Chrysin Inhibits Advanced Glycation End Products-Induced Kidney Fibrosis in Renal Mesangial Cells and Diabetic Kidneys

Abstract
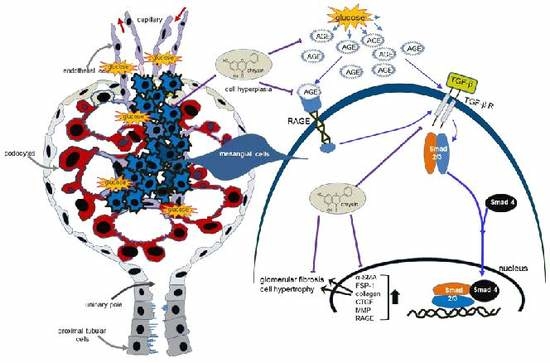
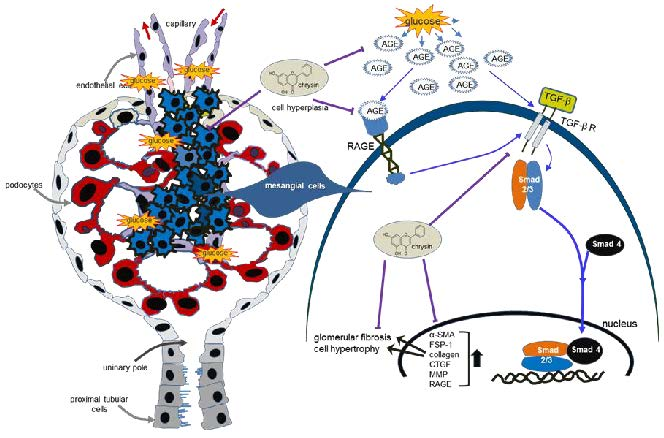
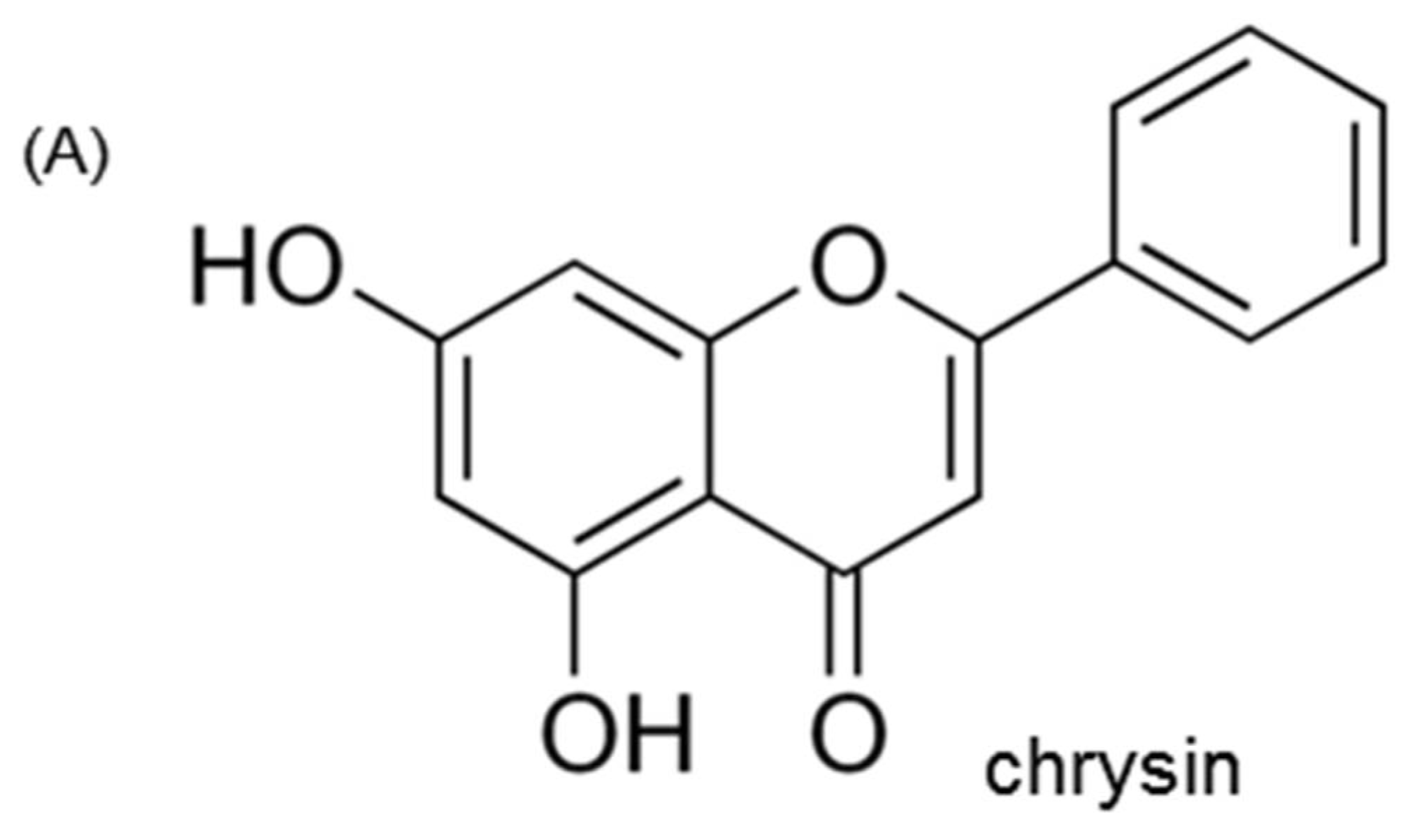
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
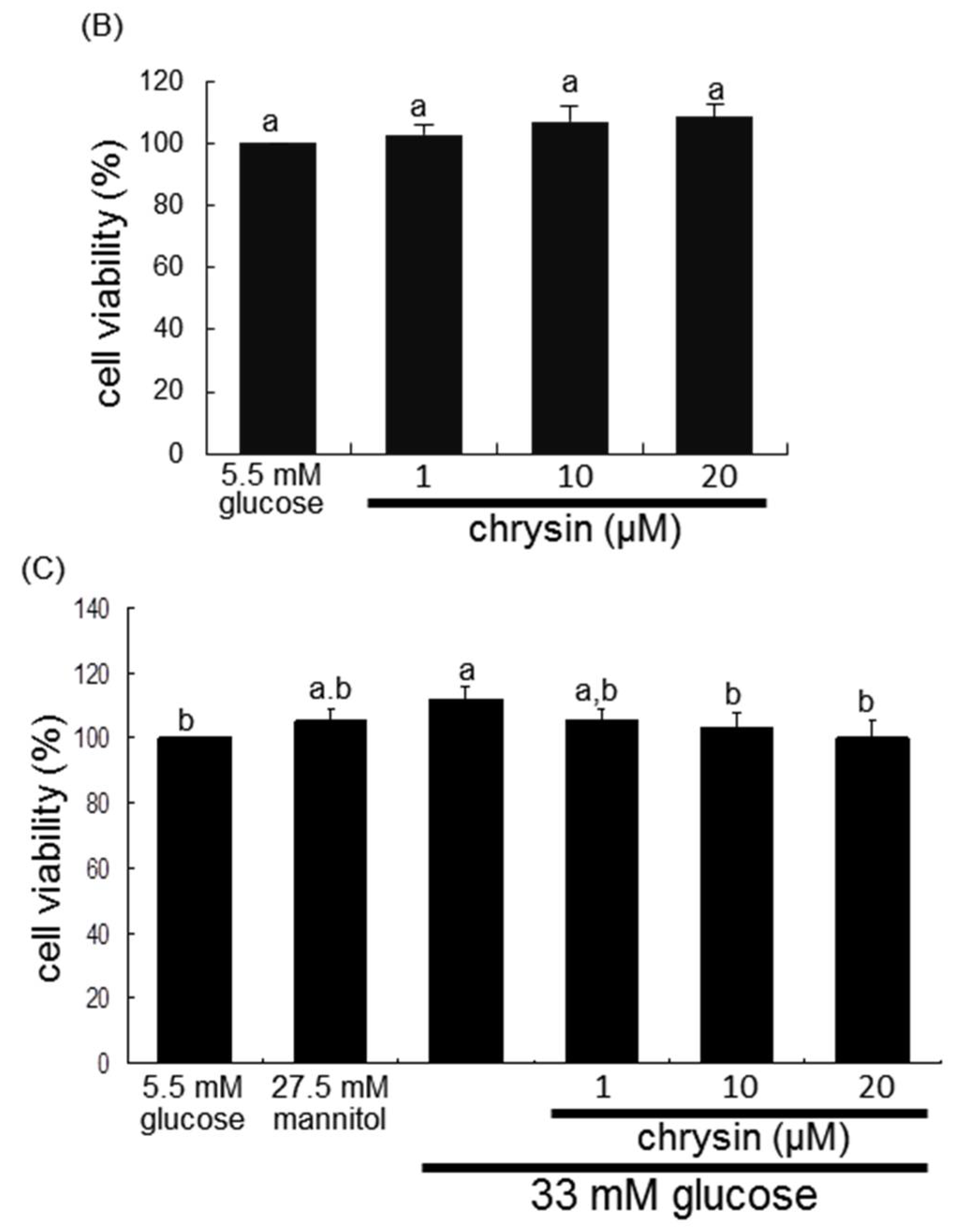
2.2. Mesangial Cell Culture and Viability
2.3. Western Blot Analysis
2.4. In Vivo Animal Experiments
2.5. Immunohistochemical Staining
2.6. PAS Staining
2.7. Masson’s Trichrome Staining
2.8. Immunocytochemical Staining
2.9. TGF-β1 Production
2.10. Data Analysis
3. Results
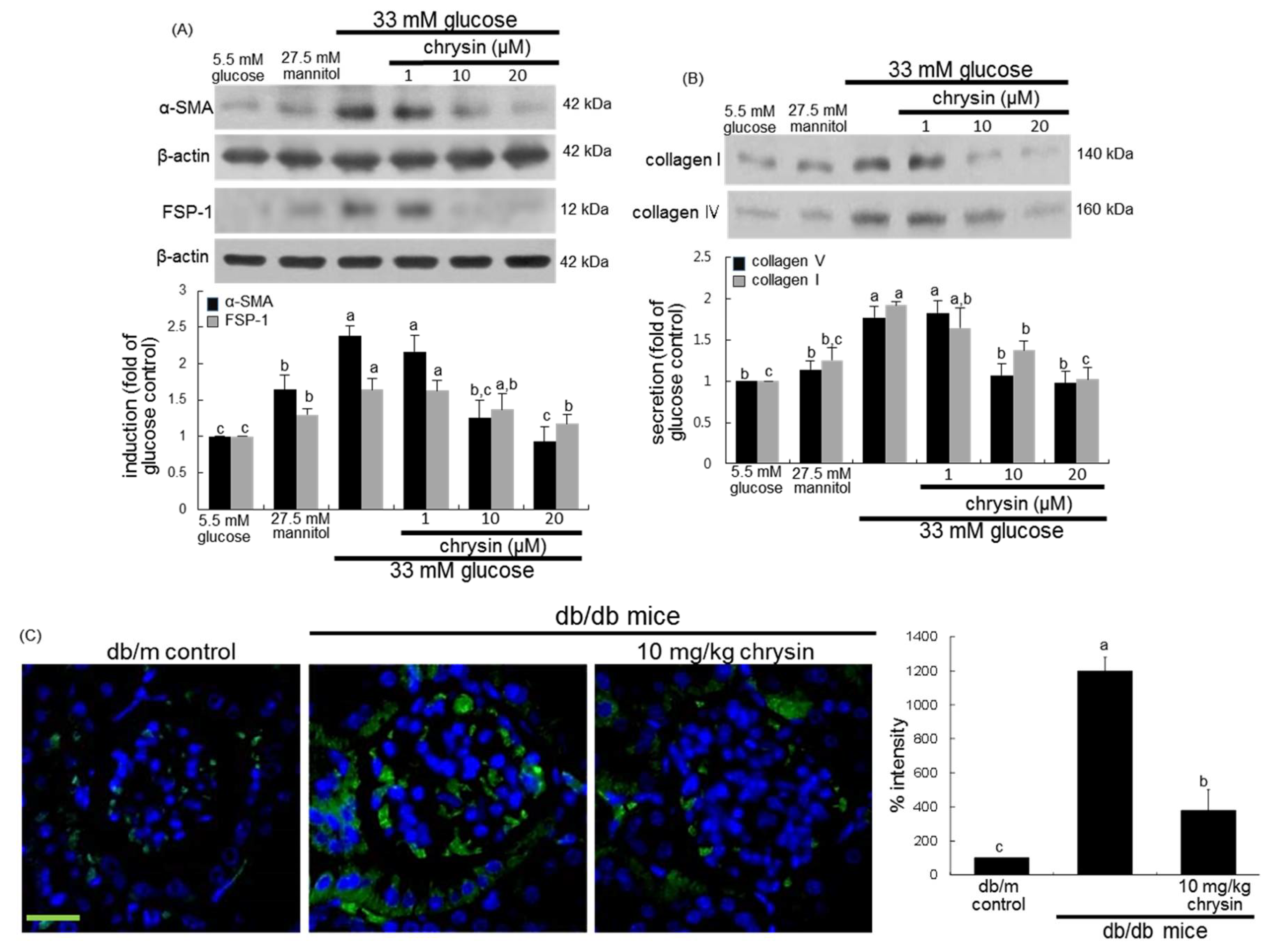
3.1. Effect of Chrysin on Glucose-Induced Mesangial Fibrosis
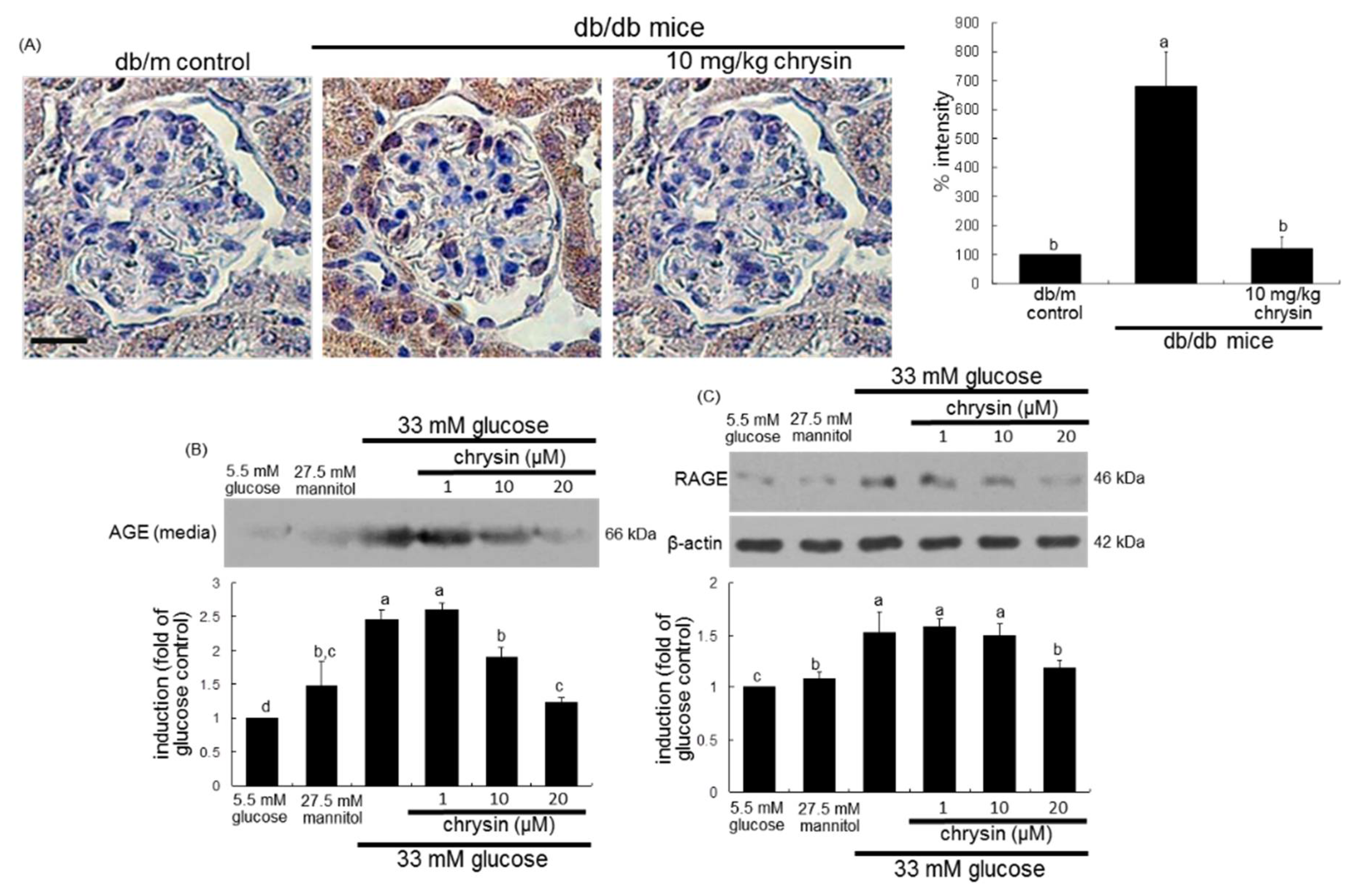
3.2. Effect of Chrysin on Glucose-Induced AGE Formation
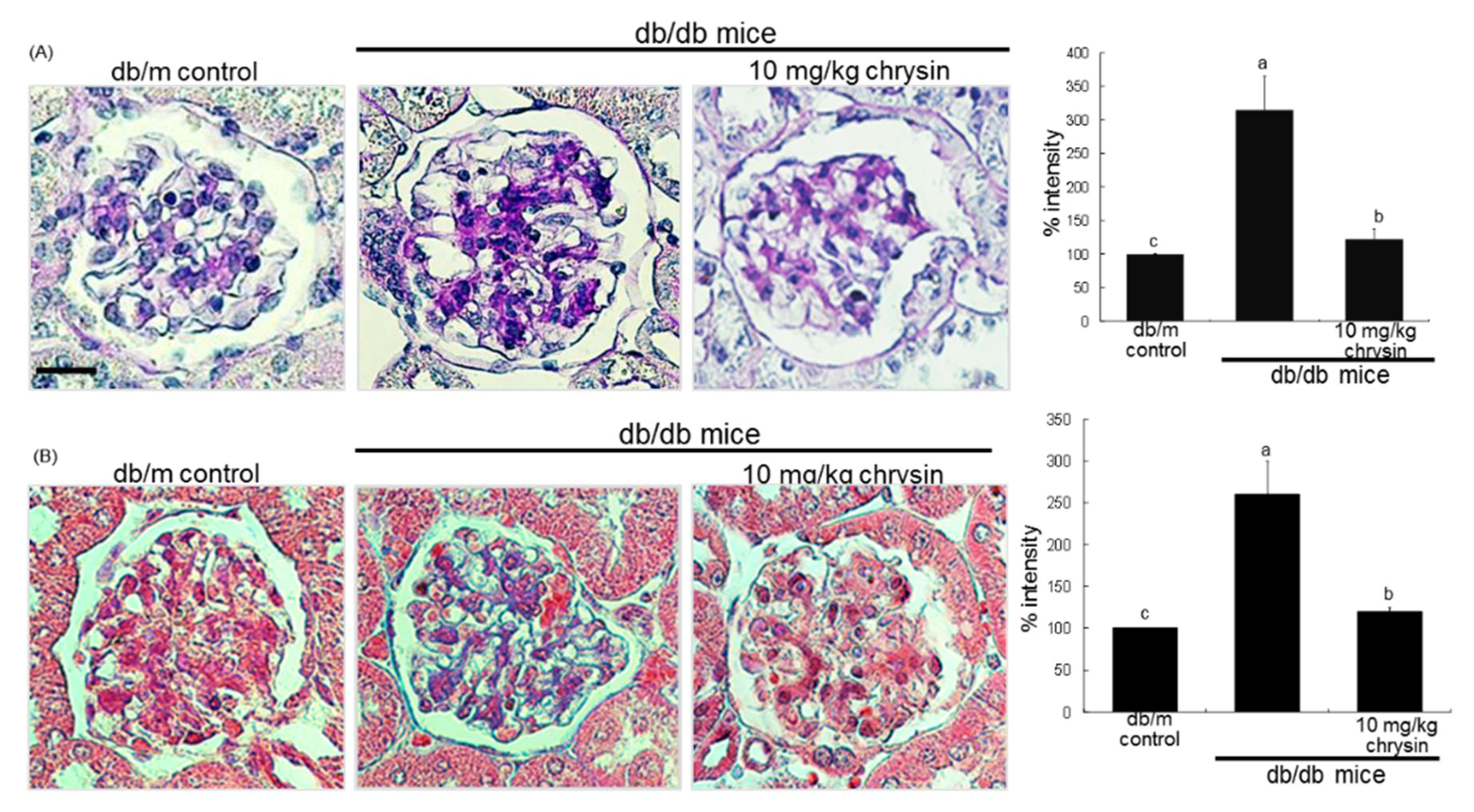
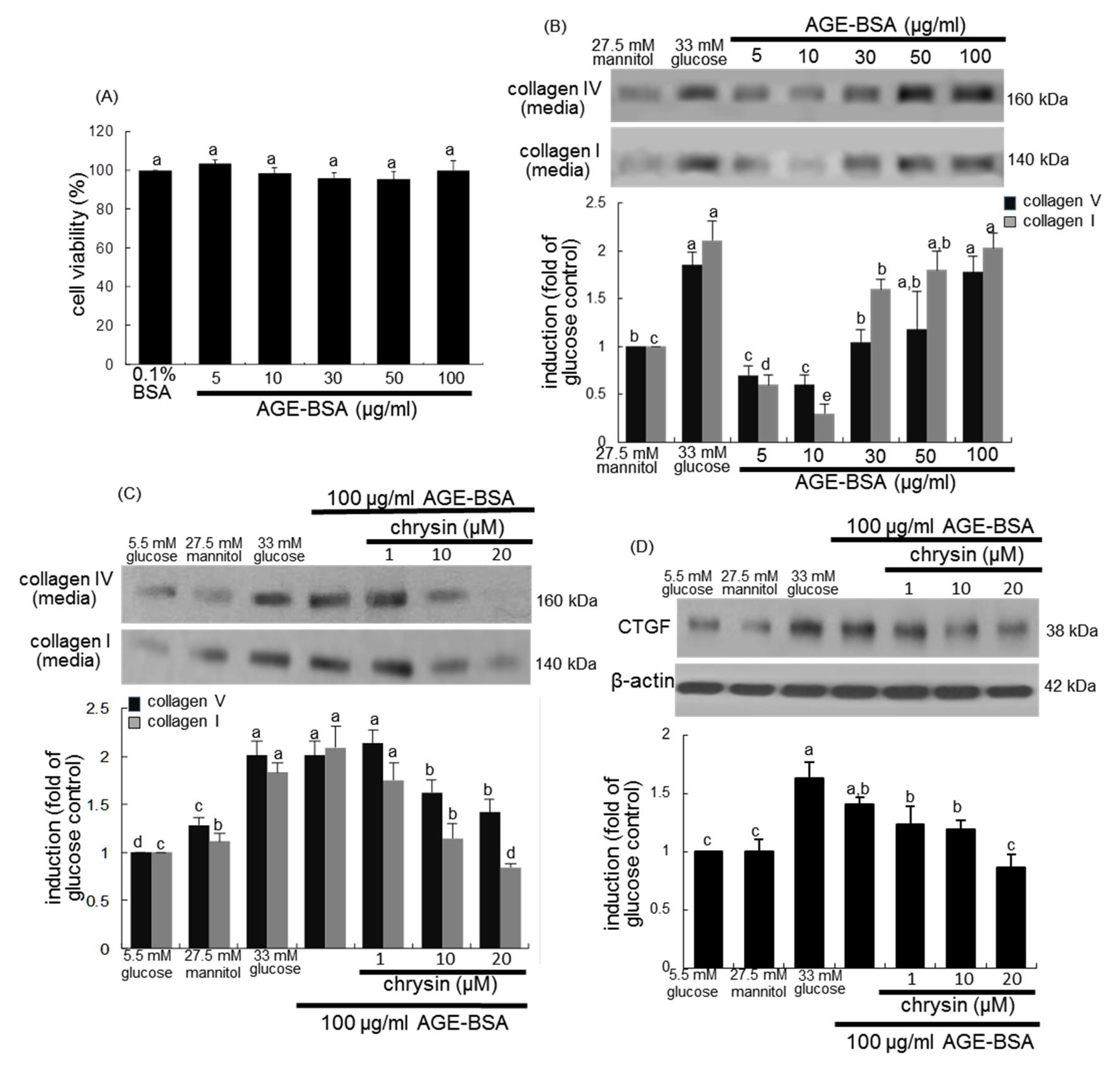
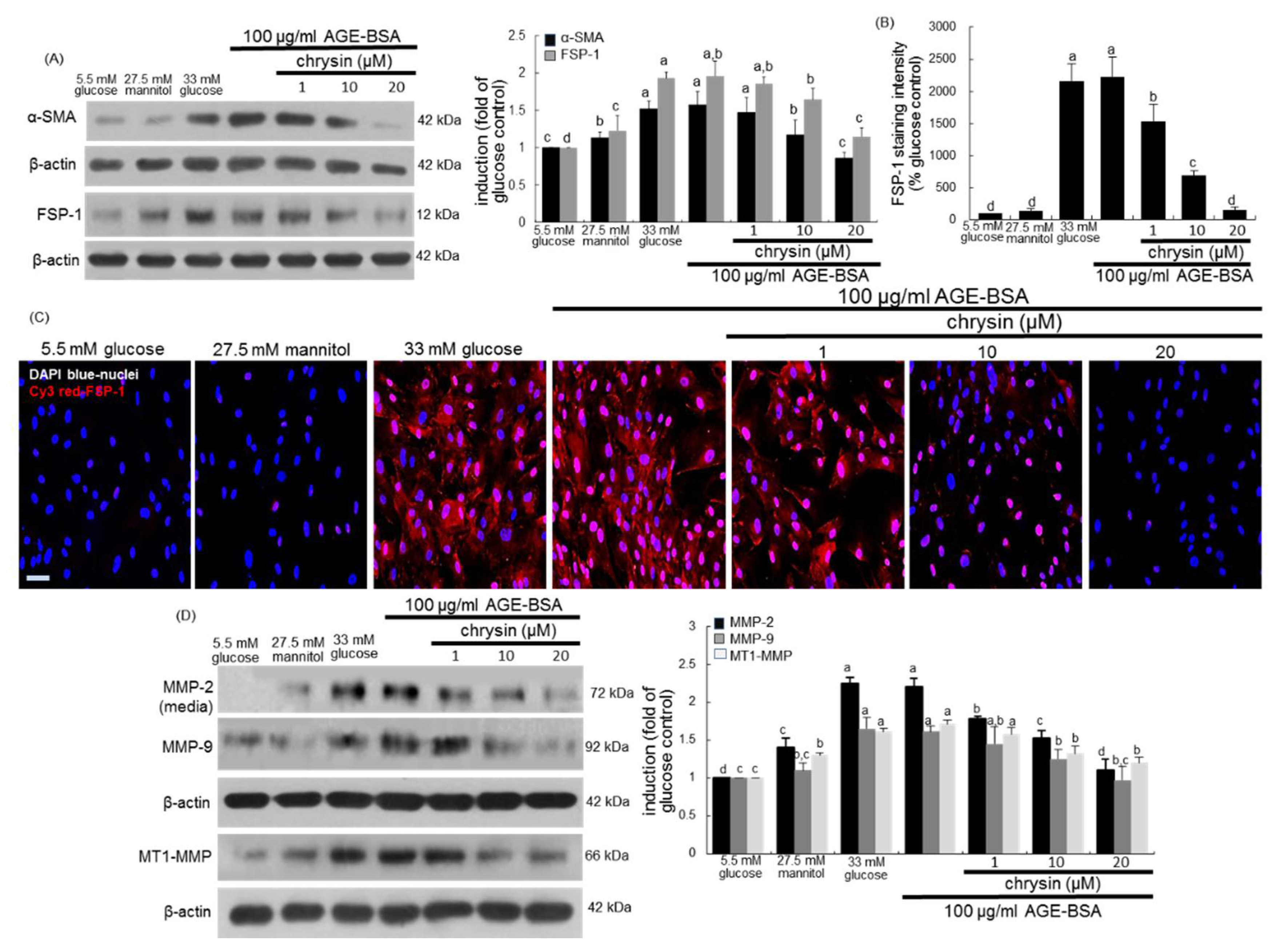
3.3. Blockade of AGEs-Induced Mesangial Expansion by Chrysin
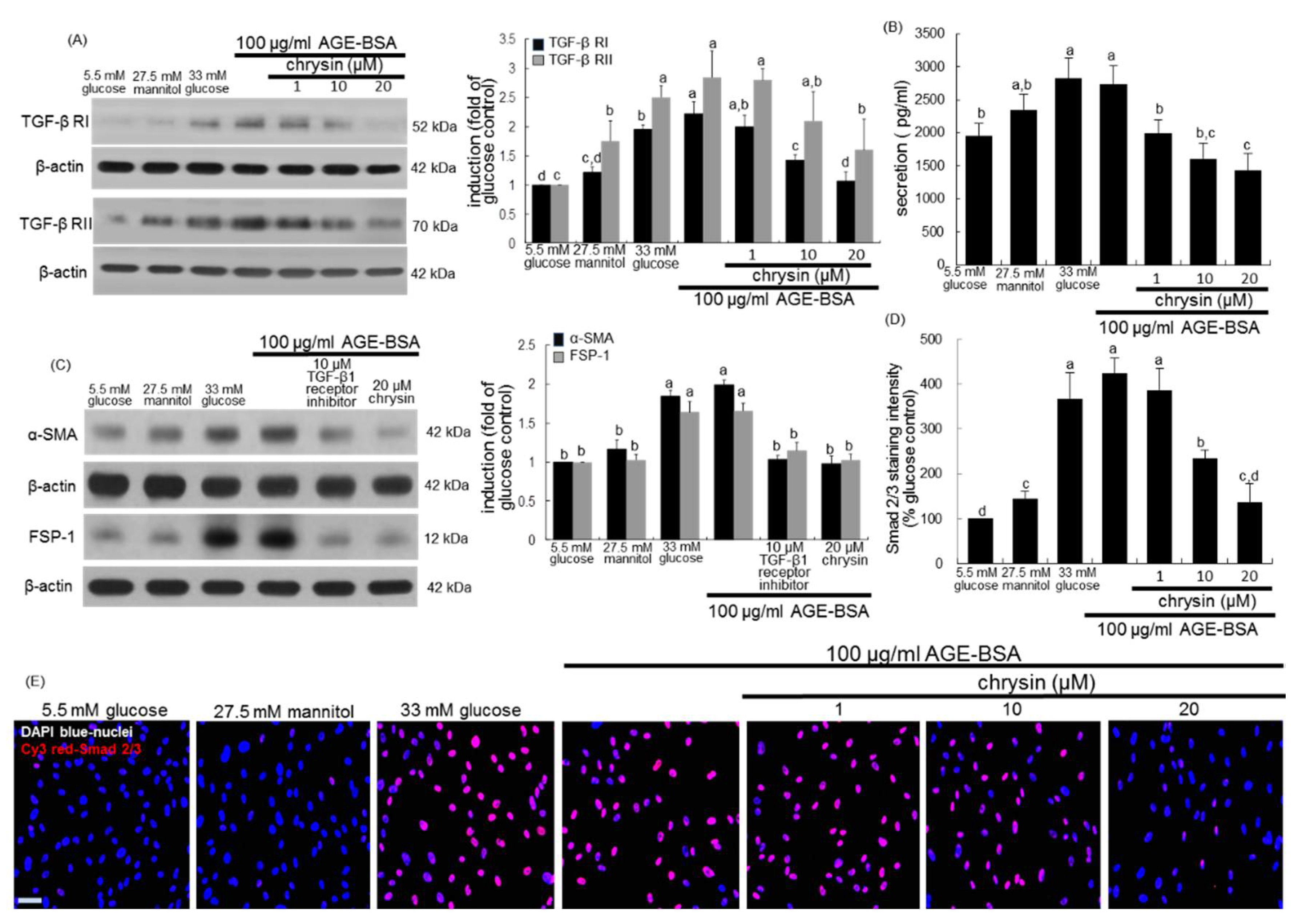
3.4. Inhibition of AGEs-Activated TGF-β/SMAD Signaling by Chrysin
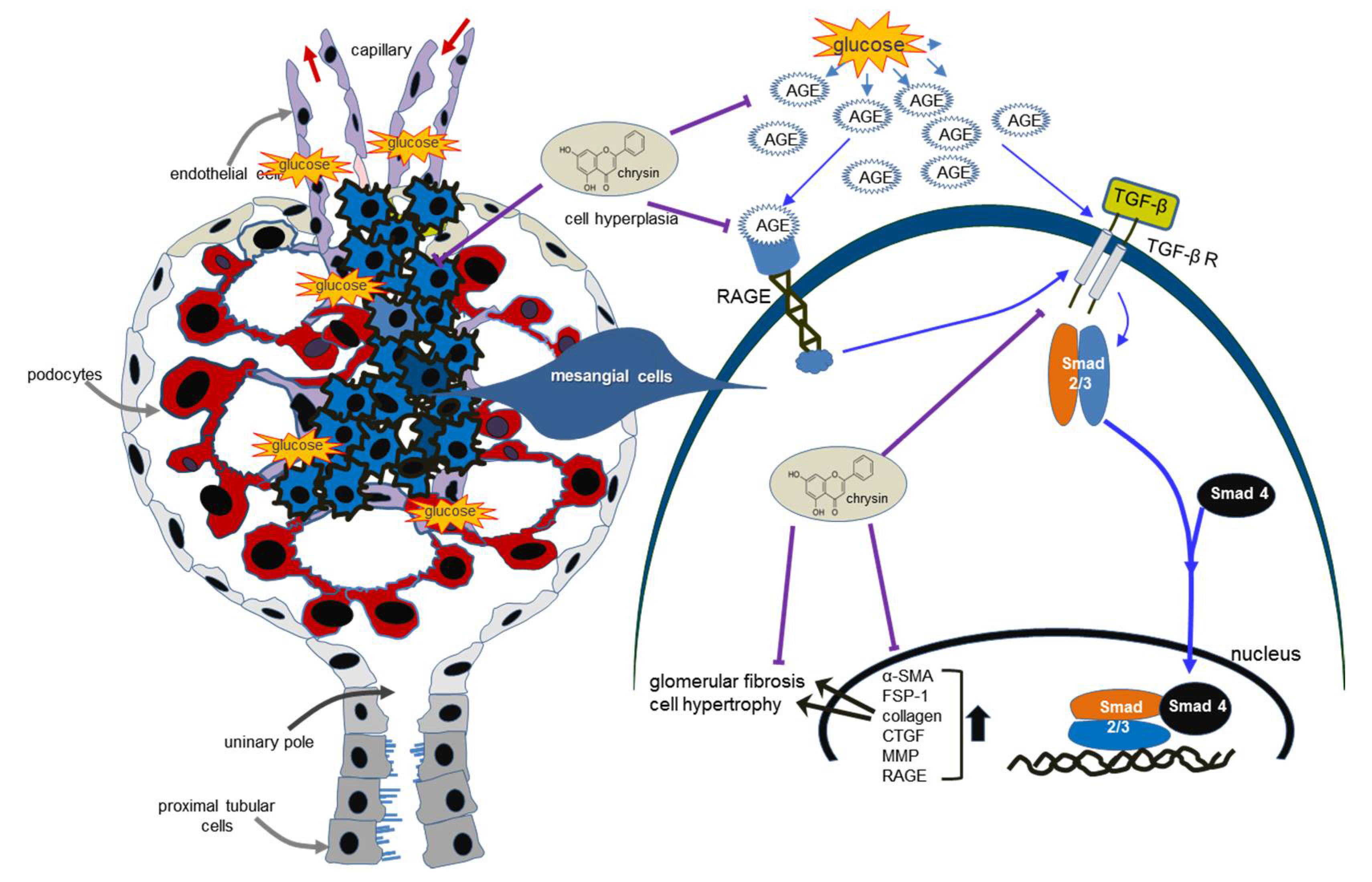
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ban, C.R.; Twigg, S.M. Fibrosis in diabetes complications: Pathogenic mechanisms and circulating and urinary markers. Vasc. Health Risk Manag. 2008, 4, 575–596. [Google Scholar] [PubMed]
- Pozzi, A.; Voziyan, P.A.; Hudson, B.G.; Zent, R. Regulation of matrix synthesis, remodeling and accumulation in glomerulosclerosis. Curr. Pharm. Des. 2009, 15, 1318–1333. [Google Scholar] [CrossRef] [PubMed]
- Min, D.; Lyons, J.G.; Bonner, J.; Twigg, S.M.; Yue, D.K.; McLennan, S.V. Mesangial cell-derived factors alter monocyte activation and function through inflammatory pathways: Possible pathogenic role in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2009, 297, F1229–F1237. [Google Scholar] [CrossRef] [PubMed]
- Fakhruddin, S.; Alanazi, W.; Jackson, K.E. Diabetes-induced reactive oxygen species: Mechanism of their generation and role in renal injury. J. Diabetes Res. 2017, 2017, 8379327. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Iturbe, B.; García García, G. The role of tubulointerstitial inflammation in the progression of chronic renal failure. Nephron Clin. Pract. 2010, 116, c81–c88. [Google Scholar] [CrossRef] [PubMed]
- Riser, B.L.; Cortes, P. Connective tissue growth factor and its regulation: A new element in diabetic glomerulosclerosis. Ren. Fail. 2001, 23, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Stockand, J.D.; Sansom, S.C. Glomerular mesangial cells: Electrophysiology and regulation of contraction. Physiol. Rev. 1998, 78, 723–744. [Google Scholar] [CrossRef] [PubMed]
- Schena, F.P.; Gesualdo, L. Pathogenetic mechanisms of diabetic nephropathy. J. Am. Soc. Nephrol. 2005, 16, S30–S33. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, M.Y.; Nam, J.S.; Kang, S.; Park, J.S.; Shin, S.; Ahn, C.W.; Kim, K.R. Hemorheological approach for early detection of chronic kidney disease and diabetic nephropathy in type 2 diabetes. Diabetes Technol. Ther. 2015, 17, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Vega, G.; Alarcón, S.; San Martín, R. The cellular and signalling alterations conducted by TGF-β contributing to renal fibrosis. Cytokine 2016, 88, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Tang, X.; Gao, P.; Guo, F.; Liu, H.; Zhao, Z.; Chen, Q.; Jiang, T.; Zhang, N.; Li, H. Sulforaphane attenuation of experimental diabetic nephropathy involves GSK-3 beta/Fyn/Nrf2 signaling pathway. J. Nutr. Biochem. 2015, 26, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Garud, M.S.; Kulkarni, Y.A. Hyperglycemia to nephropathy via transforming growth factor beta. Curr. Diabetes Rev. 2014, 10, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Sun, L.; Xiao, L.; Han, Y.; Fu, X.; Xiong, X.; Xu, X.; Liu, Y.; Yang, S.; Liu, F.; et al. Insights into the mechanisms involved in the expression and regulation of extracellular matrix proteins in diabetic nephropathy. Curr. Med. Chem. 2015, 22, 2858–2870. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, Y.S.; Wada, J.; Sun, L.; Xie, P.; Wallner, E.I.; Chen, S.; Chugh, S.; Danesh, F.R. Diabetic nephropathy: Mechanisms of renal disease progression. Exp. Biol. Med. 2008, 233, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E.A. Advanced glycosylated end products and hyperglycemia in the pathogenesis of diabetic complications. Diabetes Care 1999, 22 (Suppl. 2), B65–B71. [Google Scholar] [PubMed]
- Gruden, G.; Perin, P.C.; Camussi, G. Insight on the pathogenesis of diabetic nephropathy from the study of podocyte and mesangial cell biology. Curr. Diabetes Rev. 2005, 1, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Fukami, K.; Ueda, S.; Yamagishi, S.; Kato, S.; Inagaki, Y.; Takeuchi, M.; Motomiya, Y.; Bucala, R.; Iida, S.; Tamaki, K.; et al. AGEs activate mesangial TGF-β-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int. 2004, 66, 2137–2147. [Google Scholar] [CrossRef] [PubMed]
- Daroux, M.; Prévost, G.; Maillard-Lefebvre, H.; Gaxatte, C.; D’Agati, V.D.; Schmidt, A.M.; Boulanger, E. Advanced glycation end-products: Implications for diabetic and non-diabetic nephropathies. Diabetes Metab. 2010, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Do, M.H.; Hur, J.; Choi, J.; Kim, M.; Kim, M.J.; Kim, Y.; Ha, S.K. Eucommia ulmoides ameliorates Glucotoxicity by suppressing advanced glycation end-products in diabetic mice kidney. Nutrients 2018, 26, 10. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xiao, L.; Xiao, P.; Yang, S.; Chen, G.; Liu, F.; Kanwa, Y.S.; Sun, L. A glimpse of matrix metalloproteinases in diabetic nephropathy. Curr. Med. Chem. 2014, 21, 3244–3260. [Google Scholar] [CrossRef] [PubMed]
- McLennan, S.V.; Kelly, D.J.; Schache, M.; Waltham, M.; Dy, V.; Langham, R.G.; Yue, D.K.; Gilbert, R.E. Advanced glycation end products decrease mesangial cell MMP-7: A role in matrix accumulation in diabetic nephropathy? Kidney Int. 2007, 72, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.K.; Wang, C.C.; Lu, T.F.; Huang, K.H.; Sheu, M.L.; Liu, S.H.; Hung, K.Y. Involvement of endoplasmic reticulum stress, autophagy and apoptosis in advanced glycation end products-induced glomerular mesangial cell injury. Sci. Rep. 2016, 6, 34167. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp. Gerontol. 2011, 46, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, B.; Zhu, L.; Hao, S. A novel improved therapy strategy for diabetic nephropathy: Targeting AGEs. Organogenesis 2012, 8, 18–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasala, E.R.; Bodduluru, L.N.; Madana, R.M.; Gogoi, R.; Barua, C.C. Chemopreventive and therapeutic potential of chrysin in cancer: Mechanistic perspectives. Toxicol. Lett. 2015, 233, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Zeinali, M.; Rezaee, S.A.; Hosseinzadeh, H. An overview on immunoregulatory and anti-inflammatory properties of chrysin and flavonoids substances. Biomed. Pharmacother. 2017, 92, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Orhan, I.E.; Daglia, M.; Manayi, A.; Gortzi, O.; Nabavi, S.M. Neuroprotective effects of chrysin: From chemistry to medicine. Neurochem. Int. 2015, 90, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Park, S.H.; Choi, Y.J.; Shin, D.; Kang, Y.H. Chrysin inhibits diabetic renal tubulointerstitial fibrosis through blocking epithelial to mesenchymal transition. J. Mol. Med. 2015, 93, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Park, S.H.; Kim, Y.H.; Lee, E.J.; Antika, L.D.; Kim, D.Y.; Choi, Y.J.; Kang, Y.H. Chrysin ameliorates podocyte injury and slit diaphragm protein loss via inhibition of the PERK-eIF2α-ATF-CHOP pathway in diabetic mice. Acta Pharmacol. Sin. 2017, 38, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Park, S.H.; Kim, Y.H.; Lee, E.J.; Antika, L.D.; Kim, D.Y.; Choi, Y.J.; Kang, Y.H. Dietary compound chrysin inhibits retinal neovascularization with abnormal capillaries in db/db mice. Nutrients 2016, 8, 782. [Google Scholar] [CrossRef] [PubMed]
- Brigstock, D.R. Connective tissue growth factor (CCN2, CTGF) and organ fibrosis: Lessons from transgenic animals. J. Cell Commun. Signal. 2010, 4, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, N.; Tahira, Y.; Matsuda, H.; Matsumoto, K. Transforming growth factor-β as a treatment target in renal diseases. J. Nephrol. 2009, 22, 708–715. [Google Scholar] [PubMed]
- Muskiet, M.H.; Smits, M.M.; Morsink, L.M.; Diamant, M. The gut-renal axis: Do incretin-based agents confer renoprotection in diabetes? Nat. Rev. Nephrol. 2014, 10, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Ceranowicz, P.; Warzecha, Z.; Dembinski, A. Peptidyl hormones of endocrine cells origin in the gut-their discovery and physiological relevance. J. Physiol. Pharmacol. 2015, 66, 11–27. [Google Scholar] [PubMed]
- Tahara, A.; Tsukada, J.; Tomura, Y.; Yatsu, T.; Shibasaki, M. Effects of high glucose on AVP-induced hyperplasia, hypertrophy and type IV collagen synthesis in cultured rat mesangial cells. Endocr. Res. 2012, 37, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Matsubara, T.; Arai, H.; Doi, T. Role of Smad1 in diabetic nephropathy: Molecular mechanisms and implications as a diagnostic marker. Histol. Histopathol. 2011, 26, 531–541. [Google Scholar] [PubMed]
- Li, J.; Kang, M.K.; Kim, J.K.; Kim, J.L.; Kang, S.W.; Lim, S.S.; Kang, Y.H. Purple corn anthocyanins retard diabetes-associated glomerulosclerosis in mesangial cells and db/db mice. Eur. J. Nutr. 2012, 51, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Wolf, G. Transforming growth factor-β and the progression of renal disease. Nephrol. Dial. Transplant. 2014, 29, i37–i45. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.M.; Qi, W.; Pollock, C.A. CTGF and chronic kidney fibrosis. Front. Biosci. 2009, 1, 132–141. [Google Scholar] [CrossRef]
- Ahad, A.; Ganai, A.A.; Mujeeb, M.; Siddiqui, W.A. Chrysin, an anti-inflammatory molecule, abrogates renal dysfunction in type 2 diabetic rats. Toxicol. Appl. Pharmacol. 2014, 279, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Chaudhari, B.P.; Kakkar, P. Baicalin and chrysin mixture imparts cyto-protection against methylglyoxal induced cytotoxicity and diabetic tubular injury by modulating RAGE, oxidative stress and inflammation. Environ. Toxicol. Pharmacol. 2017, 50, 67–75. [Google Scholar] [CrossRef] [PubMed]
- El-Bassossy, H.M.; Abo-Warda, S.M.; Fahmy, A. Chrysin and luteolin attenuate diabetes-induced impairment in endothelial-dependent relaxation: Effect on lipid profile, AGEs and NO generation. Phytother. Res. 2013, 27, 1678–1684. [Google Scholar] [CrossRef] [PubMed]
- Ali, B.H.; Al Za’abi, M.; Adham, S.A.; Yasin, J.; Nemmar, A.; Schupp, N. Therapeutic Effect of Chrysin on Adenine-Induced Chronic Kidney Disease in Rats. Cell. Physiol. Biochem. 2016, 38, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Cheunsuchon, B.; Donnert, E.; Ma, L.J.; Thomas, J.W.; Neilson, E.G.; Fogo, A.B. Immunolocalization of fibroblast growth factor-1 (FGF-1), its receptor (FGFR-1) and fibroblast-specific protein-1 (FSP-1) in inflammatory renal disease. Kidney Int. 2005, 68, 2621–2628. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Guerrero, C.; Hernández-Vargas, P.; López-Franco, O.; Ortiz-Muñoz, G.; Egido, J. Mesangial cells and glomerular inflammation: From the pathogenesis to novel therapeutic approaches. Curr. Drug Targets Inflamm. Allergy 2005, 4, 341–351. [Google Scholar]
- McLennan, S.V.; Martell, S.K.; Yue, D.K. Effects of mesangium glycation on matrix metalloproteinase activities: Possible role in diabetic nephropathy. Diabetes 2002, 51, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Sporek, M.; Dumnicka, P.; Gala-Bladzinska, A.; Ceranowicz, P.; Warzecha, Z.; Dembinski, A.; Stepien, E.; Walocha, J.; Drozdz, R.; Kuzniewski, M.; et al. Angiopoietin-2 Is an early indicator of acute pancreatic-renal syndrome in patients with acute pancreatitis. Mediat. Inflamm. 2016, 2016, 5780903. [Google Scholar] [CrossRef] [PubMed]
- Sporek, M.; Gala-Błądzińska, A.; Dumnicka, P.; Mazur-Laskowska, M.; Kielczewski, S.; Walocha, J.; Ceranowicz, P.; Kuźniewski, M.; Mituś, J.; Kuśnierz-Cabala, B. Urine NGAL is useful in the clinical evaluation of renal function in the early course of acute pancreatitis. Folia Med. Cracov. 2016, 56, 13–25. [Google Scholar] [PubMed]
- Żyłka, A.; Gala-Błądzińska, A.; Dumnicka, P.; Ceranowicz, P.; Kuźniewski, M.; Gil, K.; Olszanecki, R.; Kuśnierz-Cabala, B. Is Urinary NGAL determination useful for monitoring kidney function and assessment of cardiovascular disease? A 12-month observation of patients with type 2 diabetes. Dis. Markers 2016, 2016, 8489543. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.-J.; Kang, M.-K.; Kim, D.Y.; Kim, Y.-H.; Oh, H.; Kang, Y.-H. Chrysin Inhibits Advanced Glycation End Products-Induced Kidney Fibrosis in Renal Mesangial Cells and Diabetic Kidneys. Nutrients 2018, 10, 882. https://doi.org/10.3390/nu10070882
Lee E-J, Kang M-K, Kim DY, Kim Y-H, Oh H, Kang Y-H. Chrysin Inhibits Advanced Glycation End Products-Induced Kidney Fibrosis in Renal Mesangial Cells and Diabetic Kidneys. Nutrients. 2018; 10(7):882. https://doi.org/10.3390/nu10070882
Chicago/Turabian StyleLee, Eun-Jung, Min-Kyung Kang, Dong Yeon Kim, Yun-Ho Kim, Hyeongjoo Oh, and Young-Hee Kang. 2018. "Chrysin Inhibits Advanced Glycation End Products-Induced Kidney Fibrosis in Renal Mesangial Cells and Diabetic Kidneys" Nutrients 10, no. 7: 882. https://doi.org/10.3390/nu10070882