Low Phytanic Acid-Concentrated DHA Prevents Cognitive Deficit and Regulates Alzheimer Disease Mediators in an ApoE−/− Mice Experimental Model


 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Experimental Design and Animal Model
2.3. Behavioral Studies
2.3.1. Locomotor Activity
2.3.2. Morris Water Maze
2.4. Serum Analysis
2.5. Western Blot Analysis
2.5.1. Isolation of Total Proteins
2.5.2. Western Blot Analysis
2.6. Statistical Analysis
3. Results
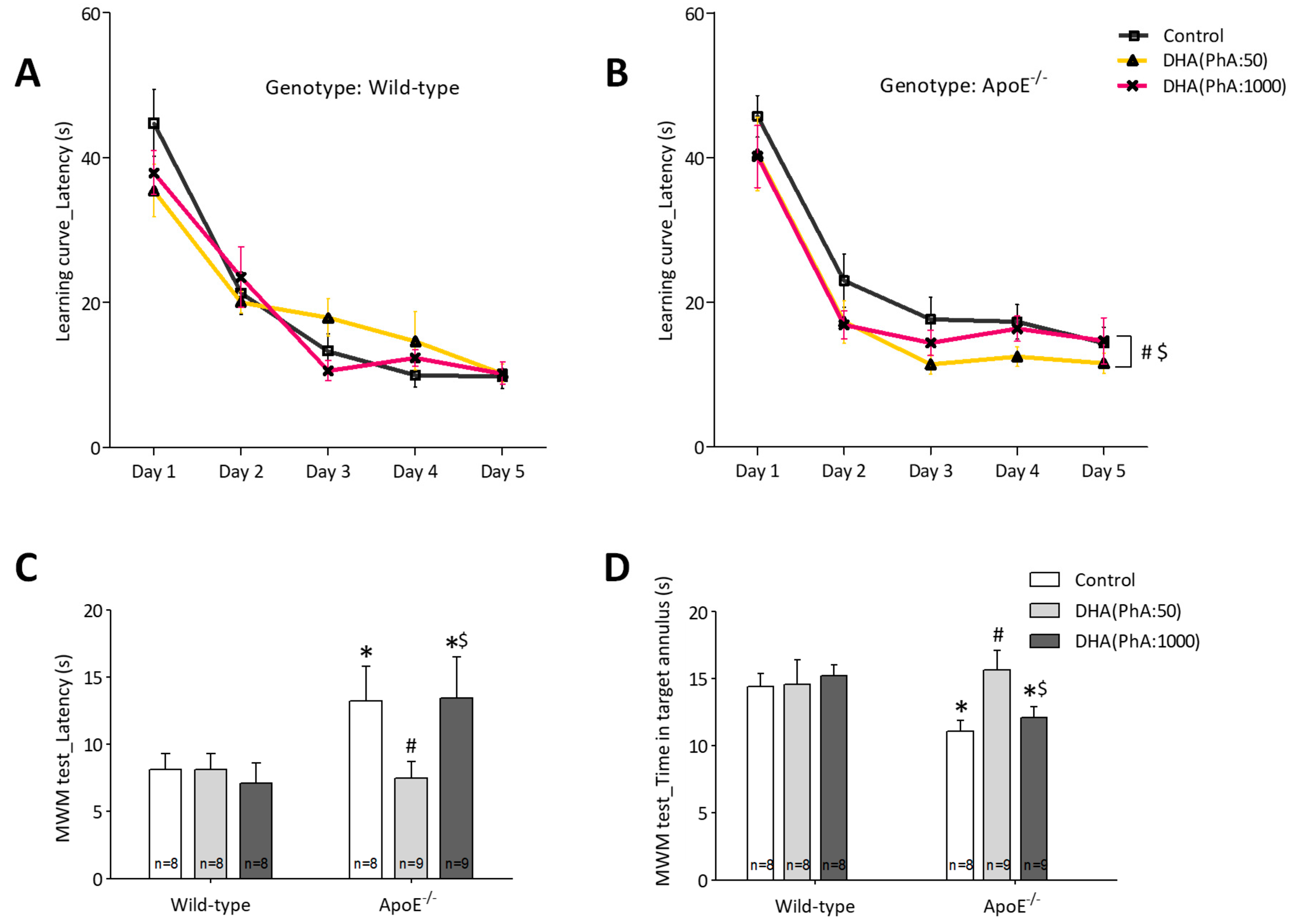
3.1. DHA (PhA:50) Improved Spatial Memory in ApoE−/− Mice Compared to DHA (PhA:1000)
3.1.1. Locomotor Activity
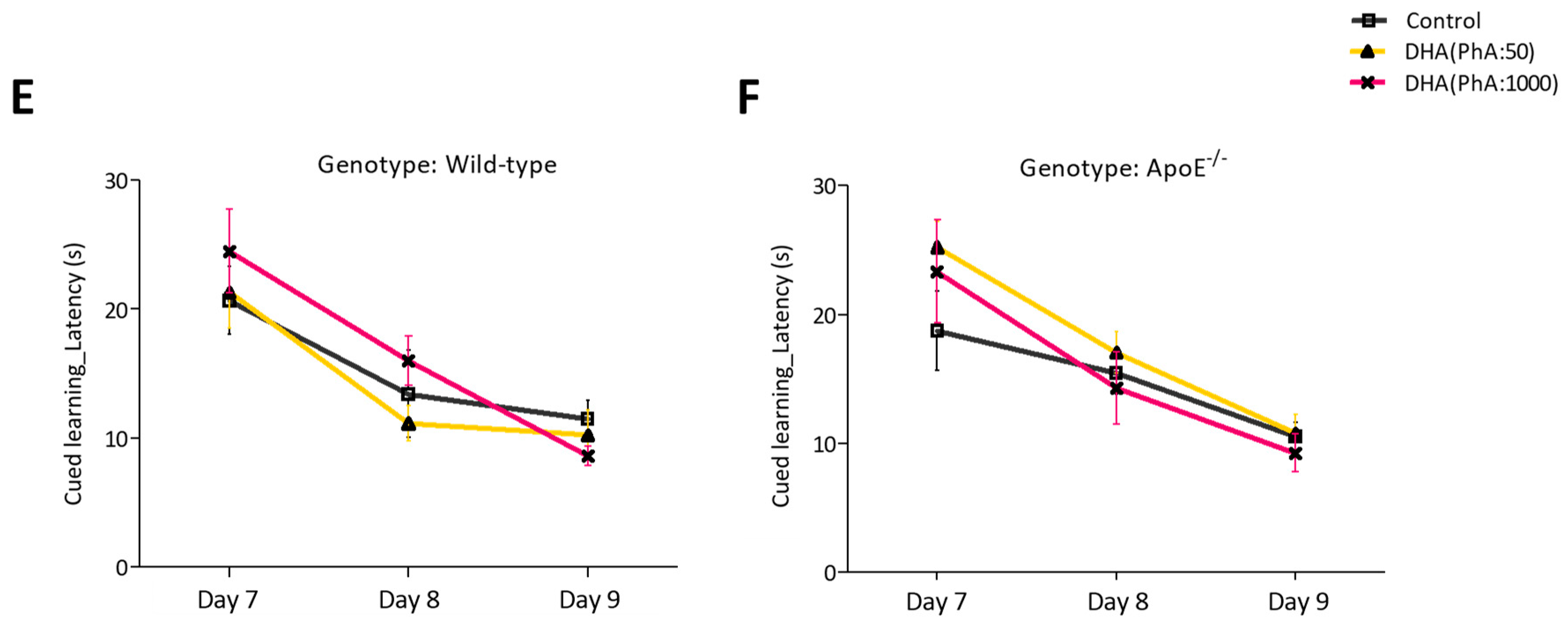
3.1.2. Learning and Memory
3.2. Effects of DHA (PhA:50) on Body Weight and Lipid Profile of ApoE−/− Mice Compared to DHA (PhA:1000)
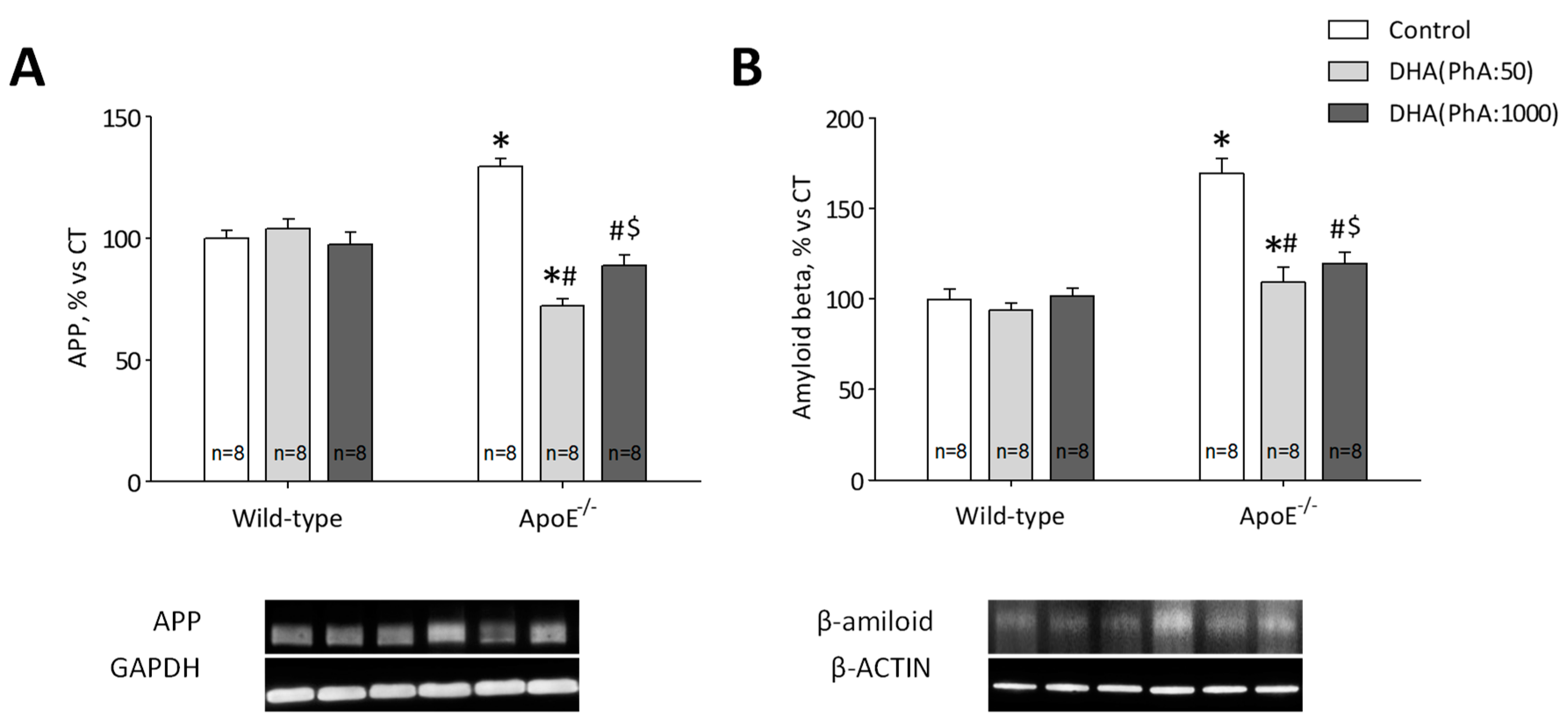
3.3. DHA (PhA:50) Decreased Hippocampal Protein Expression of APP and Aß Compared to DHA (PhA:1000) in ApoE−/− Mice
3.4. DHA (PhA:50) Decreases Tau Hyperphosphorylation in the Hippocampus of ApoE−/− Mice
3.5. DHA (PhA:50) Increases Hippocampal BDNF Protein Expression Compared to DHA (PhA:1000) in ApoE−/− Mice
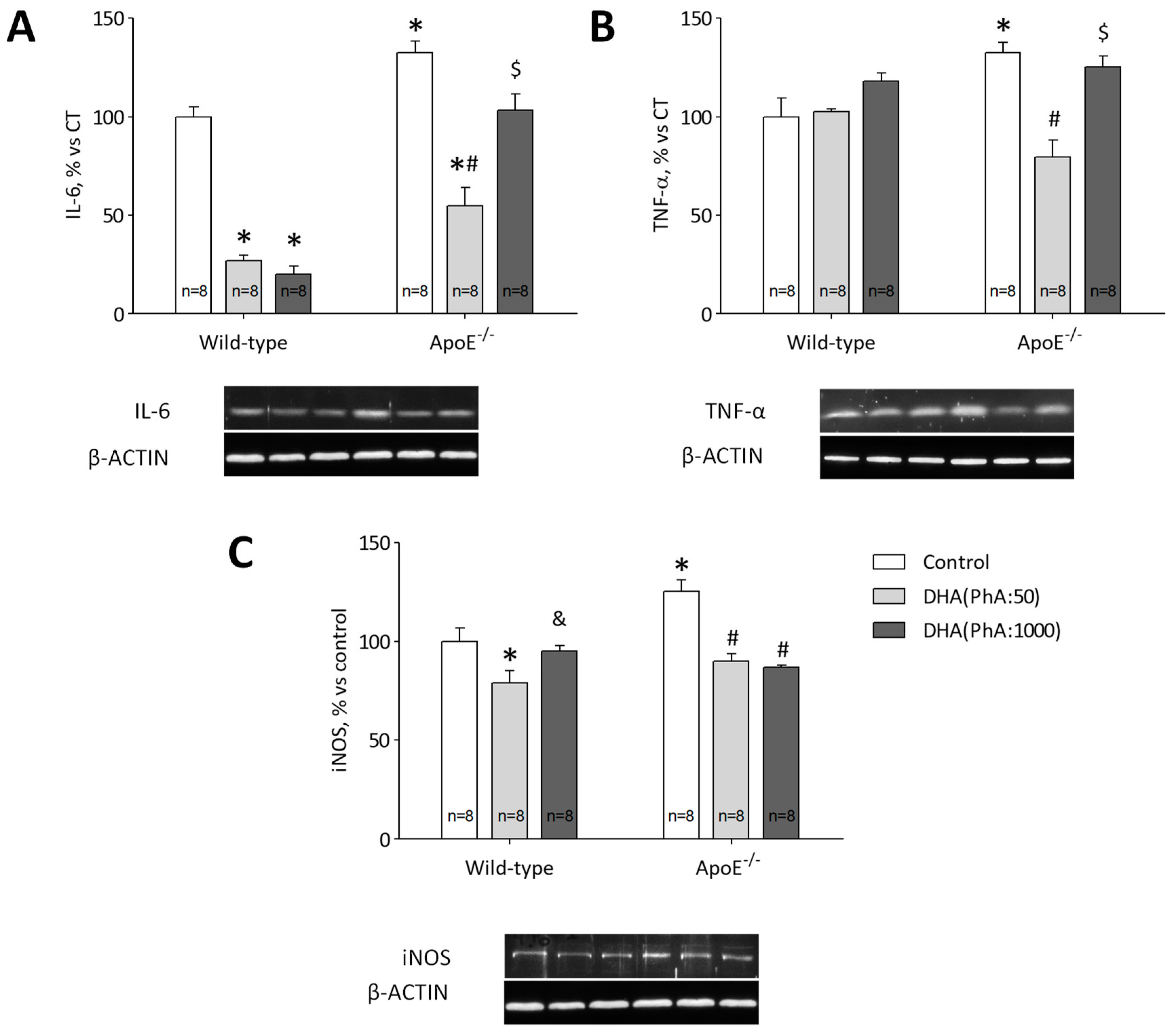
3.6. DHA (PhA:50) Exerts Anti-Inflammatory Effect on Hippocampal ApoE−/− Mice Compared to DHA (PhA:1000)
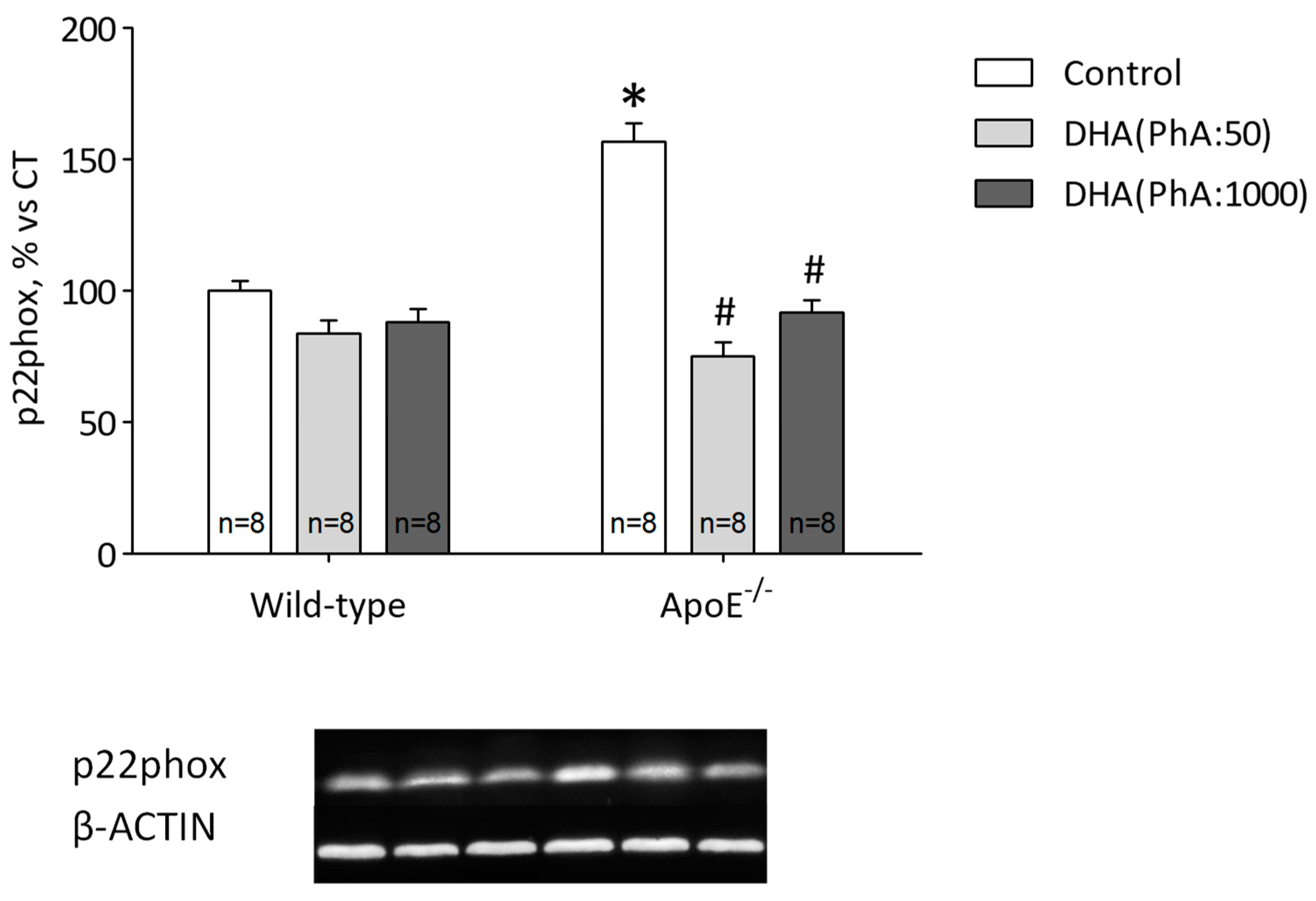
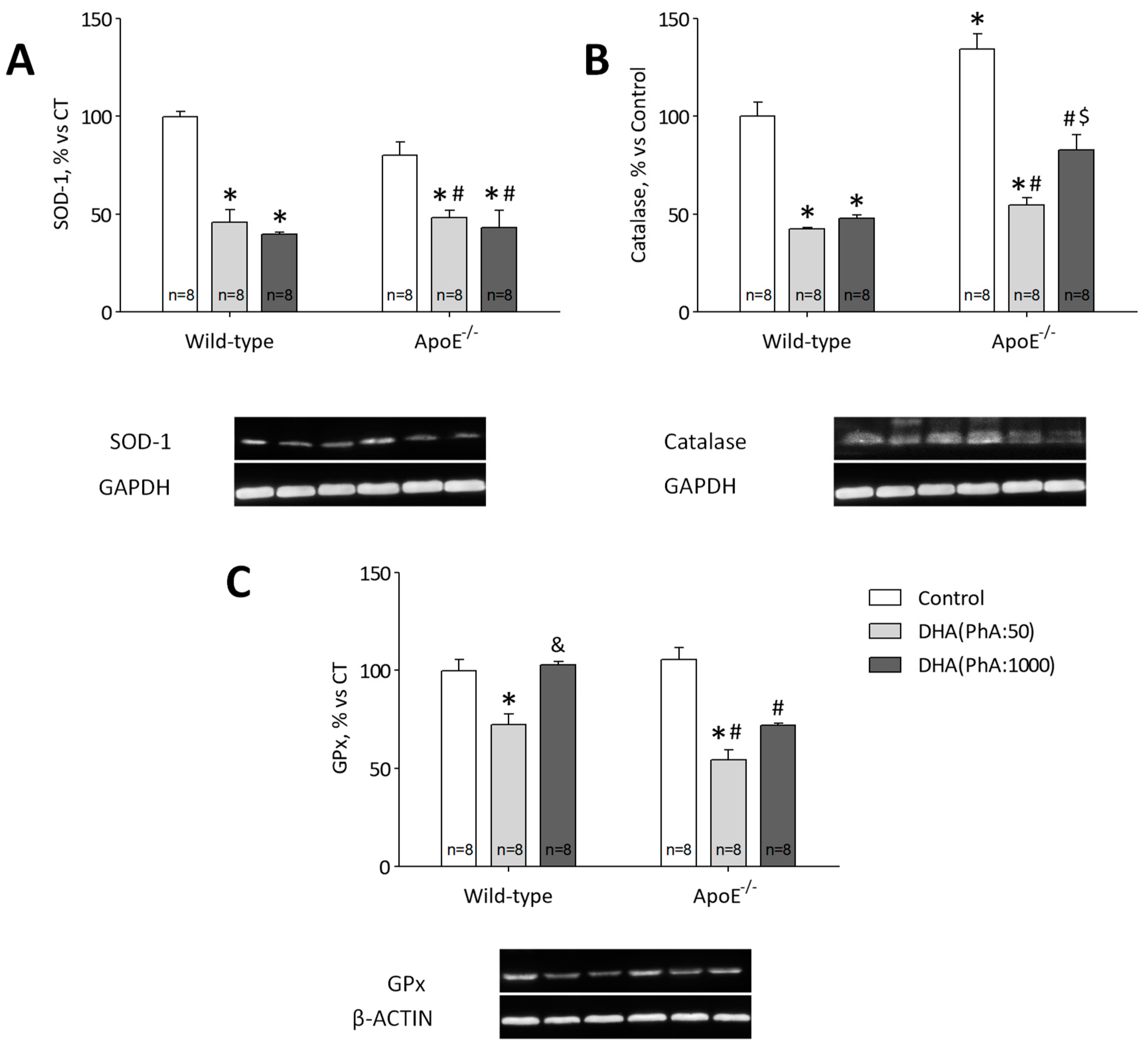
3.7. DHA (PhA:50) Decreases Hippocampal p22phox Expression and Antioxidant Response Compared to DHA (PhA:1000) in ApoE−/− Mice
3.8. DHA (PhA:50) Exerts Anti-Apoptotic Effect on Hippocampal ApoE−/− Mice Compared to DHA (PhA:1000)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A. Synapse loss in the temporal lobe in Alzheimer’s disease. Ann. Neurol. 1993, 33, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Isla, T.; Price, J.L.; McKeel, D.W., Jr.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Moolman, D.L.; Vitolo, O.V.; Vonsattel, J.P.; Shelanski, M.L. Dendrite and dendritic spine alterations in Alzheimer models. J. Neurocytol. 2004, 33, 377–387. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Grimm, M.O.; Michaelson, D.; Hartmann, T. Omega-3 fatty acids, lipids and apoE lipidation in Alzheimer’s disease: A rationale for multi-nutrient dementia prevention. J. Lipid Res. 2017, 58, 2083–2101. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B.; et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef]
- Koo, E.H.; Squazzo, S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994, 269, 17386–17389. [Google Scholar]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef]
- Sanabria-Castro, A.; Alvarado-Echeverria, I.; Monge-Bonilla, C. Molecular Pathogenesis of Alzheimer’s Disease: An Update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Giese, K.P. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Mol. Brain 2015, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Meraz-Rios, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernandez, J.; Campos-Pena, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharm. 2014, 88, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Henn, A.; Lund, S.; Hedtjarn, M.; Schrattenholz, A.; Porzgen, P.; Leist, M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 2009, 26, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Ginty, M.; Kemp, K.; Scolding, N.; Wilkins, A. Peroxisome proliferator-activated receptor-alpha agonists protect cortical neurons from inflammatory mediators and improve peroxisomal function. Eur. J. Neurosci. 2011, 33, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Glomset, J.A. Role of docosahexaenoic acid in neuronal plasma membranes. Sci. STKE 2006, 2006, pe6. [Google Scholar] [CrossRef] [PubMed]
- Galland, L. Diet and inflammation. Nutr. Clin. Pract. 2010, 25, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Anti-inflammatory Agents: Present and Future. Cell 2010, 140, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.C.; Cook-Johnson, R.J.; James, M.J.; Muhlhausler, B.S.; Stone, D.A.; Gibson, R.A. Barramundi (Lates calcarifer) desaturase with Delta6/Delta8 dual activities. Biotechnol. Lett. 2012, 34, 1283–1296. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Akbar, M.; Kim, Y.S. Phosphatidylserine-dependent neuroprotective signaling promoted by docosahexaenoic acid. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Kevala, K.; Kim, J.; Moon, H.S.; Jun, S.B.; Lovinger, D.; Kim, H.Y. Docosahexaenoic acid promotes hippocampal neuronal development and synaptic function. J. Neurochem. 2009, 111, 510–521. [Google Scholar] [CrossRef]
- Phillips, M.A.; Childs, C.E.; Calder, P.C.; Rogers, P.J. Lower omega-3 fatty acid intake and status are associated with poorer cognitive function in older age: A comparison of individuals with and without cognitive impairment and Alzheimer’s disease. Nutr. Neurosci. 2012, 15, 271–277. [Google Scholar] [CrossRef]
- Oster, T.; Pillot, T. Docosahexaenoic acid and synaptic protection in Alzheimer’s disease mice. Biochim. Biophys. Acta 2010, 1801, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Casali, B.T.; Corona, A.W.; Mariani, M.M.; Karlo, J.C.; Ghosal, K.; Landreth, G.E. Omega-3 Fatty Acids Augment the Actions of Nuclear Receptor Agonists in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 9173–9181. [Google Scholar] [CrossRef]
- Chouinard-Watkins, R.; Chen, C.T.; Metherel, A.H.; Lacombe, R.J.S.; Thies, F.; Masoodi, M.; Bazinet, R.P. Phospholipid class-specific brain enrichment in response to lysophosphatidylcholine docosahexaenoic acid infusion. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1092–1098. [Google Scholar] [CrossRef]
- McNamara, R.K. DHA deficiency and prefrontal cortex neuropathology in recurrent affective disorders. J. Nutr. 2010, 140, 864–868. [Google Scholar] [CrossRef]
- Yurko-Mauro, K.; Alexander, D.D.; Van Elswyk, M.E. Docosahexaenoic acid and adult memory: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0120391. [Google Scholar] [CrossRef] [PubMed]
- Samieri, C.; Feart, C.; Proust-Lima, C.; Peuchant, E.; Dartigues, J.F.; Amieva, H.; Barberger-Gateau, P. omega-3 fatty acids and cognitive decline: Modulation by ApoEepsilon4 allele and depression. Neurobiol. Aging 2011, 32, 2317.e2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Cederholm, T.; Salem, N., Jr.; Palmblad, J. omega-3 fatty acids in the prevention of cognitive decline in humans. Adv. Nutr. 2013, 4, 672–676. [Google Scholar] [CrossRef]
- Cole, G.M.; Ma, Q.L.; Frautschy, S.A. Omega-3 fatty acids and dementia. Prostaglandins Leukot. Essent. Fatty Acids 2009, 81, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Roso, M.B.; Olivares-Alvaro, E.; Quintela, J.C.; Ballesteros, S.; Espinosa-Parrilla, J.F.; Ruiz-Roso, B.; Lahera, V.; de Las Heras, N.; Martin-Fernandez, B. Effects of Low Phytanic Acid-Concentrated DHA on Activated Microglial Cells: Comparison with a Standard Phytanic Acid-Concentrated DHA. Neuromol. Med. 2018, 20, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, P.; Reiser, G. Brain Lipotoxicity of Phytanic Acid and Very Long-chain Fatty Acids. Harmful Cellular/Mitochondrial Activities in Refsum Disease and X-Linked Adrenoleukodystrophy. Aging Dis. 2016, 7, 136–149. [Google Scholar] [CrossRef]
- Ignatius, M.J.; Gebicke-Harter, P.J.; Skene, J.H.; Schilling, J.W.; Weisgraber, K.H.; Mahley, R.W.; Shooter, E.M. Expression of apolipoprotein E during nerve degeneration and regeneration. Proc. Natl. Acad. Sci. USA 1986, 83, 1125–1129. [Google Scholar] [CrossRef]
- Elshourbagy, N.A.; Liao, W.S.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc. Natl. Acad. Sci. USA 1985, 82, 203–207. [Google Scholar] [CrossRef]
- Sato, N.; Morishita, R. The roles of lipid and glucose metabolism in modulation of beta-amyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front. Aging Neurosci. 2015, 7, 199. [Google Scholar] [CrossRef]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Kivipelto, M.; Helkala, E.L.; Laakso, M.P.; Hanninen, T.; Hallikainen, M.; Alhainen, K.; Soininen, H.; Tuomilehto, J.; Nissinen, A. Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study. BMJ 2001, 322, 1447–1451. [Google Scholar] [CrossRef]
- Plump, A.S.; Smith, J.D.; Hayek, T.; Aalto-Setala, K.; Walsh, A.; Verstuyft, J.G.; Rubin, E.M.; Breslow, J.L. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 1992, 71, 343–353. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Li, Y.; Ng, C.T.; Song, Y.Q. Inflammation in Alzheimer’s Disease and Molecular Genetics: Recent Update. Arch. Immunol. Ther. Exp. 2015, 63, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Anstey, K.J.; Lipnicki, D.M.; Low, L.F. Cholesterol as a risk factor for dementia and cognitive decline: A systematic review of prospective studies with meta-analysis. Am. J. Geriatr. Psychiatry 2008, 16, 343–354. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Echeverry-Alzate, V.; Gine, E.; Buhler, K.M.; Calleja-Conde, J.; Olmos, P.; Gorriti, M.A.; Nadal, R.; Rodriguez de Fonseca, F.; Lopez-Moreno, J.A. Effects of topiramate on ethanol-cocaine interactions and DNA methyltransferase gene expression in the rat prefrontal cortex. Br. J. Pharmacol. 2014, 171, 3023–3036. [Google Scholar] [CrossRef]
- Gine, E.; Echeverry-Alzate, V.; Lopez-Moreno, J.A.; Rodriguez de Fonseca, F.; Perez-Castillo, A.; Santos, A. The CB1 receptor is required for the establishment of the hyperlocomotor phenotype in developmentally-induced hypothyroidism in mice. Neuropharmacology 2017, 116, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Gine, E.; Echeverry-Alzate, V.; Lopez-Moreno, J.A.; Lopez-Jimenez, A.; Torres-Romero, D.; Perez-Castillo, A.; Santos, A. Developmentally-induced hypothyroidism alters the expression of Egr-1 and Arc genes and the sensitivity to cannabinoid agonists in the hippocampus. Possible implications for memory and learning. Mol. Cell Endocrinol. 2013, 365, 119–128. [Google Scholar] [CrossRef]
- Morales-Garcia, J.A.; Echeverry-Alzate, V.; Alonso-Gil, S.; Sanz-SanCristobal, M.; Lopez-Moreno, J.A.; Gil, C.; Martinez, A.; Santos, A.; Perez-Castillo, A. Phosphodiesterase7 Inhibition Activates Adult Neurogenesis in Hippocampus and Subventricular Zone In Vitro and In Vivo. Stem. Cells 2017, 35, 458–472. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef]
- Grootendorst, J.; de Kloet, E.R.; Dalm, S.; Oitzl, M.S. Reversal of cognitive deficit of apolipoprotein E knockout mice after repeated exposure to a common environmental experience. Neuroscience 2001, 108, 237–247. [Google Scholar] [CrossRef]
- Zhou, L.; Gao, Q.; Nie, M.; Gu, J.L.; Hao, W.; Wang, L.; Cao, J.M. Degeneration and energy shortage in the suprachiasmatic nucleus underlies the circadian rhythm disturbance in ApoE−/− mice: Implications for Alzheimer’s disease. Sci. Rep. 2016, 6, 36335. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Belayev, L.; Khoutorova, L.; Obenaus, A.; Bazan, N.G. Docosahexaenoic acid confers enduring neuroprotection in experimental stroke. J. Neurol. Sci. 2014, 338, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Ozsoy, O.; Tanriover, G.; Derin, N.; Uysal, N.; Demir, N.; Gemici, B.; Kencebay, C.; Yargicoglu, P.; Agar, A.; Aslan, M. The effect of docosahexaenoic Acid on visual evoked potentials in a mouse model of Parkinson’s disease: The role of cyclooxygenase-2 and nuclear factor kappa-B. Neurotox Res. 2011, 20, 250–262. [Google Scholar] [CrossRef]
- Kuratko, C.N.; Barrett, E.C.; Nelson, E.B.; Salem, N., Jr. The relationship of docosahexaenoic acid (DHA) with learning and behavior in healthy children: A review. Nutrients 2013, 5, 2777–2810. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.R.; Jin, X.; Anzinger, J.J.; Xu, Q.; Purushothaman, S.; Fessler, M.B.; Addadi, L.; Kruth, H.S. ABCG1-mediated generation of extracellular cholesterol microdomains. J. Lipid Res. 2014, 55, 115–127. [Google Scholar] [CrossRef]
- Janssen, C.I.; Zerbi, V.; Mutsaers, M.P.; de Jong, B.S.; Wiesmann, M.; Arnoldussen, I.A.; Geenen, B.; Heerschap, A.; Muskiet, F.A.; Jouni, Z.E.; et al. Impact of dietary n-3 polyunsaturated fatty acids on cognition, motor skills and hippocampal neurogenesis in developing C57BL/6J mice. J. Nutr. Biochem. 2015, 26, 24–35. [Google Scholar] [CrossRef]
- Bouwstra, H.; Dijck-Brouwer, D.J.; Decsi, T.; Boehm, G.; Boersma, E.R.; Muskiet, F.A.; Hadders-Algra, M. Relationship between umbilical cord essential fatty acid content and the quality of general movements of healthy term infants at 3 months. Pediatr. Res. 2006, 59, 717–722. [Google Scholar] [CrossRef]
- Fewtrell, M.S.; Morley, R.; Abbott, R.A.; Singhal, A.; Isaacs, E.B.; Stephenson, T.; MacFadyen, U.; Lucas, A. Double-blind, randomized trial of long-chain polyunsaturated fatty acid supplementation in formula fed to preterm infants. Pediatrics 2002, 110, 73–82. [Google Scholar] [CrossRef]
- Linnarsson, S.; Bjorklund, A.; Ernfors, P. Learning deficit in BDNF mutant mice. Eur. J. Neurosci. 1997, 9, 2581–2587. [Google Scholar] [CrossRef]
- Molteni, E.; Butti, M.; Bianchi, A.M.; Reni, G. Activation of the prefrontal cortex during a visual n-back working memory task with varying memory load: A near infrared spectroscopy study. In Proceedings of the 2008 30th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Vancouver, BC, Canada, 20–25 August 2008; pp. 4024–4027. [Google Scholar]
- Jiang, L.H.; Shi, Y.; Wang, L.S.; Yang, Z.R. The influence of orally administered docosahexaenoic acid on cognitive ability in aged mice. J. Nutr. Biochem. 2009, 20, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J. Neurotrauma 2004, 21, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Ertley, R.N.; Lee, H.J.; DeMar, J.C., Jr.; Arnold, J.T.; Rapoport, S.I.; Bazinet, R.P. n-3 polyunsaturated fatty acid deprivation in rats decreases frontal cortex BDNF via a p38 MAPK-dependent mechanism. Mol. Psychiatry 2007, 12, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Butchart, J.; Holmes, C. Systemic and central immunity in Alzheimer’s disease: Therapeutic implications. CNS Neurosci. Ther. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Salem, N., Jr.; Vandal, M.; Calon, F. The benefit of docosahexaenoic acid for the adult brain in aging and dementia. Prostaglandins Leukot. Essent. Fatty Acids 2015, 92, 15–22. [Google Scholar] [CrossRef]
- Ronicke, S.; Kruska, N.; Kahlert, S.; Reiser, G. The influence of the branched-chain fatty acids pristanic acid and Refsum disease-associated phytanic acid on mitochondrial functions and calcium regulation of hippocampal neurons, astrocytes, and oligodendrocytes. Neurobiol. Dis. 2009, 36, 401–410. [Google Scholar] [CrossRef]
- Salemme, A.; Togna, A.R.; Mastrofrancesco, A.; Cammisotto, V.; Ottaviani, M.; Bianco, A.; Venditti, A. Anti-inflammatory effects and antioxidant activity of dihydroasparagusic acid in lipopolysaccharide-activated microglial cells. Brain Res. Bull. 2016, 120, 151–158. [Google Scholar] [CrossRef]
- He, X.; Li, Z.; Rizak, J.D.; Wu, S.; Wang, Z.; He, R.; Su, M.; Qin, D.; Wang, J.; Hu, X. Resveratrol Attenuates Formaldehyde Induced Hyperphosphorylation of Tau Protein and Cytotoxicity in N2a Cells. Front. Neurosci. 2016, 10, 598. [Google Scholar] [CrossRef] [PubMed]
- Jhang, K.A.; Park, J.S.; Kim, H.S.; Chong, Y.H. Resveratrol Ameliorates Tau Hyperphosphorylation at Ser396 Site and Oxidative Damage in Rat Hippocampal Slices Exposed to Vanadate: Implication of ERK1/2 and GSK-3beta Signaling Cascades. J. Agric. Food Chem. 2017, 65, 9626–9634. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Kuchenbecker, J.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Kaneko, K.; Okuno, M.; Wada, K.; Kashiyama, T.; Shimizu, H.; Akagi, T.; Hashikawa, T.; Nukina, N. Membrane microdomain switching: A regulatory mechanism of amyloid precursor protein processing. J. Cell Biol. 2008, 183, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Bolanos, J.P. Nitric oxide, cell bioenergetics and neurodegeneration. J. Neurochem. 2006, 97, 1676–1689. [Google Scholar] [CrossRef] [PubMed]
- De Jong, E.K.; de Haas, A.H.; Brouwer, N.; van Weering, H.R.; Hensens, M.; Bechmann, I.; Pratley, P.; Wesseling, E.; Boddeke, H.W.; Biber, K. Expression of CXCL4 in microglia in vitro and in vivo and its possible signaling through CXCR3. J. Neurochem. 2008, 105, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta 2010, 1797, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. The salutary effects of DHA dietary supplementation on cognition, neuroplasticity, and membrane homeostasis after brain trauma. J. Neurotrauma 2011, 28, 2113–2122. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, S.; Matthes, F.; Posey, K.; Kickstein, E.; Weber, S.; Hettich, M.M.; Pfurtscheller, S.; Ehninger, D.; Schneider, R.; Krauss, S. Resveratrol induces dephosphorylation of Tau by interfering with the MID1-PP2A complex. Sci. Rep. 2017, 7, 13753. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, S.; Schonfeld, P.; Reiser, G. The Refsum disease marker phytanic acid, a branched chain fatty acid, affects Ca2+ homeostasis and mitochondria, and reduces cell viability in rat hippocampal astrocytes. Neurobiol. Dis. 2005, 18, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Budd, S.L.; Castilho, R.F.; Nicholls, D.G. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 1997, 415, 21–24. [Google Scholar] [CrossRef]
- Desagher, S.; Martinou, J.C. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000, 10, 369–377. [Google Scholar] [CrossRef]
- Pan, H.C.; Kao, T.K.; Ou, Y.C.; Yang, D.Y.; Yen, Y.J.; Wang, C.C.; Chuang, Y.H.; Liao, S.L.; Raung, S.L.; Wu, C.W.; et al. Protective effect of docosahexaenoic acid against brain injury in ischemic rats. J. Nutr. Biochem. 2009, 20, 715–725. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Pedraza-Chaverri, J.; Perez-Rojas, J.M. Role of docosahexaenoic acid in the modulation of glial cells in Alzheimer’s disease. J. Neuroinflamm. 2016, 13, 61. [Google Scholar] [CrossRef]
- Chang, C.Y.; Kuan, Y.H.; Li, J.R.; Chen, W.Y.; Ou, Y.C.; Pan, H.C.; Liao, S.L.; Raung, S.L.; Chang, C.J.; Chen, C.J. Docosahexaenoic acid reduces cellular inflammatory response following permanent focal cerebral ischemia in rats. J. Nutr. Biochem. 2013, 24, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.A.; Neumann, M.A.; Lien, E.L.; Boyd, K.A.; Tu, W.C. Docosahexaenoic acid synthesis from alpha-linolenic acid is inhibited by diets high in polyunsaturated fatty acids. Prostaglandins Leukot. Essent. Fatty Acids 2013, 88, 139–146. [Google Scholar] [CrossRef]
- Lauritzen, L.; Hansen, H.S.; Jorgensen, M.H.; Michaelsen, K.F. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog. Lipid Res. 2001, 40, 1–94. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (mg/dL) | Control | DHA (PhA:50) | DHA (PhA:1000) | ApoE−/− | ApoE−/− + DHA (PhA:50) | ApoE−/− + DHA (PhA:1000) |
|---|---|---|---|---|---|---|
| Total cholesterol | 138.7 ± 22.27 | 85.33 ± 3.42 * | 77.25 ± 5.42 * | 570.4 ± 7.90 *** | 473.4 ± 23.33 ***### | 502.6 ± 15.74 ***### |
| LDL-c | 54.40 ± 16.82 | 29.40 ± 11.24 | 36.77 ± 14.28 | 361.0 ± 62.35 *** | 314.3 ± 48.40 ***### | 304.9 ± 48.62 ***### |
| Control | DHA (PhA:50) | DHA (PhA:1000) | ApoE−/− | ApoE−/− + DHA (PhA:50) | ApoE−/− + DHA (PhA:1000) | |
|---|---|---|---|---|---|---|
| Body weight (g) | 30.11 ± 0.92 | 29.58 ± 0.53 | 27.81 ± 0.52 | 30.48 ± 1.73 | 28.58 ± 0.45 | 29.32 ± 0.76 |
| Increased Body weight (g) | 10.15 ± 0.88 | 11.11 ± 0.50 | 8.71 ± 0.51 | 10.02 ± 0.76 | 8.45 ± 0.66 | 9.01 ± 0.96 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Roso, M.B.; Echeverry-Alzate, V.; Ruiz-Roso, B.; Quintela, J.C.; Ballesteros, S.; Lahera, V.; De las Heras, N.; López-Moreno, J.A.; Martín-Fernández, B. Low Phytanic Acid-Concentrated DHA Prevents Cognitive Deficit and Regulates Alzheimer Disease Mediators in an ApoE−/− Mice Experimental Model. Nutrients 2019, 11, 11. https://doi.org/10.3390/nu11010011
Ruiz-Roso MB, Echeverry-Alzate V, Ruiz-Roso B, Quintela JC, Ballesteros S, Lahera V, De las Heras N, López-Moreno JA, Martín-Fernández B. Low Phytanic Acid-Concentrated DHA Prevents Cognitive Deficit and Regulates Alzheimer Disease Mediators in an ApoE−/− Mice Experimental Model. Nutrients. 2019; 11(1):11. https://doi.org/10.3390/nu11010011
Chicago/Turabian StyleRuiz-Roso, María Belén, Víctor Echeverry-Alzate, Baltasar Ruiz-Roso, José Carlos Quintela, Sandra Ballesteros, Vicente Lahera, Natalia De las Heras, José Antonio López-Moreno, and Beatriz Martín-Fernández. 2019. "Low Phytanic Acid-Concentrated DHA Prevents Cognitive Deficit and Regulates Alzheimer Disease Mediators in an ApoE−/− Mice Experimental Model" Nutrients 11, no. 1: 11. https://doi.org/10.3390/nu11010011
APA StyleRuiz-Roso, M. B., Echeverry-Alzate, V., Ruiz-Roso, B., Quintela, J. C., Ballesteros, S., Lahera, V., De las Heras, N., López-Moreno, J. A., & Martín-Fernández, B. (2019). Low Phytanic Acid-Concentrated DHA Prevents Cognitive Deficit and Regulates Alzheimer Disease Mediators in an ApoE−/− Mice Experimental Model. Nutrients, 11(1), 11. https://doi.org/10.3390/nu11010011