Role of Citrate in Pathophysiology and Medical Management of Bone Diseases



Abstract
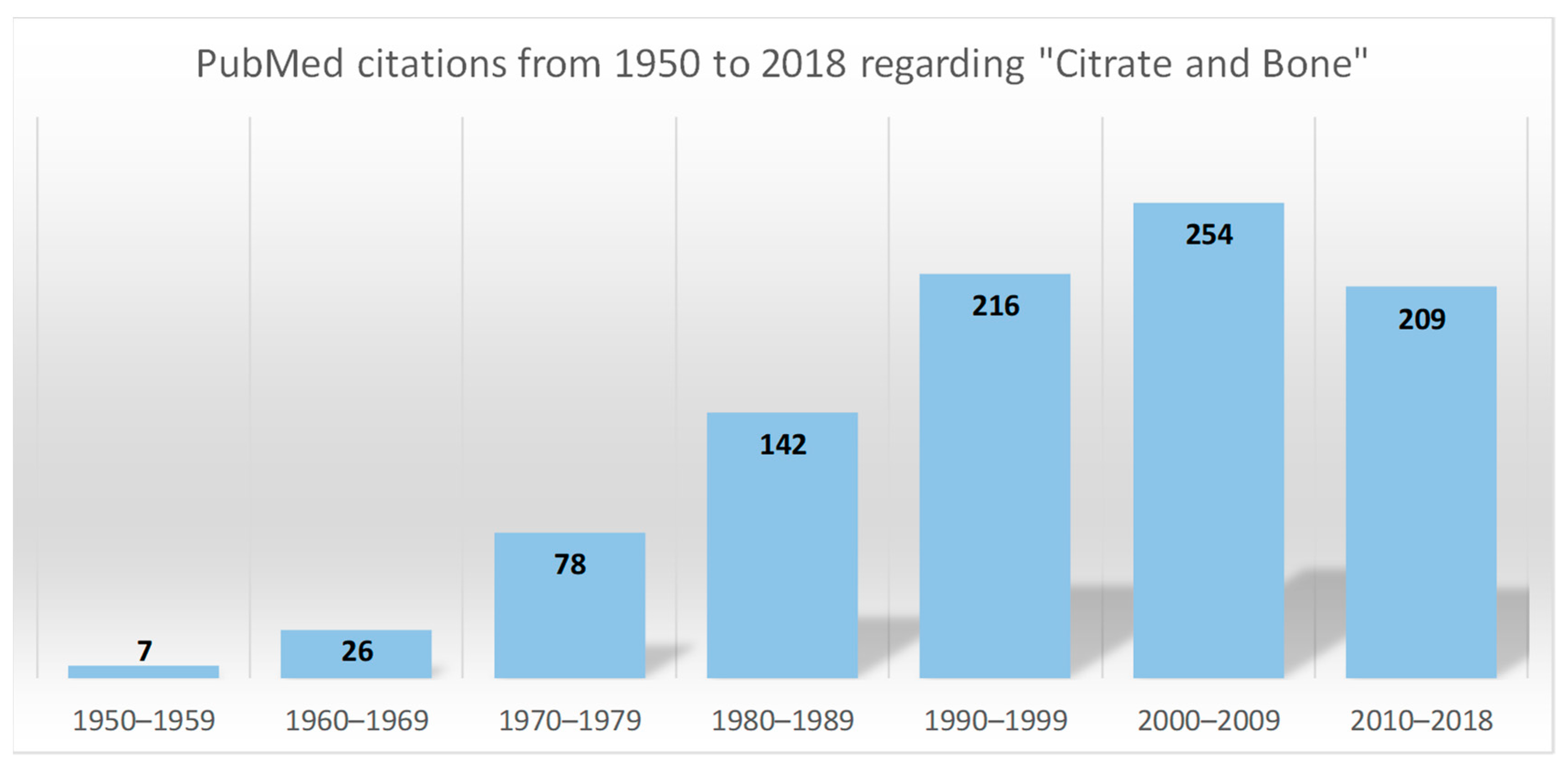
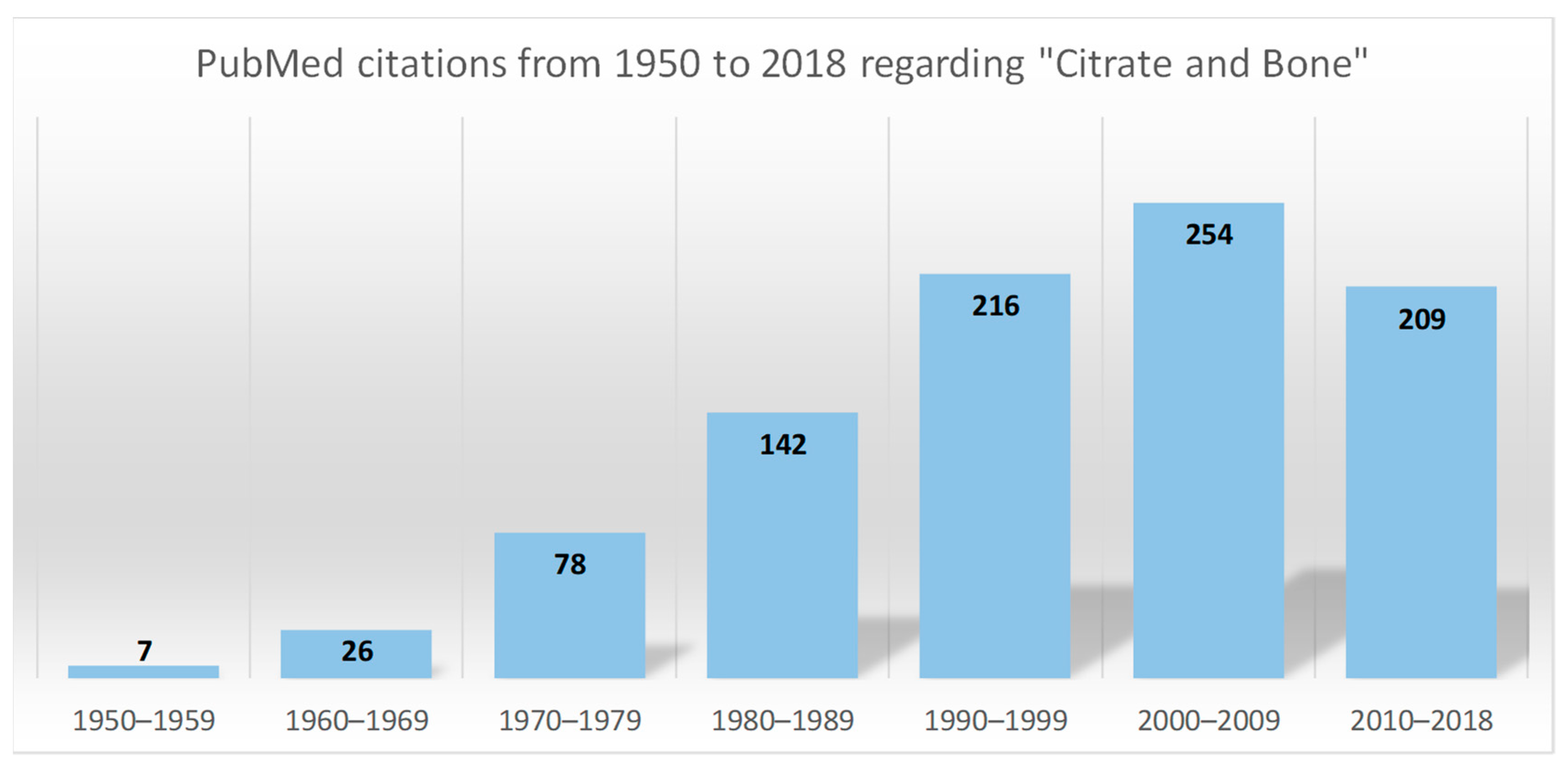
:1. Introduction
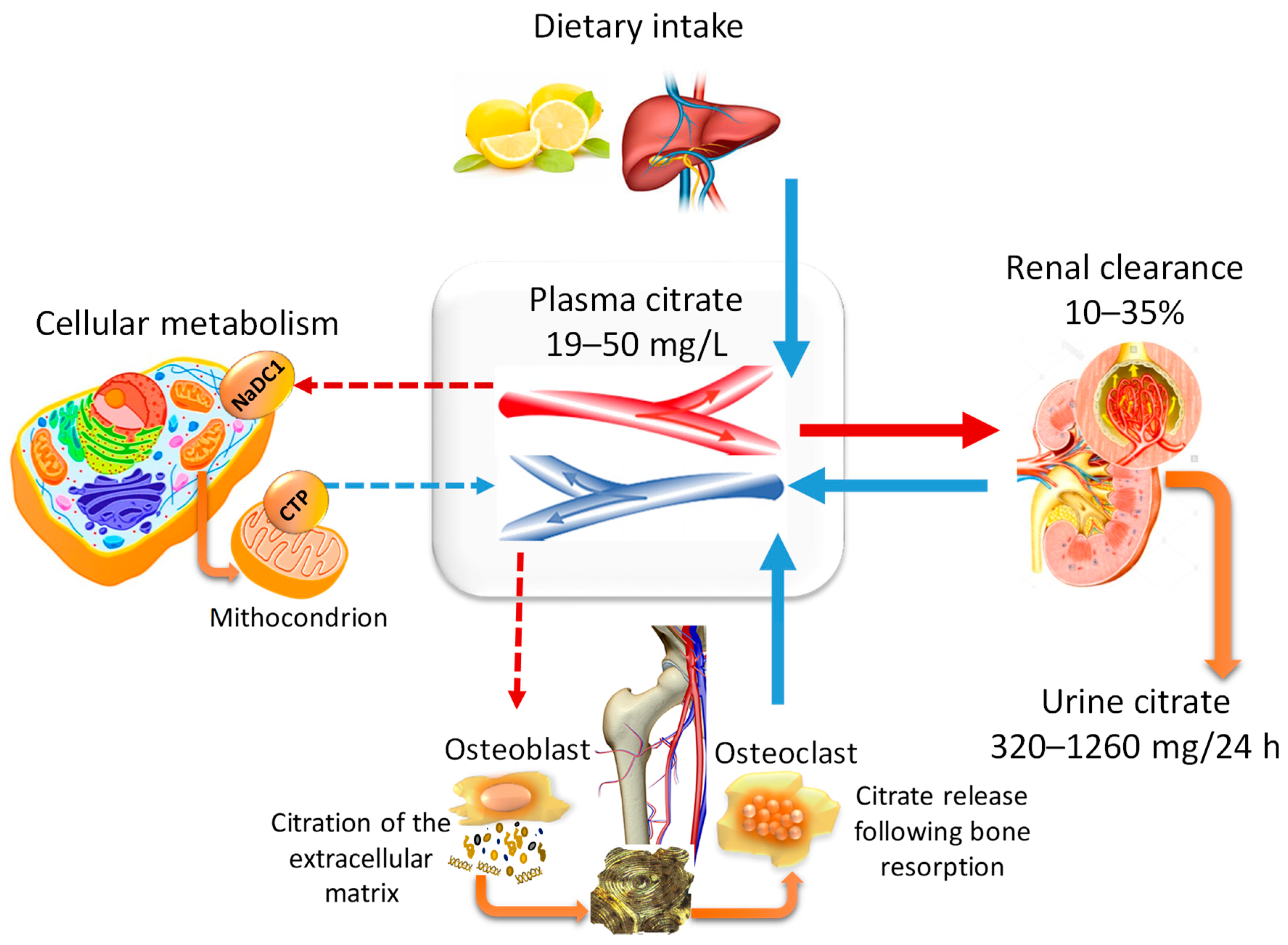
2. Citrate Homeostasis: General Physiological Concepts
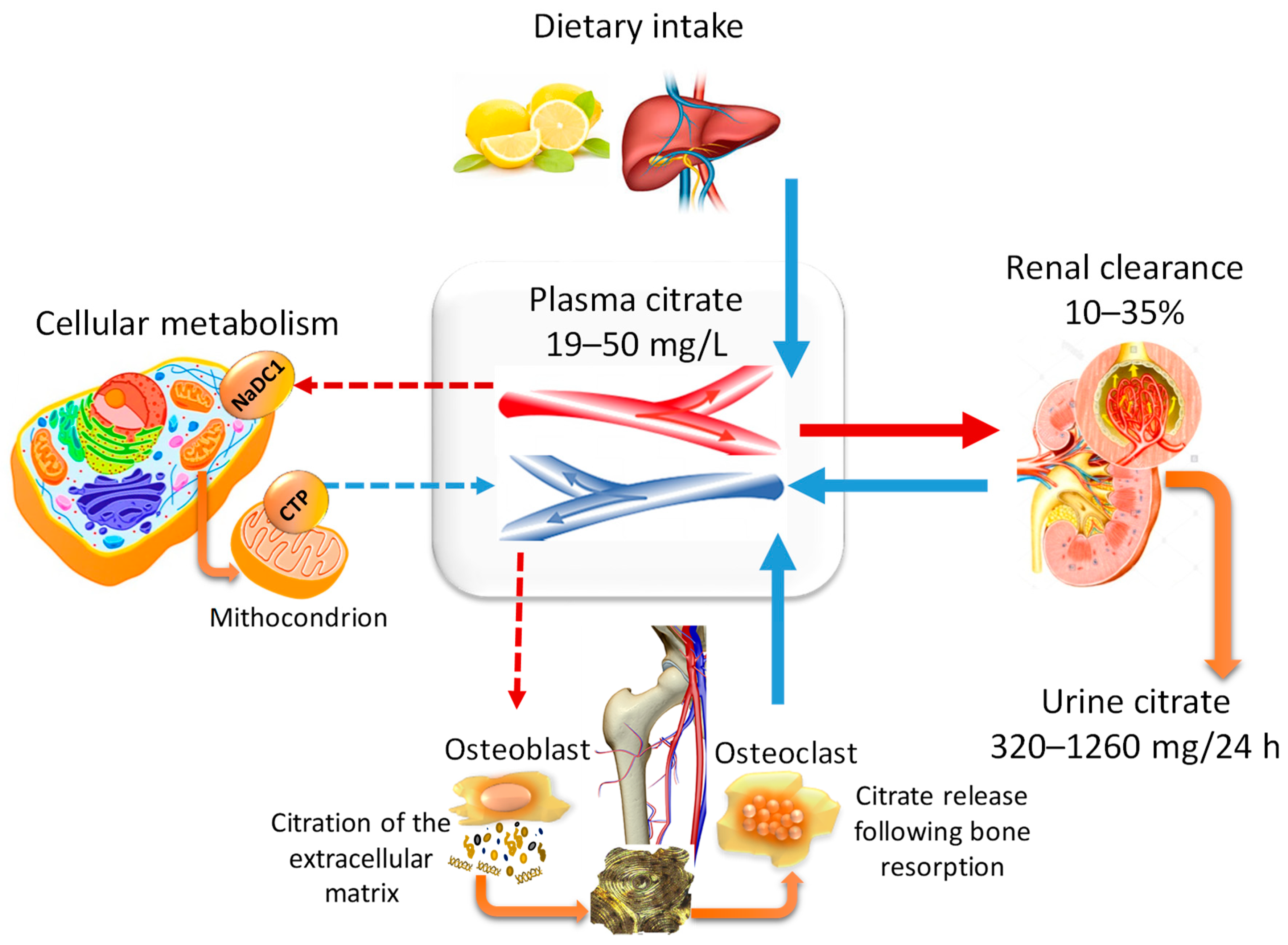
2.1. The Pillars of Citrate Homeostasis
2.2. Citraturia as A Marker of Citrate Homeostasis and Bone Health Status
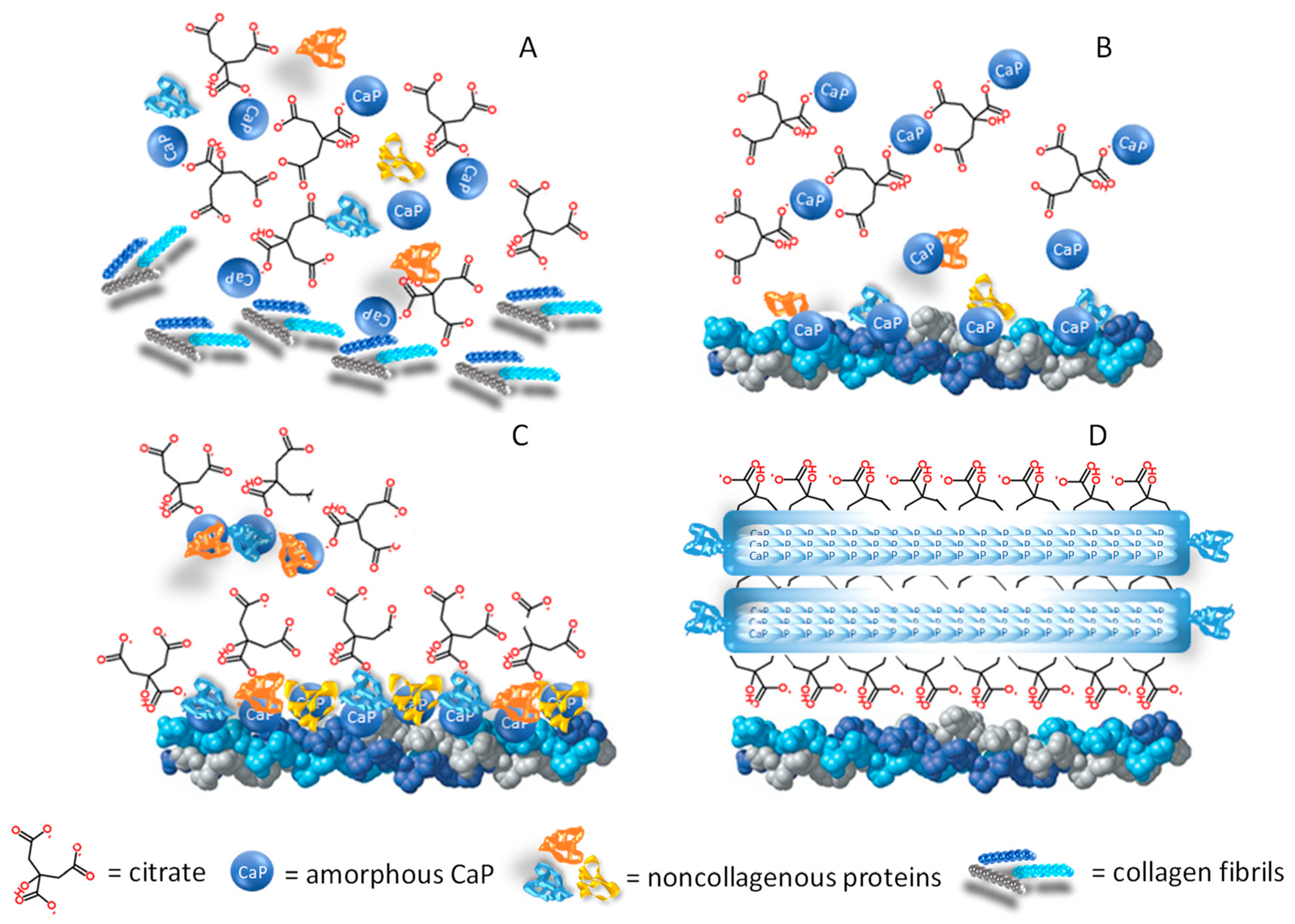
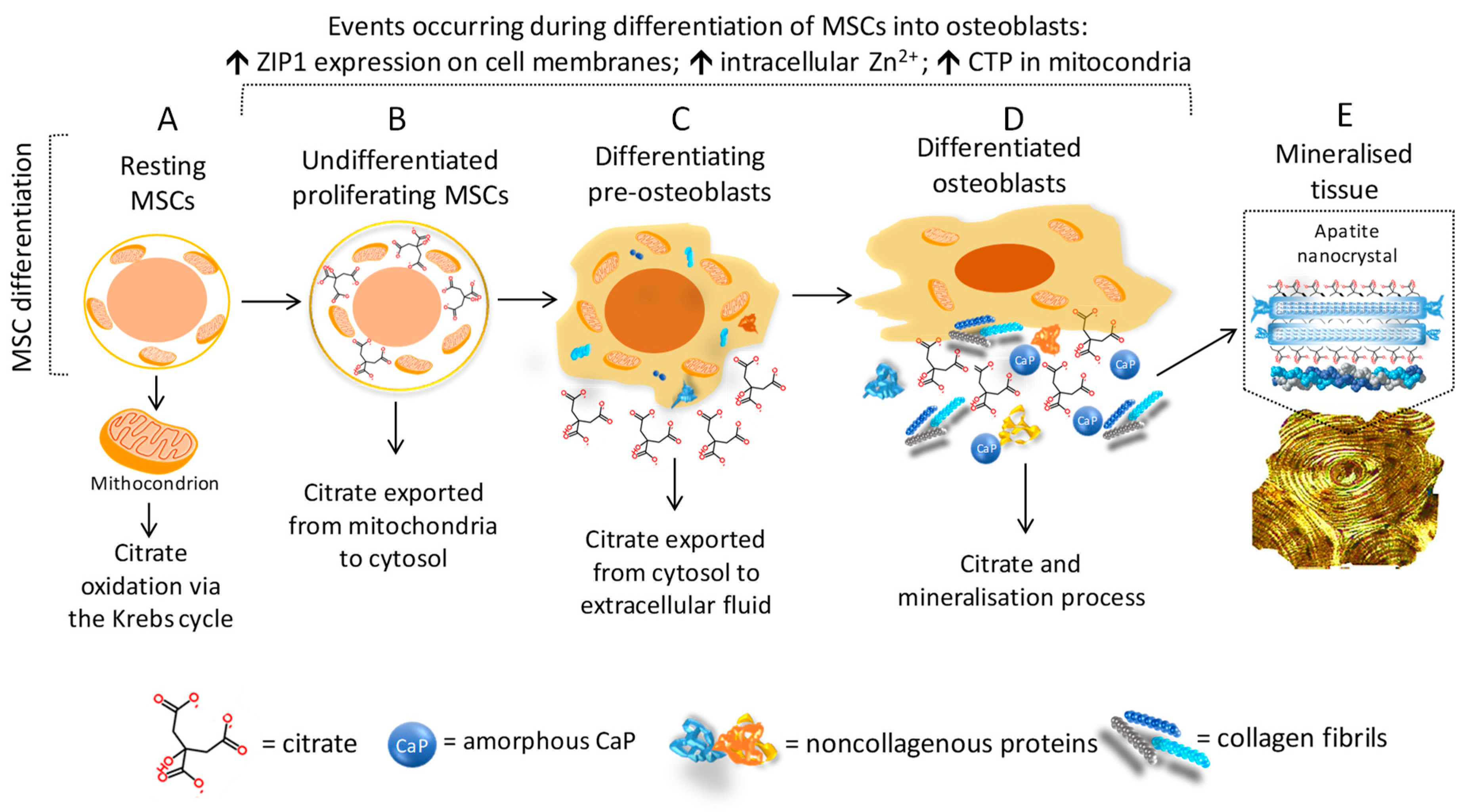
3. Citrate and Bone Tissue
3.1. Citrate and Mineral Structure
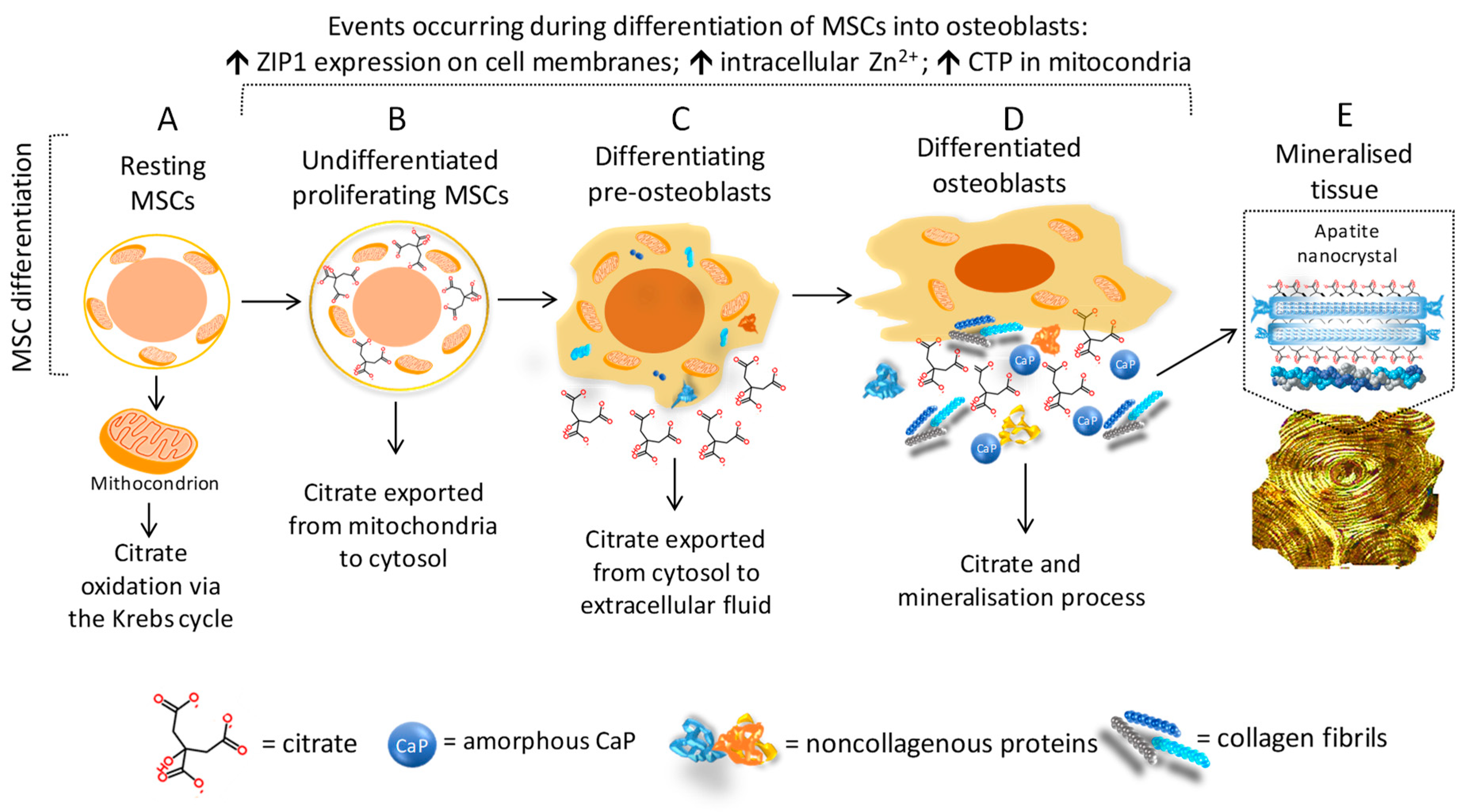
3.2. Citrate and Bone Cells
4. Citrate Pathophysiology and Bone Diseases
4.1. Bone Health Status and Alterations of Citrate Homeostasis in Kidney Diseases
4.2. Postmenopausal Osteopenia and “Net Citrate Loss”
4.3. Genetic Variations Influencing Citrate Homeostasis and Skeletal Development
5. Medical Management of Patients with Metabolic Bone Diseases Associated with Citrate Alterations
5.1. Clinical Work-Up
5.2. Dietary Modification
5.3. Citrate-Based Supplements
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Krebs, H.A.; Johnson, W.A. The role of citric acid in intermediate metabolism in animal tissues. FEBS Lett. 1980, 117 (Suppl. 1), K1–K10. [Google Scholar] [CrossRef]
- Available online: https://www.genome.jp/kegg-bin/show_pathway?map00020 (accessed on 4 July 2019).
- Iacobazzi, V.; Infantino, V. Citrate—New functions for an old metabolite. Biol. Chem. 2014, 395, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Mycielska, M.E.; Milenkovic, V.M.; Wetzel, C.H.; Rümmele, P.; Geissler, E.K. Extracellular Citrate in Health and Disease. Curr. Mol. Med. 2015, 15, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.B.; Chellaiah, M.; Zou, J.; Reynolds, M.A.; Costello, L.C. Evidence that Osteoblasts are Specialized Citrate-producing Cells that Provide the Citrate for Incorporation into the Structure of Bone. Open Bone J. 2014, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dickens, F. The citric acid content of animal tissues, with reference to its occurrence in bone and tumour. Biochem. J. 1941, 35, 1011–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haleblian, G.E.; Leitao, V.A.; Pierre, S.A.; Robinson, M.R.; Albala, D.M.; Ribeiro, A.A.; Preminger, G.M. Assessment of citrate concentrations in citrus fruit-based juices and beverages: Implications for management of hypocitraturic nephrolithiasis. J. Endourol. 2008, 22, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Penniston, K.L.; Nakada, S.Y.; Holmes, R.P.; Assimos, D.G. Quantitative assessment of citric acid in lemon juice, lime juice, and commercially-available fruit juice products. J. Endourol. 2008, 22, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Schuster, E.; Dunn-Coleman, N.; Frisvad, J.C.; Van Dijck, P.W. On the safety of Aspergillus niger—A review. Appl. Microbiol. Biotechnol. 2002, 59, 426–435. [Google Scholar] [CrossRef]
- Hu, W.; Li, W.J.; Yang, H.Q.; Chen, J.H. Current strategies and future prospects for enhancing microbial production of citric acid. Appl. Microbiol. Biotechnol. 2019, 103, 201–209. [Google Scholar] [CrossRef]
- Caudarella, R.; Vescini, F.; Buffa, A.; Stefoni, S. Citrate and mineral metabolism, kidney stones and bone disease. Front. Biosci. 2003, 8, s1084–s1106. [Google Scholar]
- Fegan, J.; Khan, R.; Poindexter, J.; Pak, C.Y. Gastrointestinal citrate absorption in nephrolithiasis. J. Urol. 1992, 147, 1212–1214. [Google Scholar] [CrossRef]
- Pajor, A.M. Sodium-coupled transporters for Krebs cycle intermediates. Annu. Rev. Physiol. 1999, 61, 663–682. [Google Scholar] [CrossRef] [PubMed]
- Poerwono, H.; Higashiyama, K.; Kubo, H.; Poernomo, A.T.; Suharjono, I.; Sudiana, I.K.; Indrayanto, G.; Brittain, H.G. Citric acid. In Analytical Profiles of Drug Substances and Excipients, 1st ed.; Brittain, H.G., Ed.; Academic Press: San Diego, CA, USA, 2001; Volume 28, pp. 1–76. [Google Scholar]
- Sakhaee, K.; Alpern, R.; Poindexter, J.; Pak, C.Y. Citraturic response to oral citric acid load. J. Urol. 1992, 147, 975–976. [Google Scholar] [CrossRef]
- Zuckerman, J.M.; Assimos, D.G. Hypocitraturia: Pathophysiology and medical management. Rev. Urol. 2009, 11, 134–144. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. Plasma citrate homeostasis, how it is regulated, and its physiological and clinical implications. An important, but neglected, relationship in medicine. HSOA J. Hum. Endocrinol. 2016, 1, 5. [Google Scholar] [CrossRef]
- Pak, C.Y.; Resnick, M. Medical therapy and new approaches to management of urolithiasis. Urol. Clin. N. Am. 2000, 27, 243–253. [Google Scholar] [CrossRef]
- Mayo Clinic Medical Laboratories. Endocrinology Catalog Bone/Minerals. Available online: https//endocrinology.testcatalog.org/show/CITR (accessed on 30 May 2019).
- Heller, H.J.; Sakhaee, K.; Moe, O.W.; Pak, C.Y. Etiological role of estrogen status in renal stone formation. J. Urol. 2002, 168, 1923–1927. [Google Scholar] [CrossRef]
- Caudarella, R.; Vescini, F. Urinary citrate and renal stone disease: The preventive role of alkali citrate treatment. Arch. Ital. Urol. 2009, 81, 182–187. [Google Scholar]
- Melnick, J.Z.; Preisig, P.A.; Moe, O.W.; Srere, P.; Alpern, R.J. Renal cortical mitochondrial aconitase is regulated in hypo- and hypercitraturia. Kidney Int. 1998, 54, 160–165. [Google Scholar] [CrossRef] [Green Version]
- Lerma, E.V. Hypocitraturia. Updated 5 October 2015. Available online: https://emedicine.medscape.com/article/444968-overview (accessed on 29 July 2019).
- Prezioso, D.; Strazzullo, P.; Lotti, T.; Bianchi, G.; Borghi, L.; Caione, P.; Carini, M.; Caudarella, R.; Ferraro, M.; Gambaro, G.; et al. Dietary treatment of urinary risk factors for renal stone formation. A review of CLU Working Group. Arch. Ital. Urol. 2015, 87, 105–120. [Google Scholar] [CrossRef]
- Dolan, E.; Sale, C. Protein and bone health across the lifespan. Proc. Nutr. Soc. 2019, 78, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Vallés, P.G.; Batlle, D. Hypokalemic Distal Renal Tubular Acidosis. Adv. Chronic Kidney Dis. 2018, 25, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Melnick, J.Z.; Preisig, P.A.; Haynes, S.; Pak, C.Y.; Sakhaee, K.; Alpern, R.J. Converting enzyme inhibition causes hypocitraturia independent of acidosis or hypokalemia. Kidney Int. 1998, 54, 1670–1674. [Google Scholar] [CrossRef] [Green Version]
- Warner, B.W.; LaGrange, C.A.; Tucker, T.; Bensalem-Owen, M.; Pais, V.M., Jr. Induction of progressive profound hypocitraturia with increasing doses of topiramate. Urology 2008, 72, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Goraya, N.; Simoni, J.; Sager, L.N.; Madias, N.E.; Wesson, D.E. Urine citrate excretion as a marker of acid retention in patients with chronic kidney disease without overt metabolic acidosis. Kidney Int. 2019, 95, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Shey, J.; Cameron, M.A.; Sakhaee, K.; Moe, O.W. Recurrent calcium nephrolithiasis associated with primary aldosteronism. Am. J. Kidney Dis. 2004, 44, e7–e12. [Google Scholar] [CrossRef]
- Dey, J.; Creighton, A.; Lindberg, J.S.; Fuselier, H.A.; Kok, D.J.; Cole, F.E.; Hamm, L. Estrogen replacement increased the citrate and calcium excretion rates in postmenopausal women with recurrent urolithiasis. J. Urol. 2002, 167, 169–171. [Google Scholar] [CrossRef]
- Brinton, R.D. The healthy cell bias of estrogen action, mitochondrial bioenergetics and neurological implications. Trends Neurosci. 2008, 31, 529–537. [Google Scholar] [CrossRef]
- Mai, Z.; Li, X.; Jiang, C.; Liu, Y.; Chen, Y.; Wu, W.; Zeng, G. Comparison of metabolic changes for stone risks in 24-h urine between non- and postmenopausal women. PLoS ONE 2019, 14, e0208893. [Google Scholar] [CrossRef]
- Granchi, D.; Caudarella, R.; Ripamonti, C.; Spinnato, P.; Bazzocchi, A.; Torreggiani, E.; Massa, A.; Baldini, N. Association between markers of bone loss and urinary lithogenic risk factors in osteopenic postmenopausal women. J. Biol. Regul. Homeost. Agents 2016, 30, S145–S151. [Google Scholar]
- Adeva, M.M.; Souto, G. Diet-induced metabolic acidosis. Clin. Nutr. 2011, 30, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Esche, J.; Johner, S.; Shi, L.; Schönau, E.; Remer, T. Urinary Citrate, an Index of Acid-Base Status, Predicts Bone Strength in Youths and Fracture Risk in Adult Females. J. Clin. Endocrinol. Metab. 2016, 101, 4914–4921. [Google Scholar] [CrossRef] [PubMed]
- Shea, M.K.; Dawson-Hughes, B. Association of Urinary Citrate With Acid-Base Status, Bone Resorption, and Calcium Excretion in Older Men and Women. J. Clin. Endocrinol. Metab. 2018, 103, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Nancollas, G.H. How to control the size and morphology of apatite nanocrystals in bone. Proc. Natl. Acad. Sci. USA 2010, 107, 22369–22370. [Google Scholar] [CrossRef] [Green Version]
- Dixon, T.F.; Perkins, H.R. Citric acid and bone metabolism. Biochem. J. 1952, 52, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.Y.; Rawal, A.; Schmidt-Rohr, K. Strongly bound citrate stabilizes the apatite nanocrystals in bone. Proc. Natl. Acad. Sci. USA 2010, 107, 22425–22429. [Google Scholar] [CrossRef] [Green Version]
- Mahamid, J.; Sharir, A.; Addadi, L.; Weiner, S. Amorphous calcium phosphate is major component of the forming fin bones of zebrafish. Indications for an amorphous precursor phase. Proc. Natl. Acad. Sci. USA 2008, 105, 12748–12753. [Google Scholar] [CrossRef]
- Bradt, J.H.; Mertig, M.; Teresiak, A.; Pompe, W. Biomimetic mineralization of collagen by combined fibril assembly and calcium phosphate formation. Chem. Mater. 1999, 11, 2694–2701. [Google Scholar] [CrossRef]
- Lotsari, A.; Rajasekharan, A.K.; Halvarsson, M.; Andersson, M. Transformation of amorphous calcium phosphate to bone-like apatite. Nat. Commun. 2018, 9, 4170. [Google Scholar] [CrossRef]
- Davies, E.; Müller, K.H.; Wong, W.C.; Pickard, C.J.; Reid, D.G.; Skepper, J.N.; Duer, M.J. Citrate bridges between mineral platelets in bone. Proc. Natl. Acad. Sci. USA 2014, 111, E1354–E1363. [Google Scholar] [CrossRef] [Green Version]
- Costello, L.C.; Chellaiah, M.; Zou, J.; Franklin, R.B.; Reynolds, M.A. The status of citrate in the hydroxyapatite/collagen complex of bone, and Its role in bone formation. J. Regen. Med. Tissue Eng. 2014, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Delgado-López, J.M.; Bertolotti, F.; Lyngsø, J.; Pedersen, J.S.; Cervellino, A.; Masciocchi, N.; Guagliardi, A. The synergic role of collagen and citrate in stabilizing amorphous calcium phosphate precursors with platy morphology. Acta Biomater. 2017, 49, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, T.A.; Boskey, A.L.; Gundberg, C.M.; Vigorita, V.J.; Devlin, V.J.; Beyer, M.M. The mineral and mechanical properties of bone in chronic experimental diabetes. J. Orthop. Res. 1988, 6, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Tran, R.T.; Yang, J.; Ameer, G.A. Citrate-Based Biomaterials and Their Applications in Regenerative Engineering. Annu. Rev. Mater. Res. 2015, 45, 277–310. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Gerhard, E.; Lu, D.; Yang, J. Citrate chemistry and biology for biomaterials design. Biomaterials 2018, 178, 383–400. [Google Scholar] [CrossRef]
- Qiu, H.; Yang, J.; Kodali, P.; Koh, J.; Ameer, G.A. A citric acid-based hydroxyapatite composite for orthopedic implants. Biomaterials 2006, 27, 5845–5854. [Google Scholar] [CrossRef]
- Xie, D.; Guo, J.; Mehdizadeh, M.; Tran, R.T.; Chen, R.; Sun, D.; Qian, G.; Jin, D.; Bai, X.; Yang, J. Development of Injectable Citrate-Based Bioadhesive Bone Implants. J. Mater. Chem. B 2015, 3, 387–398. [Google Scholar] [CrossRef]
- Chung, E.J.; Sugimoto, M.J.; Koh, J.L.; Ameer, G.A. A biodegradable tri-component graft for anterior cruciate ligament reconstruction. J. Tissue Eng. Regen. Med. 2017, 11, 704–712. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B.; Reynolds, M.A.; Chellaiah, M. The Important Role of Osteoblasts and Citrate Production in Bone Formation: “Osteoblast Citration” as a New Concept for an Old Relationship. Open Bone J. 2012, 4. [Google Scholar] [CrossRef]
- Costello, L.C.; Liu, Y.; Franklin, R.B.; Kennedy, M.C. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J. Biol. Chem. 1997, 272, 28875–28881. [Google Scholar] [CrossRef]
- Franklin, R.B.; Ma, J.; Zou, J.; Guan, Z.; Kukoyi, B.I.; Feng, P.; Costello, L.C. Human ZIP1 is a major zinc uptake transporter for the accumulation of zinc in prostate cells. J. Inorg. Biochem. 2003, 96, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Costello, L.C.; Franklin, R.B. A review of the important central role of altered citrate metabolism during the process of stem cell differentiation. J. Regen. Med. Tissue Eng. 2013, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, L.C.; Chellaiah, M.A.; Zou, J.; Reynolds, M.A.; Franklin, R.B. In vitro BMP2 stimulation of osteoblast citrate production in concert with mineralized bone nodule formation. J. Regen Med. Tissue Eng. 2015, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Granchi, D.; Ochoa, G.; Leonardi, E.; Devescovi, V.; Baglìo, S.R.; Osaba, L.; Baldini, N.; Ciapetti, G. Gene expression patterns related to osteogenic differentiation of bone marrow-derived mesenchymal stem cells during ex vivo expansion. Tissue Eng. Part C Methods 2010, 16, 511–524. [Google Scholar] [CrossRef]
- Fu, X.; Li, Y.; Huang, T.; Yu, Z.; Ma, K.; Yang, M.; Liu, Q.; Pan, H.; Wang, H.; Wang, J.; et al. Runx2/Osterix and Zinc Uptake Synergize to Orchestrate Osteogenic Differentiation and Citrate Containing Bone Apatite Formation. Adv. Sci. 2018, 5, 1700755. [Google Scholar] [CrossRef]
- Accession Number GSE12267. Available online: https://www.ncbi.nlm.nih.gov/geo (accessed on 4 July 2019).
- Inoue, K.; Zhuang, L.; Ganapathy, V. Human Na+ -coupled citrate transporter: Primary structure, genomic organization, and transport function. Biochem. Biophys. Res. Commun. 2002, 299, 465–471. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Coupling the activities of bone formation and resorption: A multitude of signals within the basic multicellular unit. Bonekey Rep. 2014, 3, 481. [Google Scholar] [CrossRef]
- Ma, C.; Tian, X.; Kim, J.P.; Xie, D.; Ao, X.; Shan, D.; Lin, Q.; Hudock, M.R.; Bai, X.; Yang, J. Citrate-based materials fuel human stem cells by metabonegenic regulation. Proc. Natl. Acad. Sci. USA 2018, 115, E11741–E11750. [Google Scholar] [CrossRef]
- Granchi, D.; Torreggiani, E.; Massa, A.; Caudarella, R.; Di Pompo, G.; Baldini, N. Potassium citrate prevents increased osteoclastogenesis resulting from acidic conditions: Implication for the treatment of postmenopausal bone loss. PLoS ONE 2017, 12, e0181230. [Google Scholar] [CrossRef]
- Arnett, T.R. Acidosis, hypoxia and bone. Arch. Biochem. Biophys. 2010, 503, 103–109. [Google Scholar] [CrossRef]
- Lindeman, R.D.; Tobin, J.D.; Shock, N.W. Association between blood pressure and the rate of decline in renal function with age. Kidney Int. 1984, 26, 861–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanis, J.A.; Johnell, O.; Oden, A.; Sembo, I.; Redlund-Johnell, I.; Dawson, A.; De Laet, C.; Jonsson, B. Long-term risk of osteoporotic fracture in Malmo. Osteoporos Int. 2000, 11, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Jassal, S.K.; von Muhlen, D.; Barrett-Connor, E. Measures of renal function, BMD, bone loss, and osteoporotic fracture in older adults: The Rancho Bernardo study. J. Bone Min. Res. 2007, 22, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO 2017 Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. 2017, 7, 1–59. [CrossRef] [PubMed]
- Malmgren, L.; McGuigan, F.E.; Berglundh, S.; Westman, K.; Christensson, A.; Akesson, K. Declining estimated glomerular filtration rate and its association with mortality and comorbidity over 10 years in elderly women. Nephron 2015, 130, 245–255. [Google Scholar] [CrossRef]
- Malmgren, L.; McGuigan, F.E.; Christensson, A.; Akesson, K. Reduced kidney function is associated with BMD, bone loss and markers of mineral homeostasis in older women: A 10-year longitudinal study. Osteoporos. Int. 2017, 28, 3463–3473. [Google Scholar] [CrossRef]
- Hocher, B.; Adamski, J. Metabolomics for clinical use and research in chronic kidney disease. Nat. Rev. Nephrol. 2017, 13, 269–284. [Google Scholar] [CrossRef]
- Hallan, S.; Afkarian, M.; Zelnick, L.R.; Kestenbaum, B.; Sharma, S.; Saito, R.; Darshi, M.; Barding, G.; Raftery, D.; Ju, W.; et al. Metabolomics and Gene Expression Analysis Reveal Down-regulation of the Citric Acid (TCA) Cycle in Non-diabetic CKD Patients. Ebiomedicine 2017, 26, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.W.; Seo, S.P.; Kim, W.T.; Kim, Y.J.; Yun, S.J.; Lee, S.C.; Kim, W.J. Effect of renal insufficiency on stone recurrence in patients with urolithiasis. J. Korean Med. Sci. 2014, 29, 1132–1137. [Google Scholar] [CrossRef]
- Krieger, N.S.; Bushinsky, D.A. The relation between bone and stone formation. Calcif. Tissue Int. 2013, 93, 374–381. [Google Scholar] [CrossRef]
- Sakhaee, K.; Maalouf, N.M.; Kumar, R.; Pasch, A.; Moe, O.W. Nephrolithiasis-associated bone disease: Pathogenesis and treatment options. Kidney Int. 2011, 79, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Denburg, M.R.; Leonard, M.B.; Haynes, K.; Tuchman, S.; Tasian, G.; Shults, J.; Copelovitch, L. Risk of fracture in urolithiasis: A population-based cohort study using thehealth improvement network. Clin. J. Am. Soc. Nephrol. 2014, 9, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.N.; Feskanich, D.; Paik, J.M.; Curhan, G.C. Nephrolithiasis and Risk of Incident Bone Fracture. J. Urol. 2016, 195, 1482–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucato, P.; Trevisan, C.; Stubbs, B.; Zanforlini, B.M.; Solmi, M.; Luchini, C.; Girotti, G.; Pizzato, S.; Manzato, E.; Sergi, G.; et al. Nephrolithiasis, bone mineral density, osteoporosis, and fractures: A systematic review and comparative meta-analysis. Osteoporos. Int. 2016, 27, 3155–3164. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.R.; Pearle, M.S.; Robertson, W.G.; Gambaro, G.; Canales, B.K.; Doizi, S.; Traxer, O.; Tiselius, H.G. Kidney stones. Nat. Rev. Dis. Primers 2016, 2, 16008. [Google Scholar] [CrossRef] [PubMed]
- Muntner, P.; Jones, T.M.; Hyre, A.D.; Melamed, M.L.; Alper, A.; Raggi, P.; Leonard, M.B. Association of serum intact parathyroid hormone with lower estimated glomerular filtration rate. Clin. J. Am. Soc. Nephrol. 2009, 4, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Han, S.G.; Oh, J.; Jeon, H.J.; Park, C.; Cho, J.; Shin, D.H. Kidney Stones and Risk of Osteoporotic Fracture in Chronic Kidney Disease. Sci. Rep. 2019, 9, 1929. [Google Scholar] [CrossRef]
- Arrabal-Polo, M.A.; Girón-Prieto, M.S.; Cano-García Mdel, C.; Poyatos-Andujar, A.; Quesada Charneco, M.; Abad-Menor, F.; Arias-Santiago, S.; Zuluaga-Gomez, A.; Arrabal-Martin, M. Retrospective review of serum and urinary lithogenic risk factors in patients with osteoporosis and osteopenia. Urology 2015, 85, 782–785. [Google Scholar] [CrossRef]
- Khosla, S.; Oursler, M.J.; Monroe, D.G. Estrogen and the skeleton. Trends Endocrinol. Metab. 2012, 23, 576–581. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wang, Y.; Dai, H.; Tian, X.; Cui, Z.K.; Chen, Z.; Hu, L.; Song, Q.; Liu, A.; Zhang, Z.; et al. Bone and plasma citrate is reduced in osteoporosis. Bone 2018, 114, 189–197. [Google Scholar] [CrossRef]
- Prochaska, M.; Taylor, E.N.; Curhan, G. Menopause and Risk of Kidney Stones. J. Urol. 2018, 200, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.T.; Clarke, B.L.; Lewiecki, E.M. The Pathophysiology and Treatment of Osteoporosis. Clin. Ther. 2015, 37, 1837–1850. [Google Scholar] [CrossRef] [PubMed]
- Rharass, T.; Lucas, S. Mechanisms in endocrinology: Bone marrow adiposity and bone, a bad romance? Eur. J. Endocrinol. 2018, 179, R165–R182. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Jeong, D.; Kang, H.K.; Jung, S.Y.; Kang, S.S.; Min, B.M. Osteoclast precursors display dynamic metabolic shifts toward accelerated glucose metabolism at an early stage of RANKL-stimulated osteoclast differentiation. Cell. Physiol. Biochem. 2007, 20, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Lemma, S.; Sboarina, M.; Porporato, P.E.; Zini, N.; Sonveaux, P.; Di Pompo, G.; Baldini, N.; Avnet, S. Energy metabolism in osteoclast formation and activity. Int. J. Biochem. Cell Biol. 2016, 79, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Granchi, D.; Caudarella, R.; Ripamonti, C.; Spinnato, P.; Bazzocchi, A.; Massa, A.; Baldini, N. Potassium Citrate Supplementation Decreases the Biochemical Markers of Bone Loss in a Group of Osteopenic Women: The Results of a Randomized, Double-Blind, Placebo-Controlled Pilot Study. Nutrients 2018, 10, 1293. [Google Scholar] [CrossRef]
- Available online: https://www.omim.org/ (accessed on 4 July 2019).
- Watanabe, T. Improving outcomes for patients with distal renal tubular acidosis: Recent advances and challenges ahead. Pediatr. Health Med. Ther. 2018, 9, 181–190. [Google Scholar] [CrossRef]
- Weinstein, D.A.; Somers, M.J.; Wolfsdorf, J.I. Decreased urinary citrate excretion in type 1a glycogen storage disease. J. Pediatr. 2001, 138, 378–382. [Google Scholar] [CrossRef]
- Kaiser, N.; Gautschi, M.; Bosanska, L.; Meienberg, F.; Baumgartner, M.R.; Spinas, G.A.; Hochuli, M. Glycemic control and complications in glycogen storage disease type I: Results from the Swiss registry. Mol. Genet. Metab. 2019, 126, 355–361. [Google Scholar] [CrossRef]
- Thevenon, J.; Milh, M.; Feillet, F.; St-Onge, J.; Duffourd, Y.; Jugé, C.; Roubertie, A.; Héron, D.; Mignot, C.; Raffo, E.; et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet. 2014, 95, 113–120. [Google Scholar] [CrossRef]
- Irizarry, A.R.; Yan, G.; Zeng, Q.; Lucchesi, J.; Hamang, M.J.; Ma, Y.L.; Rong, J.X. Defective enamel and bone development in sodium-dependent citrate transporter (NaCT) Slc13a5 deficient mice. PLoS ONE 2017, 12, e0175465. [Google Scholar] [CrossRef] [PubMed]
- Díaz, M.; García, C.; Sebastiani, G.; de Zegher, F.; López-Bermejo, A.; Ibáñez, L. Placental and cord blood methylation of genes involved in energy homeostasis: Association with fetal growth and neonatal body composition. Diabetes 2017, 66, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Cunha, T.D.S.; Heilberg, I.P. Bartter syndrome: Causes, diagnosis, and treatment. Int. J. Nephrol. Renov. Dis. 2018, 11, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.B.; Karet, F.E.; Hamdan, J.M.; DiPietro, A.; Sanjad, S.A.; Lifton, R.P. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat. Genet. 1996, 13, 183–188. [Google Scholar] [CrossRef]
- International Collaborative Study Group for Bartter-like Syndromes. Mutations in the gene encoding the inwardly-rectifying renal potassium channel, ROMK, cause the antenatal variant of Bartter syndrome: Evidence for genetic heterogeneity. Hum. Molec. Genet. 1997, 6, 17–26. [Google Scholar]
- Gross, I.; Siedner-Weintraub, Y.; Simckes, A.; Gillis, D. Antenatal Bartter syndrome presenting as hyperparathyroidism with hypercalcemia and hypercalciuria: A case report and review. J. Pediatr. Endocrinol. Metab. 2015, 28, 943–946. [Google Scholar] [CrossRef]
- Li, D.; Tian, L.; Hou, C.; Kim, C.E.; Hakonarson, H.; Levine, M.A. Association of Mutations in SLC12A1 Encoding the NKCC2 Cotransporter with Neonatal Primary Hyperparathyroidism. J. Clin. Endocrinol. Metab. 2016, 101, 2196–2200. [Google Scholar] [CrossRef]
- Hou, J. Claudins and mineral metabolism. Curr. Opin. Nephrol. Hypertens. 2016, 25, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Thorleifsson, G.; Holm, H.; Edvardsson, V.; Walters, G.B.; Styrkarsdottir, U.; Gudbjartsson, D.F.; Sulem, P.; Halldorsson, B.V.; de Vegt, F.; d’Ancona, F.C.; et al. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat. Genet. 2009, 41, 926–930. [Google Scholar] [CrossRef]
- Claverie-Martin, F. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis, clinical and molecular characteristics. Clin. Kidney J. 2015, 8, 656–664. [Google Scholar] [CrossRef]
- Bardet, C.; Courson, F.; Wu, Y.; Khaddam, M.; Salmon, B.; Ribes, S.; Thumfart, J.; Yamaguti, P.M.; Rochefort, G.Y.; Figueres, M.L.; et al. Claudin-16 Deficiency Impairs Tight Junction Function in Ameloblasts, Leading to Abnormal Enamel Formation. J. Bone Min. Res. 2016, 31, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Pajor, A.M. Sodium-coupled dicarboxylate and citrate transporters from the SLC13 family. Pflug. Arch 2014, 466, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Aruga, S.; Matsuzaki, S.; Takahashi, S.; Matsushita, K.; Kitamura, T. Associations between renal sodium-citrate cotransporter (hNaDC-1) gene polymorphism and urinary citrate excretion in recurrent renal calcium stone formers and normal controls. Int. J. Urol. 2007, 14, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Pajor, A.M.; Sun, N.N. Single nucleotide polymorphisms in the human Na+-dicarboxylate cotransporter affect transport activity and protein expression. Am. J. Physiol. Ren. Physiol. 2010, 299, F704–F711. [Google Scholar] [CrossRef] [Green Version]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; et al. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef]
- Cosso, R.; Falchetti, A. Mitochondriopathies and bone health. J. Tre. Biol. Res. 2018, 1, 1–7. [Google Scholar] [CrossRef]
- Brommage, R.; Liu, J.; Hansen, G.M.; Kirkpatrick, L.L.; Potter, D.G.; Sands, A.T.; Zambrowicz, B.; Powell, D.R.; Vogel, P. High-throughput screening of mouse gene knockouts identifies established and novel skeletal phenotypes. Bone Res. 2014, 2, 14034. [Google Scholar] [CrossRef] [Green Version]
- Lorentzon, M.; Branco, J.; Brandi, M.L.; Bruyère, O.; Chapurlat, R.; Cooper, C.; Cortet, B.; Diez-Perez, A.; Ferrari, S.; Gasparik, A.; et al. Algorithm for the Use of Biochemical Markers of Bone Turnover in the Diagnosis, Assessment and Follow-Up of Treatment for Osteoporosis. Adv. Ther. 2019. [Google Scholar] [CrossRef]
- Kanis, J.A.; Cooper, C.; Rizzoli, R.; Reginster, J.Y.; Scientific Advisory Board of the European Society for Clinical and Economic Aspects of Osteoporosis (ESCEO) and the Committees of Scientific Advisors and National Societies of the International Osteoporosis Foundation (IOF). European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos. Int. 2019, 30, 3–44. [Google Scholar] [CrossRef]
- Ripamonti, C.; Lisi, L.; Buffa, A.; Gnudi, S.; Caudarella, R. The Trabecular Bone Score Predicts Spine Fragility Fractures in Postmenopausal Caucasian Women Without Osteoporosis Independently of Bone Mineral Density. Med. Arch. 2018, 72, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Damasiewicz, M.J.; Nickolas, T.L. Rethinking Bone Disease in Kidney Disease. JBMR Plus 2018, 2, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.; Drüeke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G. Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006, 69, 1945–1953. [Google Scholar] [CrossRef] [PubMed]
- Jorgetti, V.; Drüeke, T.B.; Ott, S.M. Role of proton receptor OGR1 in bone response to metabolic acidosis? Kidney Int. 2016, 89, 529–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachman, A.; Bernstein, D.S. Diet and osteoporosis. Lancet 1968, 1, 958–959. [Google Scholar] [CrossRef]
- Nicoll, R.; McLaren Howard, J. The acid-ash hypothesis revisited: A reassessment of the impact of dietary acidity on bone. J. Bone Min. Metab. 2014, 32, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Frassetto, L.A.; Todd, K.M.; Morris, R.C., Jr.; Sebastian, A. Estimation of net endogenous noncarbonic acidproduction in humans from diet potassium and protein contents. Am. J. Clin. Nutr. 1998, 68, 576–583. [Google Scholar] [CrossRef]
- Remer, T.; Manz, F. Estimation of the renal net acid excretion by adults consuming diets containing variable amounts of protein. Am. J. Clin. Nutr. 1994, 59, 1356–1361. [Google Scholar] [CrossRef]
- Pachaly, M.A.; Baena, C.P.; Buiar, A.C.; de Fraga, F.S.; Carvalho, M. Effects of non-pharmacological interventions on urinary citrate levels: A systematic review and meta-analysis. Nephrol. Dial. Transpl. 2016, 31, 1203–1211. [Google Scholar] [CrossRef]
- Frassetto, L.; Banerjee, T.; Powe, N.; Sebastian, A. Acid Balance, Dietary Acid Load, and Bone Effects-A Controversial Subject. Nutrients 2018, 10, 517. [Google Scholar] [CrossRef]
- Fenton, T.R.; Lyon, A.W.; Eliasziw, M.; Tough, S.C.; Hanley, D.A. Meta-analysis of the effect of the acid-ash hypothesis of osteoporosis on calcium balance. J. Bone Min. Res. 2009, 24, 1835–1840. [Google Scholar] [CrossRef]
- Bonjour, J.P. Nutritional disturbance in acid-base balance and osteoporosis: A hypothesis that disregards the essential homeostatic role of the kidney. Br. J. Nutr. 2013, 110, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Carnauba, R.A.; Baptistella, A.B.; Paschoal, V.; Hübscher, G.H. Diet-Induced Low-Grade Metabolic Acidosis and Clinical Outcomes: A Review. Nutrients 2017, 9, 538. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, H.P.; Kinsella, R.; Duque, G. Osteosarcopenia: Where bone, muscle, and fat collide. Osteoporos. Int. 2017, 28, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Cases, A.; Cigarrán-Guldrís, S.; Mas, S.; Gonzalez-Parra, E. Vegetable-Based Diets for Chronic Kidney Disease? It Is Time to Reconsider. Nutrients 2019, 11, 1263. [Google Scholar] [CrossRef] [PubMed]
- Domrongkitchaiporn, S.; Stitchantrakul, W.; Kochakarn, W. Causes of hypocitraturia in recurrent calcium stone formers: Focusing on urinary potassium excretion. Am. J. Kidney Dis. 2006, 48, 546–554. [Google Scholar] [CrossRef]
- Odvina, C.V. Comparative value of orange juice versus lemonade in reducing stone-forming risk. Clin. J. Am. Soc. Nephrol. 2006, 1, 1269–1274. [Google Scholar] [CrossRef]
- Goldfarb, D.S.; Asplin, J.R. Effect of grapefruit juice on urinary lithogenicity. J. Urol. 2001, 166, 263–267. [Google Scholar] [CrossRef]
- Macdonald, H.M.; Black, A.J.; Aucott, L.; Duthie, G.; Duthie, S.; Sandison, R.; Hardcastle, A.C.; Lanham New, S.A.; Fraser, W.D.; Reid, D.M. Effect of potassium citrate supplementation or increased fruit and vegetable intake on bone metabolism in healthy postmenopausal women: A randomized controlled trial. Am. J. Clin. Nutr. 2008, 88, 465–474. [Google Scholar] [CrossRef]
- Phillips, R.; Hanchanale, V.S.; Myatt, A.; Somani, B.; Nabi, G.; Biyani, C.S. Citrate salts for preventing and treating calcium containing kidney stones in adults. Cochrane Database Syst. Rev. 2015, 10, CD010057. [Google Scholar] [CrossRef]
- Kern, A.; Grimsby, G.; Mayo, H.; Baker, L.A. Medical and dietary interventions for preventing recurrent urinary stones in children. Cochrane Database Syst. Rev. 2017, 11, CD011252. [Google Scholar] [CrossRef]
- Lencel, P.; Magne, D. Inflammaging: The driving force in osteoporosis? Med. Hypotheses 2011, 76, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Lambert, H.; Frassetto, L.; Moore, J.B.; Torgerson, D.; Gannon, R.; Burckhardt, P.; Lanham-New, S. The effect of supplementation with alkaline potassium salts on bone metabolism: A meta-analysis. Osteoporos. Int. 2015, 26, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Sakhaee, K.; Maalouf, N.M.; Abrams, S.A.; Pak, C.Y. Effects of potassium alkali and calcium supplementation on bone turnover in postmenopausal women. J. Clin. Endocrinol. Metab. 2005, 90, 3528–3533. [Google Scholar] [CrossRef] [PubMed]
- Karp, H.J.; Ketola, M.E.; Lamberg-Allardt, C.J. Acute effects of calcium carbonate, calcium citrate and potassium citrate on markers of calcium and bone metabolism in young women. Br. J. Nutr. 2009, 102, 1341–1347. [Google Scholar] [CrossRef] [Green Version]
- Dawson-Hughes, B.; Dallal, G.E.; Krall, E.A.; Sadowski, L.; Sahyoun, N.; Tannenbaum, S. A Controlled Trial of the Effect of Calcium Supplementation on Bone Density in Postmenopausal Women. N. Engl. J. Med. 1990, 323, 878–883. [Google Scholar] [CrossRef]
- Dawson-Hughes, B.; Harris, S.S.; Krall, E.A.; Dallal, G.E. Effect of Calcium and Vitamin D Supplementation on Bone Density in Men and Women 65 Years of Age Or Older. N. Engl. J. Med. 1997, 337, 670–676. [Google Scholar] [CrossRef]
- Ruml, L.A.; Sakhaee, K.; Peterson, R.; Adams-Huet, B.; Pak, C.Y. The Effect of Calcium Citrate on Bone Density in the Early and Mid-Postmenopausal Period: A Randomized Placebo-Controlled Study. Am. J. Ther. 1999, 6, 303–311. [Google Scholar] [CrossRef]
- Sellmeyer, D.E.; Schloetter, M.; Sebastian, A. Potassium Citrate Prevents Increased Urine Calcium Excretion and Bone Resorption Induced by a High Sodium Chloride Diet. J. Clin. Endocrinol. Metab. 2002, 87, 2008–2012. [Google Scholar] [CrossRef]
- Dawson-Hughes, B.; Harris, S.S. Calcium Intake Influences the Association of Protein Intake with Rates of Bone Loss in Elderly Men and Women. Am. J. Clin. Nutr. 2002, 75, 773–779. [Google Scholar] [CrossRef]
- Marangella, M.; Di Stefano, M.; Casalis, S.; Berutti, S.; D’Amelio, P.; Isaia, G.C. Effects of potassium citrate supplementation on bone metabolism. Calcif. Tissue Int. 2004, 74, 330–335. [Google Scholar] [CrossRef]
- Kenny, A.M.; Prestwood, K.M.; Biskup, B.; Robbins, B.; Zayas, E.; Kleppinger, A.; Burleson, J.A.; Raisz, L.G. Comparison of the Effects of Calcium Loading with Calcium Citrate Or Calcium Carbonate on Bone Turnover in Postmenopausal Women. Osteoporos. Int. 2004, 15, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Jehle, S.; Zanetti, A.; Muser, J.; Hulter, H.N.; Krapf, R. Partial Neutralization of the Acidogenic Western Diet with Potassium Citrate Increases Bone Mass in Postmenopausal Women with Osteopenia. J. Am. Soc. Nephrol. 2006, 17, 3213–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.D.; Need, A.G.; Tucker, G.; Slobodian, P.; O’Loughlin, P.D.; Nordin, B.E. Suppression of parathyroid hormone and bone resorption by calcium carbonate and calcium citrate in postmenopausal women. Calcif. Tissue Int. 2008, 83, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Jehle, S.; Hulter, H.N.; Krapf, R. Effect of potassium citrate on bone density, microarchitecture, and fracture risk in healthy older adults without osteoporosis: A randomized controlled trial. J. Clin. Endocrinol. Metab. 2013, 98, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Moseley, K.F.; Weaver, C.M.; Appel, L.; Sebastian, A.; Sellmeyer, D.E. Potassium Citrate Supplementation Results in Sustained Improvement in Calcium Balance in Older Men and Women. J. Bone Min. Res. 2013, 28, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Gregory, N.S.; Kumar, R.; Stein, E.M.; Alexander, E.; Christos, P.; Bockman, R.S.; Rodman, J.S. Potassium Citrate Decreases Bone Resorption in Postmenopausal Women with Osteopenia: A Randomized, Double-Blind Clinical Trial. Endocr. Pr. 2015, 21, 1380–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Sathiyakumar, V.; Kapoor, K.; Jones, S.R.; Banach, M.; Martin, S.S.; Toth, P.P. Novel Therapeutic Targets for Managing Dyslipidemia. Trends Pharm. Sci. 2018, 39, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Liu, Z.; Li, S.; Zhu, X.; Lin, Y.; Han, J.; Duan, Z.; Jia, L.; Gui, B. Citrate attenuates vascular calcification in chronic renal failure rats. APMIS 2017, 125, 452–458. [Google Scholar] [CrossRef]
- Michalczuk, M.; Urban, B.; Porowski, T.; Wasilewska, A.; Bakunowicz-Łazarczyk, A. Citrate usage in the leading causes of blindness: New possibilities for the old metabolite. Metabolomics 2018, 14, 82. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cause | Annotation |
|---|---|
| Acid-base status [16,23] |
|
| Hypokalemia [16,23] |
|
| Diet [24,25] |
|
| Distal renal tubular acidosis (dRTA) [26] |
|
| Chronic diarrheal syndrome [16,23] |
|
| Medications [16,23,27,28] |
|
| Strenuous physical exercise [23] |
|
| Hyperuricosuria [23] |
|
| Active urinary tract infection [23] |
|
| Chronic kidney disease (CKD) [29] |
|
| Primary hyperaldosteronism [30] |
|
| Menopause [31,32,33,34] |
|
| Genetic defects [16] |
|
| Gene/Locus Name | Gene/Locus | Cytogenetic Location | MIM Number: Phenotype | Inheritance |
|---|---|---|---|---|
| Solute carrier family 4, anion exchanger, member 1 (erythrocyte membrane protein band 3, Diego blood group) | SLC4A1, AE1, EPB3, SPH4, SAO, CHC | 17q21.31 | 179800: Distal renal tubular acidosis | Autosomal dominant |
| Solute carrier family 4, anion exchanger, member 1 (erythrocyte membrane protein band 3, Diego blood group) | SLC4A1, AE1, EPB3, SPH4, SAO, CHC | 17q21.31 | 611590: Distal renal tubular acidosis | Autosomal recessive |
| Glucose-6-phosphatase, catalytic | G6PC, G6PT | 17q21.31 | 232200: Glycogen storage disease Ia | Autosomal recessive |
| Solute carrier family 13 (sodium-dependent citrate transporter), member 5 | SLC13A5, NACT, INDY | 17p13.1 | 615905: Early infantile, epileptic encephalopathy, 25 | Autosomal recessive |
| Solute carrier family 12 (sodium/potassium/chloride transporters), member 1 | SLC12A1, NKCC2 | 15q21.1 | 60167: Bartter syndrome, type 1 | Autosomal recessive |
| Claudin 16 (paracellin 1) | CLDN16, PCLN1, HOMG3 | 3q28 | 248250: Renal hypomagnesemia 3 | Autosomal recessive |
| Reference | Study Design; Population | Intervention (Dose/Day) (I) Control (C) | Other Supplements (Dose/Day) and/or Controlled Dietary Intake | Follow Up and Outcomes | BTM Changes (Intragroup) | Changes in BTM and BMD Induced by Intervention (Intergroup) | Conclusion |
|---|---|---|---|---|---|---|---|
| Dawson-Hughes, 1990 [141] | RCT, controlled vs placebo, double-blind; ≥6 months postmenopausal women (early, <5 years: n = 67; late, >5 years: n = 169); age ≥ 65 years | I 1: Ca citrate malate (500 mg Ca), n = 78 I 2: Ca carbonate (500 mg Ca), n = 78 C: Placebo (n = 80) | Controlled Ca intake | Baseline, 18, 24, 36 months; BTM (BAP) and BMD | I 1: 24 months ↓ BAP I 2: 24 months ↓ BAP C: 24 months ↓ BAP | BTM I 1 vs. C: 36 months ↓ BAP, related to the Ca intake I 2 vs. C: 36 months ↓ BAP, related to the Ca intake BMD I 1 vs. C: 12, 24 months ↑ only in late postmenopause and Ca intake ≤400 mg/day I 2 vs C: ↓ in both groups | Adequate Ca intake is essential in preventing postmenopausal bone loss; Ca citrate is more effective than Ca carbonate. |
| Dawson-Hughes, 1997 [142] | RCT, controlled vs. placebo, double-blind; healthy subjects living in a community (176 M/ 213 F); age ≥ 65 years | I: Ca citrate malate (500 mg Ca) & Vit D3 (700 IU), n = 187 C: Placebo, n = 202 | Controlled Ca intake | Baseline, 6, 12, 18, 24, 30, 36 months; BTM (OC, u-NTX) and BMD | I: n.s C: n.s. | BTM I vs. C: 36 months ↓ OC BMD I vs. C: 36 months ↑ | Ca and vitamin D supplementation leads to a moderate reduction in bone loss and may substantially reduce the risk of nonvertebral fractures among elderly subjects who live in the community. |
| * Ruml, 1999 [143] | RCT, controlled vs. placebo; postmenopausal women (90% ≤5 years) | I: Ca citrate (800 mg Ca), n = 25 C: Placebo, n = 31 | Baseline, 12, 24 months BTM (BAP, OC, u-NTX, u- OH proline) and BMD | I: all BTMs are ↓, at unspecified time points | BMD I: 24 months, stable | Ca citrate supplementation averted bone loss and stabilised bone density in early postmenopausal women. | |
| Sellmeyer, 2002 [144] | RCT, controlled vs. placebo, double-blind; ≥2 years postmenopausal women; age I: 65 ± 8 years; C: 63 ± 8 years | I: K citrate (90 mmol), n = 26 C: Placebo, n = 26 | Ca carbonate (500 mg); controlled salt intake | Baseline, 1 months; BTM (OC, u-NTX) | I: n.s. C: 1 month ↓ OC, ↑ u-NTX | BTM I vs. C: 1 month, ↓ u-NTX | K citrate prevents increased bone resorption due to high salt intake. |
| Dawson-Hughes, 2002 [145] | RCT, controlled vs. placebo, double-blind; healthy subjects (161 M/ 181F); normal BMD; age ≥ 65 years | I: Ca citrate malate (500 mg Ca), n = 158 C: Placebo, n = 184 | Vitamin D3 (700 IU); controlled protein intake | Baseline, 18, 36 months; BTM (OC, u-NTX) and BMD | I: 36 months ↓ u-NTX, related to the protein intake; C: n.s. | BTM I vs C: 36 months, ↓ u-NTX BMD I vs C: 36 month, ↑ related to the protein intake | BMD may be improved by increasing protein intake as long as an adequate intake of Ca and vitamin D is assumed. |
| Marangella, 2004 [146] | Controlled vs. untreated; postmenopausal women; T score: <−1.0; age: 43–72 years | I: K citrate 37-74 mEq (≈1 mEq/kg), n = 30 C: No treatment, n = 24 | Controlled Ca intake | Baseline, 3 months BTM (BAP, OC, u-OH proline, u-DPD) | I: 3 months ↓ OC, u-OH proline, u-DPD; C: 3 months ↑ OC | not shown | K citrate decreases bone resorption thereby contrasting the potential adverse effects caused by chronic acidemia. The implication for the prevention and treatment of postmenopausal osteoporosis has to be confirmed. |
| Kenny, 2004 [147] | RCT, crossover, open label, 2 phases; 3 months/phase with a washout period of 2 weeks between phases; postmenopausal women; T score: <−1.0 and >−3.5; age: 73 ± 5 years | I 1: Ca citrate (1000 mg Ca), n = 20; I 2: Ca carbonate (1000 mg Ca), n = 20 | Vitamin D3 (900 IU); controlled Ca intake | Baseline, 1, 3 months (each phase) BTM (BAP, OC, NTX, u-CTX, u-NTX, u-DPD) | I 1: 3 months ↓ NTX, u-CTX, u-NTX, u-DPD I 2: n.s | Ca citrate inhibits bone resorption more than Ca carbonate. | |
| Sakhae, 2005 [139] | RCT, crossover, placebo controlled, double-blind, 4 phases; 2 weeks/phase with a washout period of 2 weeks between phases; postmenopausal women; age: 48–76 years | I 1: K citrate (40 mEq), n = 18 I 2: Ca citrate (800 mg), n = 18 I 3: K citrate (40 mEq) and Ca citrate (800 mg), n = 18 C (1st phase): Placebo, n = 18 | Rigid diet with fixed content of protein, Ca, P, Na, K and fluids | Baseline and at the end of each phase; BTM (BAP, CTX, OC, u-NTX, u-OH proline) | I 1: n.s I 2: ↓ CTX, u-OH proline I 3: I: ↓ CTX, u-OH proline, u NTX | I 3 vs I 1: ↓ u NTX | In postmenopausal women, combined treatment with K citrate and Ca citrate decreases bone resorption by providing an alkali load and increasing absorbed Ca. |
| Jehle, 2006 [148] | RCT, controlled; ≥5 years postmenopausal women; T score −1/−4; age: ≤70 years | I: K citrate (30 mEq), n = 82 C: KCl (30 mmol), n = 79 | Ca carbonate (500 mg), Vitamin D3 (400 IU); free, nonvegetarian diet | Baseline, 3, 6, 9, 12 months; BTM (BAP, CTX, OC, u-DPD, u-PD) and BMD | I: 3 months, ↓ u-DPD, u-PD; 6 months, ↑ BAP and ↓ OC, u-DPD, u-PD; 9 months, ↓ u-DPD, u-PD; 12 months, ↑ BAP and ↓ OC, u-DPD, u-PD; C: 3 months, ↓ OC, u-DPD, u-PD; 6 months, ↑ BAP, u-DPD, u-PD and ↓ OC; 9 months, ↑ u-DPD, u-PD and ↓ OC; 12 months, ↑ BAP, u-DPD, u-PD and ↓ OC | BTM I vs C: 3 months, ↓ u-DPD BMD I vs C: 12 months ↑ | In postmenopausal women, bone mass can be increased significantly by K citrate. The effect on bone resorption seems to be unrelated to K intake. |
| Macdonald, 2008 [134] | RCT, controlled vs. placebo, double-blind for I1 e I2; ≥5 years postmenopausal women; age: 49–54 years | I 1: K citrate (55.5 mEq), n = 70 I 2: K citrate (18.5 mEq), n = 70 I 3: Diet (300 g fruit = 18.5 mEq alkali), n = 66 C: Placebo, n = 70 | Food diary (free nonvegetarian diet) | Baseline, 3, 6, 12, 18, 24 months; BTM (CTX, P1NP, u-DPD) and BMD | I 1: n.s. I 2: n.s. I 3: n.s. C: n.s | BTM I 1, I 2, I 3 vs. C: n.s BMD I 1, I 2, I 3 vs. C: n.s | In healthy postmenopausal women, neither K citrate at 18.5 or 55.6 mEq/d, nor 300 g self-selected fruit and vegetables influenced bone turnover or prevented BMD loss over 2 years. |
| Thomas, 2008 [149] | RCT, crossover, double-blind, 2 phases; postmenopausal women for 2 to 6 years; age: 50–60 years | I 1: Ca carbonate (1000 mg Ca), n = 12 I 2: 2) Ca citrate (500 mg Ca), n = 13 | Controlled Ca intake | Baseline, 7 days; BTM (CTX) | I 1: 7 days, ↓ CTX I 2: 7 days, ↓ CTX | Ca citrate is at least as effective as Ca carbonate in decreasing PTH and CTX cross-links, at half the dose. All changes are numerically superior after Ca citrate supplementation. | |
| Karp, 2009 [140] | RCT, controlled, 24 h study sessions; women of child-bearing age: 22–30 years | I 1: Ca carbonate (1000 mg Ca), n = 12 I 2: Ca citrate (Ca: 1000 mg; citrate: 3145 mg), n = 12 I 3: K citrate (K: 57 mEq; citrate: 3145 mg), n = 12 | 4-day diary to estimate food habits before starting the study session; the meals served during each study session were identical | Baseline, 2, 4, 6, 8, 20, 24 h; BTM (BAP, u-NTX) and BMD | I 1: 24 h, ↓ u-NTX I 2: n.s I 3: 24 h, ↓ u-NTX | K citrate supplementation decreases urinary Ca excretion and reduces bone resorption even when the diet is not acidogenic, and reduces the bone resorption marker despite low Ca intake. | |
| Jehle, 2013 [150] | RCT, controlled vs. placebo, double-blind; healthy subjects (79 M/122 F); T score > −2.5; age 65–80 years; women were past the perimenopausal peak turnover | I: K citrate (60 mEq), n = 101 C: Placebo, n = 100 | Ca carbonate (500 mg), Vitamin D3 (400 IU); free nonvegetarian diet | Baseline, 6, 12, 18, 24; BTM (BAP, P1NP, u-NTX) and BMD | I: 6, 12 months, ↓ u-NTX; 18, 24 months, ↑ P1NP C: n.s. | BTM I vs. C: 6 months, ↓ u-NTX BMD I vs. C: 12, 18, 24 months ↑ | K citrate administered in a background of vitamin D and Ca supplementation is well tolerated and constitutes an inexpensive intervention to improve BMD and bone microarchitecture in healthy elderly people. |
| Moseley, 2013 [151] | RCT, controlled vs. placebo, double blind; healthy subjects (17 M/35 F); age ≥ 55 years; women were ≥5 years postmenopause | I 1: K citrate (60 mmol), n = 17 I 2: K citrate (90 mmol), n = 17 C: Placebo, n = 18 | Ca citrate (630 mg), Vitamin D3 (400 IU); controlled Ca, Na, P, protein, fat intake | Baseline, 6 months; BTM, (BAP, CTX) | BTM I 1, I2 vs. C: 6 months, ↓ CTX | K citrate decreases markers of bone resorption over 6 months, but a significant improvement in Ca balance is obtained with 90 mmol/day. This dose is well tolerated. | |
| Gregory, 2015 [152] | RCT, controlled vs. placebo, double-blind; ≥2 year postmenopausal women s; T score: <−1.0 > −2.5, or <−2.5 unable to take any other medication; age I: 65.1 ± 5.9 years; C: 66.1 ± 7.1 years | I: K citrate (40 mEq), n = 42 C: Placebo, n = 41 | Ca citrate (630 mg), Vitamin D3 (400 IU); free nonvegetarian diet | Baseline, 1, 3, 6, 12 months; BTM (BAP, OC, P1NP, u-NTX) and BMD | I: 1 month, ↓ P1NP; 3, 6, 12 months, ↓ P1NP, u-NTX C. I: 6 months, ↓ P1NP u-NTX; 12 months, ↓ P1NP | BTM I vs. C: n.s BMD I: 12 months, stable | In postmenopausal osteopenia, K citrate improves the effect of supplementation with Ca citrate and Vitamin D, as proven by the more rapid decrease in BTM levels. |
| Granchi, 2018 [91] | RCT, controlled vs. placebo, double-blind; ≥5 years postmenopausal women; T score: <−1.0 and >−2.5; age I: 60.8 ± 1.0 years; C: 58.2 ± 1.1 years | I: K citrate (30 mEq), n = 20 C: Placebo, n = 20 | Ca carbonate (500 mg), Vitamin D3 (400 IU); free nonvegetarian diet | Baseline, 3, 6 months; BTM (BAP, CTX, P1NP, TRAcP) | I: 6 months, ↓ BAP, CTX C: 3, 6 months, ↓ BAP, CTX | BTM I vs. C: 6 months ↓ BAP, CTX in subjects with low excretion of K and/or citrate, and/or low urine pH | In postmenopausal osteopenia, K citrate improves the effects of supplementation with Ca carbonate and vitamin D, but only in women with low K and/or citrate excretion and/or low urine pH. |
| Highlights |
|---|
|
|
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granchi, D.; Baldini, N.; Ulivieri, F.M.; Caudarella, R. Role of Citrate in Pathophysiology and Medical Management of Bone Diseases. Nutrients 2019, 11, 2576. https://doi.org/10.3390/nu11112576
Granchi D, Baldini N, Ulivieri FM, Caudarella R. Role of Citrate in Pathophysiology and Medical Management of Bone Diseases. Nutrients. 2019; 11(11):2576. https://doi.org/10.3390/nu11112576
Chicago/Turabian StyleGranchi, Donatella, Nicola Baldini, Fabio Massimo Ulivieri, and Renata Caudarella. 2019. "Role of Citrate in Pathophysiology and Medical Management of Bone Diseases" Nutrients 11, no. 11: 2576. https://doi.org/10.3390/nu11112576
APA StyleGranchi, D., Baldini, N., Ulivieri, F. M., & Caudarella, R. (2019). Role of Citrate in Pathophysiology and Medical Management of Bone Diseases. Nutrients, 11(11), 2576. https://doi.org/10.3390/nu11112576