Equol Pretreatment Protection of SH-SY5Y Cells against Aβ (25–35)-Induced Cytotoxicity and Cell-Cycle Reentry via Sustaining Estrogen Receptor Alpha Expression


Abstract
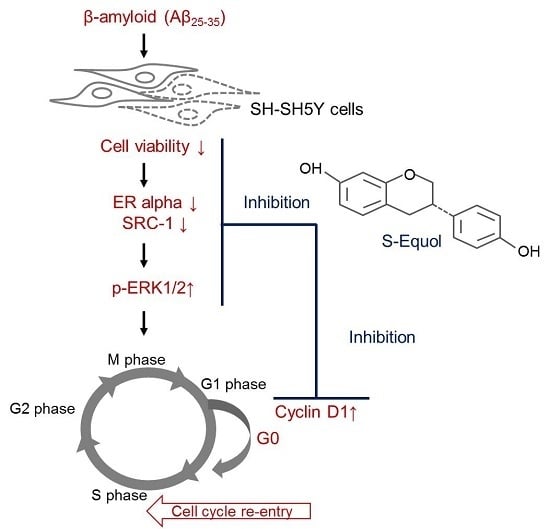
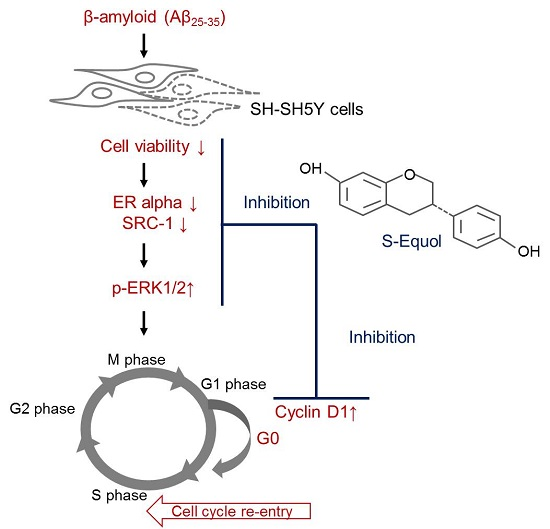{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Treatments
2.3. Cell Viability Analysis
2.4. Protein Extraction and Quantification
2.5. Cell-Cycle Analysis
2.6. Western Blot Analysis
2.7. Statistical Analysis
3. Results
3.1. Cell Viability
3.2. Estrogen Receptor Alpha (ERα) Protein Expression
3.3. SRC-1 Protein Expression
3.4. Cell Cycle
3.5. Cyclin D1 Protein Expression
3.6. Activation of ERK 1/2
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nagy, Z. Cell cycle regulatory failure in neurones: Causes and consequences. Neurobiol. Aging 2000, 21, 761–769. [Google Scholar] [CrossRef]
- Frade, J.M.; Ovejero-Benito, M.C. Neuronal cell cycle: The neuron itself and its circumstances. Cell Cycle 2015, 14, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K. The involvement of cell cycle events in the pathogenesis of Alzheimer’s disease. Alzheimer’s Res. Ther. 2010, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Clementi, M.E.; Marini, S.; Coletta, M.; Orsini, F.; Giardina, B.; Misiti, F. Abeta(31–35) and Abeta(25–35) fragments of amyloid beta-protein induce cellular death through apoptotic signals: Role of the redox state of methionine-35. FEBS Lett. 2005, 579, 2913–2918. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.J.; Du, S. The protective effect of curcumin on Abeta induced aberrant cell cycle reentry on primary cultured rat cortical neurons. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 445–454. [Google Scholar] [PubMed]
- Frasca, G.; Carbonaro, V.; Merlo, S.; Copani, A.; Sortino, M.A. Integrins mediate beta-amyloid-induced cell-cycle activation and neuronal death. J. Neurosci. Res. 2008, 86, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Frasca, G.; Chiechio, S.; Vancheri, C.; Nicoletti, F.; Copani, A.; Angela Sortino, M. Beta-amyloid-activated cell cycle in SH-SY5Y neuroblastoma cells: Correlation with the MAP kinase pathway. J. Mol. Neurosci. 2004, 22, 231–236. [Google Scholar] [CrossRef]
- Mannella, P.; Brinton, R.D. Estrogen receptor protein interaction with phosphatidylinositol 3-kinase leads to activation of phosphorylated Akt and extracellular signal-regulated kinase 1/2 in the same population of cortical neurons: A unified mechanism of estrogen action. J. Neurosci. 2006, 26, 9439–9447. [Google Scholar] [CrossRef]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors alpha (ERalpha) and beta (ERbeta): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.E.; Gatch, M.B.; Simpkins, J.W. Estrogen neuroprotection against the neurotoxic effects of ethanol withdrawal: Potential mechanisms. Exp. Biol. Med. 2005, 230, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.C. Role of estrogen receptor alpha and beta expression and signaling on cognitive function during aging. Hippocampus 2012, 22, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Bean, L.A.; Ianov, L.; Foster, T.C. Estrogen receptors, the hippocampus, and memory. Neuroscientist 2014, 20, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.C.; Pike, C.J. Selective estrogen receptor modulators differentially regulate Alzheimer-like changes in female 3xTg-AD mice. Endocrinology 2008, 149, 2607–2611. [Google Scholar] [CrossRef]
- Zhao, L.; Wu, T.W.; Brinton, R.D. Estrogen receptor subtypes alpha and beta contribute to neuroprotection and increased Bcl-2 expression in primary hippocampal neurons. Brain Res. 2004, 1010, 22–34. [Google Scholar] [CrossRef]
- Kim, H.; Bang, O.Y.; Jung, M.W.; Ha, S.D.; Hong, H.S.; Huh, K.; Kim, S.U.; Mook-Jung, I. Neuroprotective effects of estrogen against beta-amyloid toxicity are mediated by estrogen receptors in cultured neuronal cells. Neurosci. Lett. 2001, 302, 58–62. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, F.; Jiang, S.; Siedlak, S.L.; Shen, L.; Perry, G.; Wang, X.; Tang, B.; Zhu, X. Estrogen receptor-alpha is localized to neurofibrillary tangles in Alzheimer’s disease. Sci. Rep. 2016, 6, 20352. [Google Scholar] [CrossRef]
- Gonzalez-Arenas, A.; Hansberg-Pastor, V.; Hernandez-Hernandez, O.T.; Gonzalez-Garcia, T.K.; Henderson-Villalpando, J.; Lemus-Hernandez, D.; Cruz-Barrios, A.; Rivas-Suarez, M.; Camacho-Arroyo, I. Estradiol increases cell growth in human astrocytoma cell lines through ERalpha activation and its interaction with SRC-1 and SRC-3 coactivators. Biochim. Biophys. Acta 2012, 1823, 379–386. [Google Scholar] [CrossRef]
- Mueller, S.O.; Simon, S.; Chae, K.; Metzler, M.; Korach, K.S. Phytoestrogens and their human metabolites show distinct agonistic and antagonistic properties on estrogen receptor alpha (ERalpha) and ERbeta in human cells. Toxicol. Sci. 2004, 80, 14–25. [Google Scholar] [CrossRef]
- Setchell, K.D.; Clerici, C. Equol: Pharmacokinetics and biological actions. J. Nutr. 2010, 140, 1363s–1368s. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.; Zhao, X.; Shoaf, S.E.; Ragland, K. The pharmacokinetics of S-(-)equol administered as SE5-OH tablets to healthy postmenopausal women. J. Nutr. 2009, 139, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Morabito, N.; Crisafulli, A.; Vergara, C.; Gaudio, A.; Lasco, A.; Frisina, N.; D’Anna, R.; Corrado, F.; Pizzoleo, M.A.; Cincotta, M.; et al. Effects of genistein and hormone-replacement therapy on bone loss in early postmenopausal women: A randomized double-blind placebo-controlled study. J. Bone Miner. Res. 2002, 17, 1904–1912. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Ji, E.; Shin, D.; Jin, J.; Yeo, J.H.; Kim, S.Y. Equol, a Dietary Daidzein Gut Metabolite Attenuates Microglial Activation and Potentiates Neuroprotection In Vitro. Nutrients 2017, 9, 207. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.F.; Sortino, M.A. Estrogen and Alzheimer’s disease: Still an attractive topic despite disappointment from early clinical results. Eur. J. Pharmacol. 2017, 817, 51–58. [Google Scholar] [CrossRef]
- Christensen, A.; Pike, C.J. Age-dependent regulation of obesity and Alzheimer-related outcomes by hormone therapy in female 3xTg-AD mice. PLoS ONE 2017, 12, e0178490. [Google Scholar] [CrossRef]
- Dong, Y.; Yang, N.; Liu, Y.; Li, Q.; Zuo, P. The neuroprotective effects of phytoestrogen alpha-zearalanol on beta-amyloid-induced toxicity in differentiated PC-12 cells. Eur. J. Pharmacol. 2011, 670, 392–398. [Google Scholar] [CrossRef]
- Zhao, L.; Mao, Z.; Brinton, R.D. A select combination of clinically relevant phytoestrogens enhances estrogen receptor beta-binding selectivity and neuroprotective activities in vitro and in vivo. Endocrinology 2009, 150, 770–783. [Google Scholar] [CrossRef]
- Valles, S.L.; Borras, C.; Gambini, J.; Furriol, J.; Ortega, A.; Sastre, J.; Pallardo, F.V.; Vina, J. Oestradiol or genistein rescues neurons from amyloid beta-induced cell death by inhibiting activation of p38. Aging Cell 2008, 7, 112–118. [Google Scholar] [CrossRef]
- Hwang, C.J.; Yun, H.M.; Park, K.R.; Song, J.K.; Seo, H.O.; Hyun, B.K.; Choi, D.Y.; Yoo, H.S.; Oh, K.W.; Hwang, D.Y.; et al. Memory Impairment in Estrogen Receptor alpha Knockout Mice Through Accumulation of Amyloid-beta Peptides. Mol. Neurobiol. 2015, 52, 176–186. [Google Scholar] [CrossRef]
- Tang, Y.; Min, Z.; Xiang, X.J.; Liu, L.; Ma, Y.L.; Zhu, B.L.; Song, L.; Tang, J.; Deng, X.J.; Yan, Z.; et al. Estrogen-related receptor alpha is involved in Alzheimer’s disease-like pathology. Exp. Neurol. 2018, 305, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Tetel, M.J.; Acharya, K.D. Nuclear receptor coactivators: Regulators of steroid action in brain and behaviour. J. Neuroendocrinol. 2013, 25, 1209–1218. [Google Scholar] [CrossRef]
- Cicatiello, L.; Addeo, R.; Sasso, A.; Altucci, L.; Petrizzi, V.B.; Borgo, R.; Cancemi, M.; Caporali, S.; Caristi, S.; Scafoglio, C.; et al. Estrogens and progesterone promote persistent CCND1 gene activation during G1 by inducing transcriptional derepression via c-Jun/c-Fos/estrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin D1 to its own gene promoter. Mol. Cell. Biol. 2004, 24, 7260–7274. [Google Scholar] [CrossRef] [PubMed]
- JavanMoghadam, S.; Weihua, Z.; Hunt, K.K.; Keyomarsi, K. Estrogen receptor alpha is cell cycle-regulated and regulates the cell cycle in a ligand-dependent fashion. Cell Cycle 2016, 15, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Negis, Y.; Unal, A.Y.; Korulu, S.; Karabay, A. Cell cycle markers have different expression and localization patterns in neuron-like PC12 cells and primary hippocampal neurons. Neurosci. Lett. 2011, 496, 135–140. [Google Scholar] [CrossRef]
- Sumrejkanchanakij, P.; Tamamori-Adachi, M.; Matsunaga, Y.; Eto, K.; Ikeda, M.A. Role of cyclin D1 cytoplasmic sequestration in the survival of postmitotic neurons. Oncogene 2003, 22, 8723–8730. [Google Scholar] [CrossRef][Green Version]
- Quelle, D.E.; Ashmun, R.A.; Shurtleff, S.A.; Kato, J.Y.; Bar-Sagi, D.; Roussel, M.F.; Sherr, C.J. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 1993, 7, 1559–1571. [Google Scholar] [CrossRef]
- Modi, P.K.; Komaravelli, N.; Singh, N.; Sharma, P. Interplay between MEK-ERK signaling, cyclin D1, and cyclin-dependent kinase 5 regulates cell cycle reentry and apoptosis of neurons. Mol. Biol. Cell 2012, 23, 3722–3730. [Google Scholar] [CrossRef]
- Jackman, J.; O’Connor, P.M. Methods for synchronizing cells at specific stages of the cell cycle. Curr. Protoc. Cell Biol. 2001. [Google Scholar] [CrossRef]
- Malik, B.; Currais, A.; Andres, A.; Towlson, C.; Pitsi, D.; Nunes, A.; Niblock, M.; Cooper, J.; Hortobagyi, T.; Soriano, S. Loss of neuronal cell cycle control as a mechanism of neurodegeneration in the presenilin-1 Alzheimer’s disease brain. Cell Cycle 2008, 7, 637–646. [Google Scholar] [CrossRef]
- Copani, A.; Condorelli, F.; Caruso, A.; Vancheri, C.; Sala, A.; Giuffrida Stella, A.M.; Canonico, P.L.; Nicoletti, F.; Sortino, M.A. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 1999, 13, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and biological activities of amyloid beta peptide 25–35. Curr. Protein Pept. Sci. 2010, 11, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Barrio-Alonso, E.; Hernandez-Vivanco, A.; Walton, C.C. Cell cycle reentry triggers hyperploidization and synaptic dysfunction followed by delayed cell death in differentiated cortical neurons. Sci. Rep. 2018, 8, 14316. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, J.L.; Mize, A.L.; Wade, C.B.; Harris, J.A.; Shapiro, R.A.; Dorsa, D.M. Estrogen-mediated neuroprotection against beta-amyloid toxicity requires expression of estrogen receptor alpha or beta and activation of the MAPK pathway. J. Neurochem. 2002, 82, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, P.; Schieb, H.; Kamrowski-Kruck, H.; Otto, M.; Chiasserini, D.; Parnetti, L.; Herukka, S.K.; Schuchhardt, J.; Wiltfang, J.; Klafki, H.W. Evidence for Elevated Cerebrospinal Fluid ERK1/2 Levels in Alzheimer Dementia. Int. J. Alzheimer’s Dis. 2011, 2011, 739847. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, M.; Ferreira, A. PD98059 prevents neurite degeneration induced by fibrillar beta-amyloid in mature hippocampal neurons. J. Neurochem. 2000, 74, 125–133. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, M.-C.; Lin, S.-H.; Hidayah, K.; Lin, C.-I. Equol Pretreatment Protection of SH-SY5Y Cells against Aβ (25–35)-Induced Cytotoxicity and Cell-Cycle Reentry via Sustaining Estrogen Receptor Alpha Expression. Nutrients 2019, 11, 2356. https://doi.org/10.3390/nu11102356
Tsai M-C, Lin S-H, Hidayah K, Lin C-I. Equol Pretreatment Protection of SH-SY5Y Cells against Aβ (25–35)-Induced Cytotoxicity and Cell-Cycle Reentry via Sustaining Estrogen Receptor Alpha Expression. Nutrients. 2019; 11(10):2356. https://doi.org/10.3390/nu11102356
Chicago/Turabian StyleTsai, Meng-Chao, Shyh-Hsiang Lin, Kiswatul Hidayah, and Ching-I Lin. 2019. "Equol Pretreatment Protection of SH-SY5Y Cells against Aβ (25–35)-Induced Cytotoxicity and Cell-Cycle Reentry via Sustaining Estrogen Receptor Alpha Expression" Nutrients 11, no. 10: 2356. https://doi.org/10.3390/nu11102356
APA StyleTsai, M.-C., Lin, S.-H., Hidayah, K., & Lin, C.-I. (2019). Equol Pretreatment Protection of SH-SY5Y Cells against Aβ (25–35)-Induced Cytotoxicity and Cell-Cycle Reentry via Sustaining Estrogen Receptor Alpha Expression. Nutrients, 11(10), 2356. https://doi.org/10.3390/nu11102356