Ahwah, S.M.; Pardini, R.S. Docosahexaenoic acid alters epidermal growth factor receptor-related signaling by disrupting its lipid raft association. Carcinogenesis 2010, 31, 1523–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duraisamy, Y.; Lambert, D.; O’Neill, C.A.; Padfield, P.J. Differential incorporation of docosahexaenoic acid into distinct cholesterol-rich membrane raft domains. Biochem. Biophys. Res. Commun. 2007, 360, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and physiological relevance of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Rissanen, S.; Grzybek, M.; Orłowski, A.; Róg, T.; Cramariuc, O.; Levental, I.; Eggeling, C.; Sezgin, E.; Vattulainen, I. Phase partitioning of GM1 and its bodipy-labeled analog determine their different binding to cholera toxin. Front Physiol. 2017, 8, 252. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Vind-Kezunovic, D.; Karvinen, S.; Gniadecki, R. Ligand-independent activation of the EGFR by lipid raft disruption. J. Investig. Dermatol. 2006, 126, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Radeva, G.; Sharom, F.J. Isolation and characterization of lipid rafts with different properties from RBL-2H3 (rat basophilic leukaemia) cells. Biochem. J. 2004, 380, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Cheepala, S.; Roberts, J.N.; Syson-Chan, K.; DiGiovanni, J.; Clifford, J.L. Active Stat3 is required for survival of human squamous cell carcinoma cells in serum-free conditions. Mol. Cancer 2006, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Lee, C.-K. “What does Stat3 do?”. J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Page, B.D.; Ball, D.P.; Gunning, P.T. Signal transducer and activator of transcription 3 inhibitors: A patent review. Expert Opin. Ther. Pat. 2011, 21, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [PubMed]
- Jianqiang, W.M.; Patmore, D.M.; Jousma, E.; Eaves, D.W.; Breving, K.; Patel, A.V.; Schwartz, E.B.; Fuchs, J.R.; Cripe, T.P.; Stemmer-Rachamimov, A.O. EGFR-STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene 2014, 33, 173–180. [Google Scholar]
- Levental, I.; Veatch, S. The continuing mystery of lipid rafts. J. Mol. Biol. 2016, 428, 4749–4764. [Google Scholar] [CrossRef] [PubMed]
- Pton, R.G.; Richards, A.A. Lipid rafts and caveolae as portals for endocytosis: New insights and common mechanisms. Traffic 2003, 4, 724–738. [Google Scholar]
- Holmgren, J. Comparison of the tissue receptor for Vibrio cholerae and Escherichia coli endotoxins by means of gangliosides and natural choleric toxoid. Infect. Immun. 1973, 8, 851–859. [Google Scholar] [PubMed]
- Park, E.K.; Lee, E.J.; Lee, S.H.; Koo, K.H.; Sung, J.Y.; Hwang, E.H.; Park, J.H.; Kim, C.W.; Jeong, K.C.; Park, B.K. Induction of apoptosis by the ginsenoside Rh2 by internalization of lipid rafts and caveolae and inactivation of Akt. Br. J. Pharmacol. 2010, 160, 1212–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, H.F.; Barhoumi, R.; Chapkin, R.S. Alteration of EGFR spatiotemporal dynamics suppresses signal transduction. PLoS ONE 2012, 7, e39682. [Google Scholar] [CrossRef] [PubMed]
- Stunig, T.M.; Huber, J.; Leitinger, N.; Imre, E.M.; Angelisova, P.; Nowotny, P.; Waldhausl, W. Polyunsaturated eicosapentaenoic acid displaces proteins from membrane rafts by altering raft lipid composition. J. Biol. Chem. 2001, 276, 37335–37340. [Google Scholar] [CrossRef] [PubMed]
- Ewaschuk, J.B.; Newell, M.; Field, C.J. Docosahexaenoic acid improves chemotherapy efficacy by inducing CD95 translocation to lipid rafts in ER(-) breast cancer cells. Lipids 2012, 47, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Turk, H.F.; Monk, J.M.; Fan, Y.Y.; Callaway, E.S.; Weeks, B.; Chapkin, R.S. Inhibitory effects of omega-3 fatty acids on injury-induced epidermal growth factor receptor transactivation contribute to delayed wound healing. Am. J. Physiol. Cell Physiol. 2013, 304, C905–C917. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.; Kundu, J.K.; Shin, J.W.; Na, H.K.; Surh, Y.J. Docosahexaenoic acid inhibits UVB-induced activation of NF-κB and expression of COX-2 and NOX-4 in HR-1 hairless mouse skin by blocking MSK1 signaling. PLoS ONE 2011, 6, e28065. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Das, S. Identification of cytotoxic mediators and their putative role in the signaling pathways during docosahexaenoic acid (DHA)-induced apoptosis of cancer cells. Apoptosis 2016, 21, 1408–1421. [Google Scholar] [CrossRef] [PubMed]
- Song, E.A.; Kim, H. Docosahexaenoic acid induces oxidative DNA damage and apoptosis, and enhances the chemosensitivity of cancer cells. Int. J. Mol. Sci. 2016, 17, 1257. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Kim, H. Anti-cancer mechanism of docosahexaenoic acid in pancreatic carcinogenesis: A mini-review. J. Cancer Prev. 2017, 22, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Trombino, S.; Oliva, F.; Piccioni, E.; Monego, G.; Resci, F.; Boninsegna, A.; Picci, N.; Ranelletti, F.O.; Calviello, G. Docosahexaenoic acid induces apoptosis in lung cancer cells by increasing MKP-1 and down-regulating p-ERK1/2 and p-p38 expression. Apoptosis 2008, 13, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Zhou, W.; Liu, B.; Wang, X.; Chen, B. DHA induces apoptosis of human malignant breast cancer tissues by the TLR-4/PPAR-α pathways. Oncol Lett. 2018, 15, 2967–2977. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Jing, K.; Jeong, S.; Kim, N.; Song, K.S.; Heo, J.Y.; Park, J.H.; Seo, K.S.; Han, J.; Park, J.I. The omega-3 polyunsaturated fatty acid DHA induces simultaneous apoptosis and autophagy via mitochondrial ROS-mediated Akt-mTOR signaling in prostate cancer cells expressing mutant p53. Biomed. Res. Int. 2013, 2013, 568671. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, M.; Lim, J.W.; Kim, H. Docoxahexaenoic Acid Induces Apoptosis of Pancreatic Cancer Cells by Suppressing Activation of STAT3 and NF-?B. Nutrients 2018, 10, 1621. https://doi.org/10.3390/nu10111621
Park M, Lim JW, Kim H. Docoxahexaenoic Acid Induces Apoptosis of Pancreatic Cancer Cells by Suppressing Activation of STAT3 and NF-?B. Nutrients. 2018; 10(11):1621. https://doi.org/10.3390/nu10111621
Chicago/Turabian StylePark, Mirae, Joo Weon Lim, and Hyeyoung Kim. 2018. "Docoxahexaenoic Acid Induces Apoptosis of Pancreatic Cancer Cells by Suppressing Activation of STAT3 and NF-?B" Nutrients 10, no. 11: 1621. https://doi.org/10.3390/nu10111621
APA StylePark, M., Lim, J. W., & Kim, H. (2018). Docoxahexaenoic Acid Induces Apoptosis of Pancreatic Cancer Cells by Suppressing Activation of STAT3 and NF-?B. Nutrients, 10(11), 1621. https://doi.org/10.3390/nu10111621