Kidney Response to the Spectrum of Diet-Induced Acid Stress

{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Daily H+ Challenge
3. Maintenance of Normal Acid–Base Homeostasis
3.1. Buffers
3.2. H+ Sequestration in Interstitial Fluid
3.3. Urine H+ Excretion
3.4. Kidney Cytokine Response to Systemic H+ Challenge
4. Other Organ Contributors to Systemic Acid–Base Status
5. General Dietetic Management Strategies for the Spectrum of H+ Stress
Dietary H+ Reduction
6. Considerations Regarding Management of the Spectrum of H+ Stress
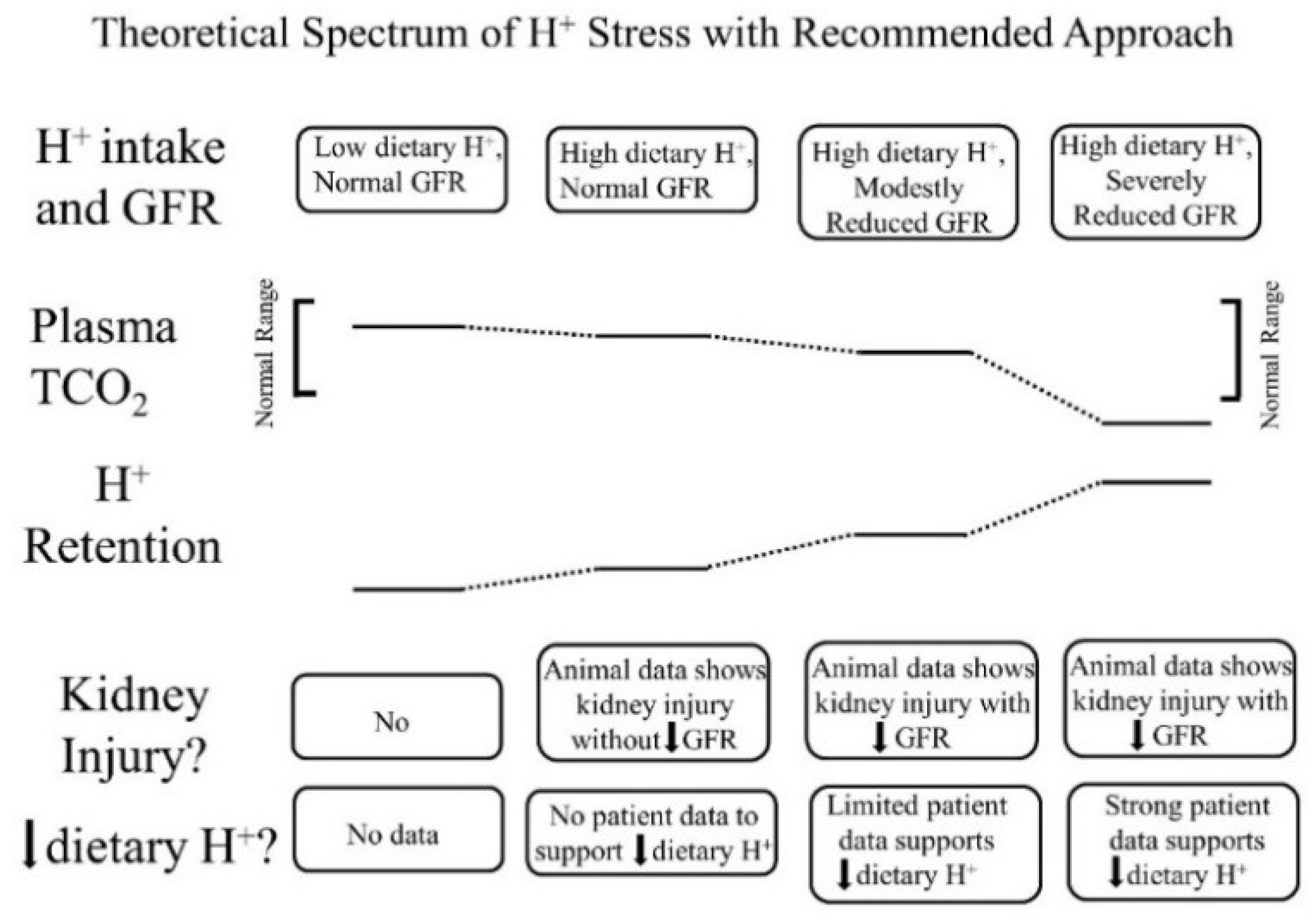
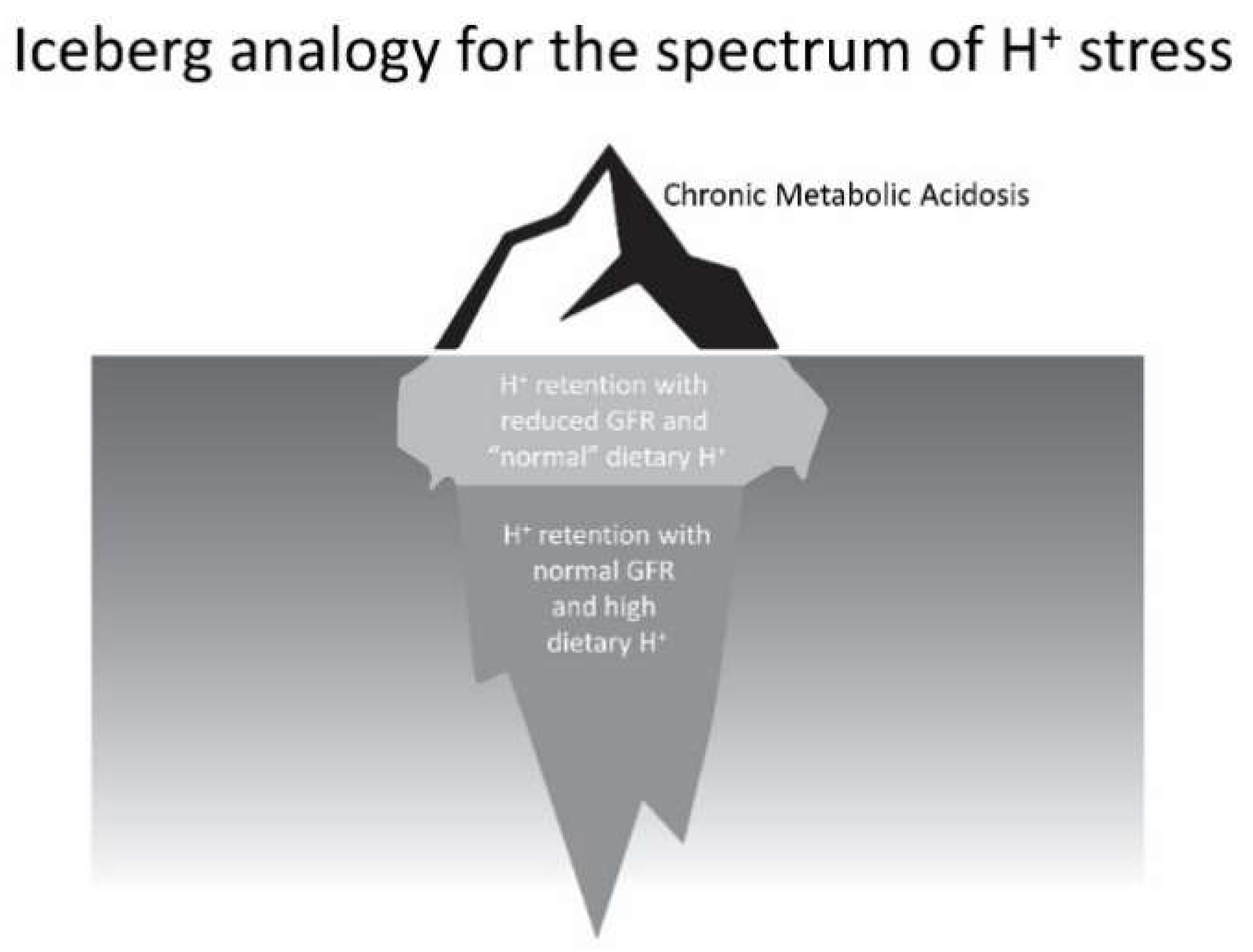
- Low dietary H+ intake, normal GFR: Animals ingesting a plant protein, base-producing diet have low kidney levels of endothelin [35], angiotensin II [16], and aldosterone [42], likely because of their reduced need to increase H+ excretion compared to animals eating an animal protein, H+-producing diet. Because the kidney levels of these substances are low, comparably little to no kidney injury occurs [35]. These animal data support the lack of need for additional dietary H+ reduction for analogous patients fitting this construct.
- High dietary H+ intake, normal GFR: Such animals achieve steady-state H+ excretion which avoids progressive H+ retention, yet they have steady-state H+ retention [11,17] without any significant change in plasma TCO2 [11,17,18]. These animals also have increased levels of endothelin [18], angiotensin II [16], and aldosterone [42], each of which help to mediate increased H+ excretion in response to this increment in dietary H+. Despite increased kidney levels of these substances, 96 weeks of these H+-producing diets in animals did not decrease GFR but did increase kidney tubulo-interstitial fibrosis (TIF) [35], a feature of progressive nephropathy. High H+ diets might decrease serum [HCO3]/pH slightly, but such small decreases remain within normal limits for clinical laboratories and so would not be evident clinically [22]. Although epidemiological studies support the idea that high H+ diets increase the risk of developing CKD [56] or type 2 diabetes [57], there are no published interventional data examining the long-term effect of high dietary H+ on kidney or other organ function in patients with baseline normal GFR to inform a management recommendation. That being said, such patients would theoretically benefit from reducing their high dietary H+ content.
- High dietary H+ intake, modestly decreased GFR, but no metabolic acidosis based on plasma acid–base parameters: Such animal CKD models have H+ retention yet no significant decrease in plasma TCO2 [15,16,17,42]. They also have increased kidney levels of endothelin [42], angiotensin II [16,43], and aldosterone [42], and they have progressive GFR decline characterized by TIF without dietary H+ reduction [16,42,43]. Analogous CKD patients with modestly reduced eGFR have H+ retention but with plasma TCO2, similar to comparable patients with normal eGFR [23,24] and have increased urine excretion of endothelin and aldosterone [23]. Small-scale interventional studies have shown that such CKD patients have progressive eGFR decline without dietary H+ reduction and that dietary H+ reduction reduces urine excretion of endothelin and aldosterone [23], reduces indices of kidney injury [44], and slows eGFR decline [40]. Larger scale studies are required to determine if dietary H+ reduction should be standard-of-care for CKD patients with modestly-reduced GFR but no metabolic acidosis based on serum acid-base parameters.
- High dietary H+ intake, severely decreased GFR with metabolic acidosis: Analogous animal CKD models fitting this construct have increased kidney levels of endothelin [14] with progressive GFR decline that is slowed by dietary H+ reduction [41]. Analogous CKD patients with severely reduced eGFR have increased urine endothelin excretion [46], and small-scale interventional studies have shown progressive GFR decline that is ameliorated by dietary H+ reduction [46,47,49]. Current guidelines recommend treatment with Na+-based oral alkali for amelioration of the disturbed bone and muscle metabolism caused by the metabolic acidosis of CKD [48] and, as mentioned, recent studies support the proposal that this intervention also slows eGFR decline. Other small-scale studies have provided support for the benefit of base-producing fruits and vegetables in treating metabolic acidosis in CKD [49,53], but larger scale studies must confirm these studies before such interventions, which carry the risk of hyperkalemia due to the high potassium content of plant-based foods, becomes a standard recommendation for treating the metabolic acidosis of CKD. These fruit and vegetable interventions, however, might be effective adjuncts to Na+-based alkali to improve metabolic acidosis, particularly given that the fruit and vegetable intervention has additional salutary effects like better blood pressure control [49,53].
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Lemann, J., Jr.; Bushinsky, D.A.; Hamm, L.L. Bone buffering of acid and base in humans. Am. J. Physiol. 2003, 285, F811–F832. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E. Physiologic and Pathophysiologic Renal Consequences of H+-Stimulated Endothelin Secretion. Am. J. Kid Dis. 2000, 35, lii–liv. [Google Scholar] [CrossRef]
- Kovesdy, C.P.; Anderson, J.E.; Kalantar-Zadeh, K. Association of serum bicarbonate levels with mortality in patients with non-dialysis-dependent CKD. Nephrol. Dial. Transplant. 2009, 24, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Raphael, K.; Wei, G.; Baird, B.; Greene, T.; Beddhu, S. Higher plasma bicarbonate levels within the normal range are associated with better survival and renal outcomes in African Americans. Kid Int. 2011, 79, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Relman, A.S.; Lennon, E.J.; Lemann, J., Jr. Endogenous production of fixed acid and the measurement of the net balance of acid in normal subjects. J. Clin. Investig. 1961, 40, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Frassetto, L.A.; Morris, R.C., Jr.; Sebastian, A. Dietary sodium chloride intake independently predicts the degree of hyperchloremic metabolic acidosis in healthy humans consuming a net acid-producing diet. Am. J. Physiol. Renal Physiol. 2007, 293, F521–F525. [Google Scholar] [CrossRef] [PubMed]
- Remer, T. Influence of nutrition on acid-base: Balance-metabolic aspects. Eur. J. Nutr. 2001, 40, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Remer, T.; Manz, F. Potential renal acid load of foods and its influence on urine pH. J. Am. Dietet. Assoc. 1995, 95, 791–797. [Google Scholar] [CrossRef]
- Gausseres, N.; Catala, I.; Mahe, S.; Luengo, C.; Bornet, F.; Guy-Grand, B.; Tome, D. Whole-body protein turnover in humans fed a soy protein-rich diet. Eur. J. Clin. Nutr. 1997, 51, 308–311. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Swan, R.C.; Pitts, R.F. Neutralization of infused acid by nephrectomized dogs. J. Clin. Investig. 1955, 34, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E. Dietary acid increases blood and renal cortical acid content in rats. Am. J. Physiol. 1998, 274, F97–F103. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Kaissling, B. Structural organization of the mammalian kidney. In The Kidney. Physiology and Pathophysiology; Seldin, D., Giebisch, G., Eds.; Raven Press: New York, NY, USA, 1992; p. 207. [Google Scholar]
- Siragy, H.M.; Ibrahim, M.M.; Jaffa, A.A.; Mayfield, R.; Margolis, H.S. Rat renal interstitial bradykinin, prostaglandin E2, and cyclic guanosine 3’,5’-monophosphate. Effects of altered sodium intake. Hypertension 1994, 23, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E. Endogenous endothelins mediate augmented acidification in remnant kidneys. J. Am. Soc. Nephrol. 2001, 12, 1826–1835. [Google Scholar] [PubMed]
- Wesson, D.E.; Simoni, J. Increased tissue acid mediates progressive GFR decline in animals with reduced nephron mass. Kid Int. 2009, 75, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Jo, C.-H.; Simoni, J. Angiotensin II receptors mediate increased distal nephron acidification caused by acid retention. Kid Int. 2012, 82, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Pruszynski, J.; Cai, W.; Simoni, J. Acid retention with reduced glomerular filtration rate increases urine biomarkers of kidney and bone injury. Kid Int. 2017, 91, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E. Endogenous endothelins mediate increased distal tubule acidification induced by dietary acid in rats. J. Clin. Investig. 1997, 99, 2203–2211. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Simoni, J.; Hacker, C.; Duran, M.-J.; Wesson, D.E. Increased endothelin activity mediates augmented distal nephron acidification induced by dietary protein. J. Am. Soc. Nephrol. 2004, 15, 2266–2275. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Simoni, J.; Wesson, D.E. Endothelin-induced increased aldosterone activity mediates augmented distal nephron acidification as a result of dietary protein. J. Am. Soc. Nephrol. 2005, 16, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Simoni, J.; Prabhakar, S. Endothelin-induced increased nitric oxide activity mediates augmented distal nephron acidification as a result of dietary protein. J. Am. Soc. Nephrol. 2006, 17, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, I.; Maher, T.; Hulter, H.N.; Schambelan, M.; Sebastian, A. Effect of diet on plasma acid-base composition in normal humans. Kid Int. 1983, 24, 670–680. [Google Scholar] [CrossRef]
- Wesson, D.E.; Simoni, J.; Broglio, K.; Sheather, S. Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. Am. J. Physiol. Renal Physiol. 2011, 300, F830–F837. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E. Assessing acid retention in humans. Am. J. Physiol. Renal Physiol. 2011, 301, F1140–F1142. [Google Scholar] [CrossRef]
- Vallet, M.; Metzger, M.; Haymann, J.-P.; Flamant, M.; Gauci, C.; Thervet, E.; Boffa, J.-J.; Vrtovsnik, F.; Froissart, M.; Stengel, S.; et al. Urinary ammonia and long-term outcomes in chronic kidney disease. Kid Int. 2015, 88, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Goraya, N.; Simoni, J.; Sager, L.N.; Pruszynski, J.; Wesson, D.E. Acid retention in chronic kidney disease is inversely related to GFR. Am. J. Physiol. Renal Physiol. 2018. [CrossRef] [PubMed]
- Adeva, M.M.; Souto, G. Diet-induced metabolic acidosis. Clin. Nutr. 2011, 30, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Ebah, L.M.; Wiig, H.; Dawidowska, I.; O’Toole, C.; Summers, A.; Nikam, M.; Jayanti, A.; Coupes, B.; Brenchley, P.; Mitra, S. Subcutaneous interstitial pressure and volume characteristics in renal impairment associated with edema. Kid Int. 2013, 84, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yang, L.V.; Tieg, B.C.; Arend, L.J.; McGraw, D.W.; Penn, R.B.; Petrovic, S. Deletion of the pH sensor GPR4 decreases renal acid excretion. J. Am. Soc. Nephrol. 2010, 21, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Lemann, J., Jr.; Lennon, E.J.; Goodman, A.D.; Litzow, J.R.; Relman, A.S. The net balance of acid in subjects given large loads of acid or alkali. J. Clin. Investig. 1965, 44, 507–517. [Google Scholar] [CrossRef] [PubMed]
- MacClean, A.J.; Hayslett, J.P. Adaptive change in ammonia excretion in renal insufficiency. Kid Int. 1980, 17, 595–606. [Google Scholar] [CrossRef]
- Goodman, A.D.; Lemann, J., Jr.; Lennon, E.J.; Relman, A.S. Production, excretion, and net balance of fixed acid in patients with renal acidosis. J. Clin. Investig. 1965, 44, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Dolson, G.M. Endothelin-1 increases rat distal tubule acidification in vivo. Am. J. Physiol. 1997, 273, F586–F594. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.L.; Mogelesky, T.C.; Chou, M.; Jeng, A.Y. Enhanced expression of renal endothelin-converting enzyme-1 and endothelin-A-receptor mRNA in rats with interstitial fibrosis following ureter ligation. J. Cardiovasc. Pharmacol. 2000, 36 (Suppl. 1), S255–S259. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Nathan, T.; Rose, T.; Simoni, J.; Tran, R.M. Dietary protein induces endothelin-mediated kidney injury through enhanced intrinsic acid production. Kid Int. 2007, 71, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Green, E.L.; Kren, S.; Hostetter, T.H. Role of aldosterone in the remnant kidney model in the rat. J. Clin. Investig. 1996, 98, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.Y.; Chertow, G.M. Elevations of serum phosphorus and potassium due to mild to moderate chronic renal insufficiency. Nephrol. Dial. Transplant. 2002, 17, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Moranne, O.; Friossart, M.; Rossert, J.; Gauci, C.; Boffa, J.; Haymann, J.P.; Mrad, M.B.; Jacquot, C.; Houillier, P.; Stengel, B.; et al. Timing of onset of CKD-related metabolic complications. J. Am. Soc. Nephrol. 2009, 20, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Appel, L.J.; Wright, J.T.; Greene, T.; Kusek, J.W.; Lewis, J.B.; Wang, X.; Lipkowitz, M.S.; Norris, K.C.; Bakris, G.L.; Rahman, M.; et al. Long-term effects of renin-angiotensin-system-blocking therapy and a low blood pressure goal on progression of hypertensive chronic kidney disease in African Americans. Arch. Int. Med. 2008, 168, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Simoni, J.; Sheather, S.; Broglio, K.; Rajab, M.H.; Wesson, D.E. Daily oral sodium bicarbonate preserves glomerular filtration rate by slowing its decline in early hypertensive nephropathy. Kid Int. 2010, 78, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Phisitkul, S.; Hacker, C.; Simoni, J.; Tran, R.M.; Wesson, D.E. Dietary protein causes a decline in the glomerular filtration rate of the remnant kidney mediated by metabolic acidosis and endothelin receptors. Kid Int. 2008, 73, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Simoni, J. Acid retention during kidney failure induces endothelin and aldosterone production which lead to progressive GFR decline, a situation ameliorated by alkali diet. Kid Int. 2010, 78, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Jo, C.-H.; Simoni, J. Angiotensin II-mediated GFR decline in subtotal nephrectomy is due to acid retention associated with reduced GFR. Nephrol. Dial. Transplant. 2015, 30, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Goraya, N.; Simoni, J.; Jo, C.-H.; Wesson, D.E. Dietary acid reduction with fruits and vegetables or sodium bicarbonate reduces kidney injury in subjects with moderately reduced GFR due to hypertensive nephropathy. Kid Int. 2012, 81, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Hostetter, M.K.; Hostetter, T.H. Pathophysiology of chronic tubulo-interstitial disease in rats-Interactions of dietary acid load, ammonia, and complement component C3. J. Clin. Investig. 1985, 76, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Phisitkul, S.; Khanna, A.; Simoni, J.; Broglio, K.; Sheather, S.; Rajab, H.; Wesson, D.E. Amelioration of metabolic acidosis in subjects with low GFR reduces kidney endothelin production, reduces kidney injury, and better preserves GFR. Kid Int. 2010, 77, 617–623. [Google Scholar] [CrossRef] [PubMed]
- De Brito-Ashurst, I.; Varagunam, M.; Raferty, M.J.; Yaqoob, M. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J. Am. Soc. Nephrol. 2009, 20, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- KDIGO Guidelines. Chapter 3: Management of progression and complications of CKD. Kid Int. Suppl. 2013, 3, 73–90. [Google Scholar]
- Goraya, N.; Simoni, J.; Jo, C.-H.; Wesson, D.E. Treatment of metabolic acidosis in individuals with stage 3 CKD with fruits and vegetables or oral NaHCO3 reduces urine angiotensinogen and preserves GFR. Kid Int. 2014, 86, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.T.; Bakris, G.; Greene, T.; Agodoa, L.Y.; Appel, L.J.; Charleston, J.; Cheek, D.; Douglas-Baltimore, J.G.; Gassman, J.; Glassock, R.; et al. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease. JAMA 2002, 288, 2421–2431. [Google Scholar] [CrossRef] [PubMed]
- Lemann, J.; Litzow, J.R.; Lennon, E.J. Studies of the mechanism by which chronic metabolic acidosis augments urinary calcium excretion in man. J. Clin. Investig. 1967, 46, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Bushinsky, D.A. Net calcium efflux from live bone during chronic metabolic but not respiratory acidosis. Am. J. Physiol. 1989, 256, F836–F842. [Google Scholar] [CrossRef] [PubMed]
- Goraya, N.; Simoni, J.; Jo, C.-H.; Wesson, D.E. Comparison of treating the metabolic acidosis of CKD stage 4 hypertensive kidney disease with fruits and vegetables or sodium bicarbonate. Clin. J. Am. Soc. Nephrol. 2013, 8, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Barsotti, G.; Morelli, E.; Cupisti, A.; Meola, M.; Dani, L.; Giovannetti, S. A low-nitrogen low phosphorous vegan diet for patients with chronic renal failure. Nephron 1996, 74, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.; Califano, J.R.; Butzin, C.A. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kid Int. 1990, 38, 937–941. [Google Scholar] [CrossRef]
- Banerjee, T.; Crews, D.; Wesson, D.E.; Tilea, A.; Saran, R.; Burrows, N.; Williams, D.; Powe, N. Dietary acid load and chronic kidney disease among adults in the United States. BMC Nephrol. 2014, 15, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Fagherazzi, G.; Villier, A.; Bonnet, F.; Lajous, M.; Balkau, B.; Bouton-Rualt, M.C.; Clavel-Chapelon, F. Dietary acid load and risk of type 2 diabetes: The E3N-EPIC cohort study. Diabetolgia 2014, 57, 313–320. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goraya, N.; Wesson, D.E. Kidney Response to the Spectrum of Diet-Induced Acid Stress. Nutrients 2018, 10, 596. https://doi.org/10.3390/nu10050596
Goraya N, Wesson DE. Kidney Response to the Spectrum of Diet-Induced Acid Stress. Nutrients. 2018; 10(5):596. https://doi.org/10.3390/nu10050596
Chicago/Turabian StyleGoraya, Nimrit, and Donald E. Wesson. 2018. "Kidney Response to the Spectrum of Diet-Induced Acid Stress" Nutrients 10, no. 5: 596. https://doi.org/10.3390/nu10050596
APA StyleGoraya, N., & Wesson, D. E. (2018). Kidney Response to the Spectrum of Diet-Induced Acid Stress. Nutrients, 10(5), 596. https://doi.org/10.3390/nu10050596