Atorvastatin and Vitamin E Accelerates NASH Resolution by Dietary Intervention in a Preclinical Guinea Pig Model

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
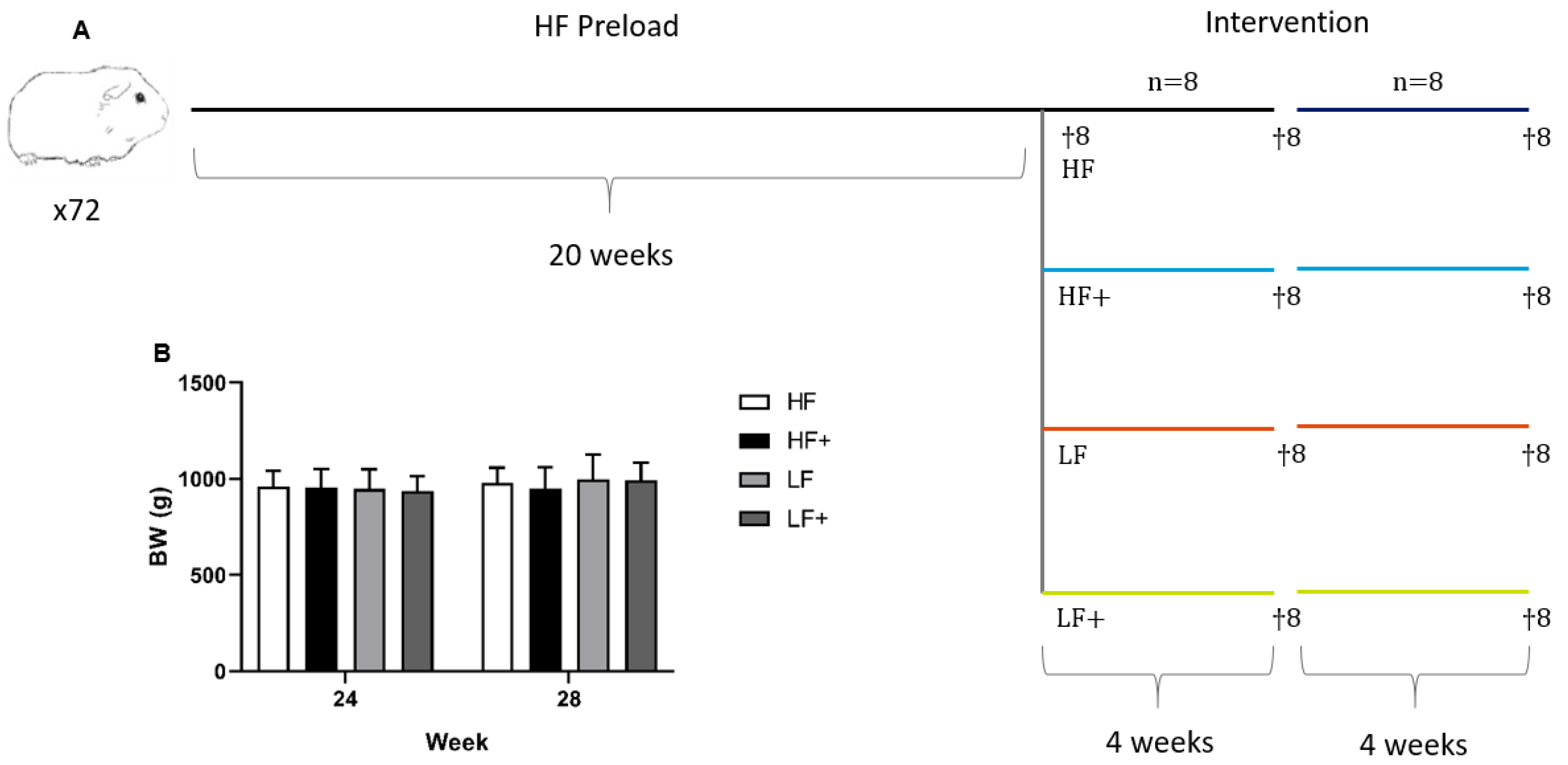
2.1. Animals and Experimental Design
2.2. Plasma Samples
2.3. Liver Samples
2.4. Histology
2.5. qPCR
2.6. Statistical Analyses
3. Results
3.1. Plasma and Hepatic Vitamin E Status
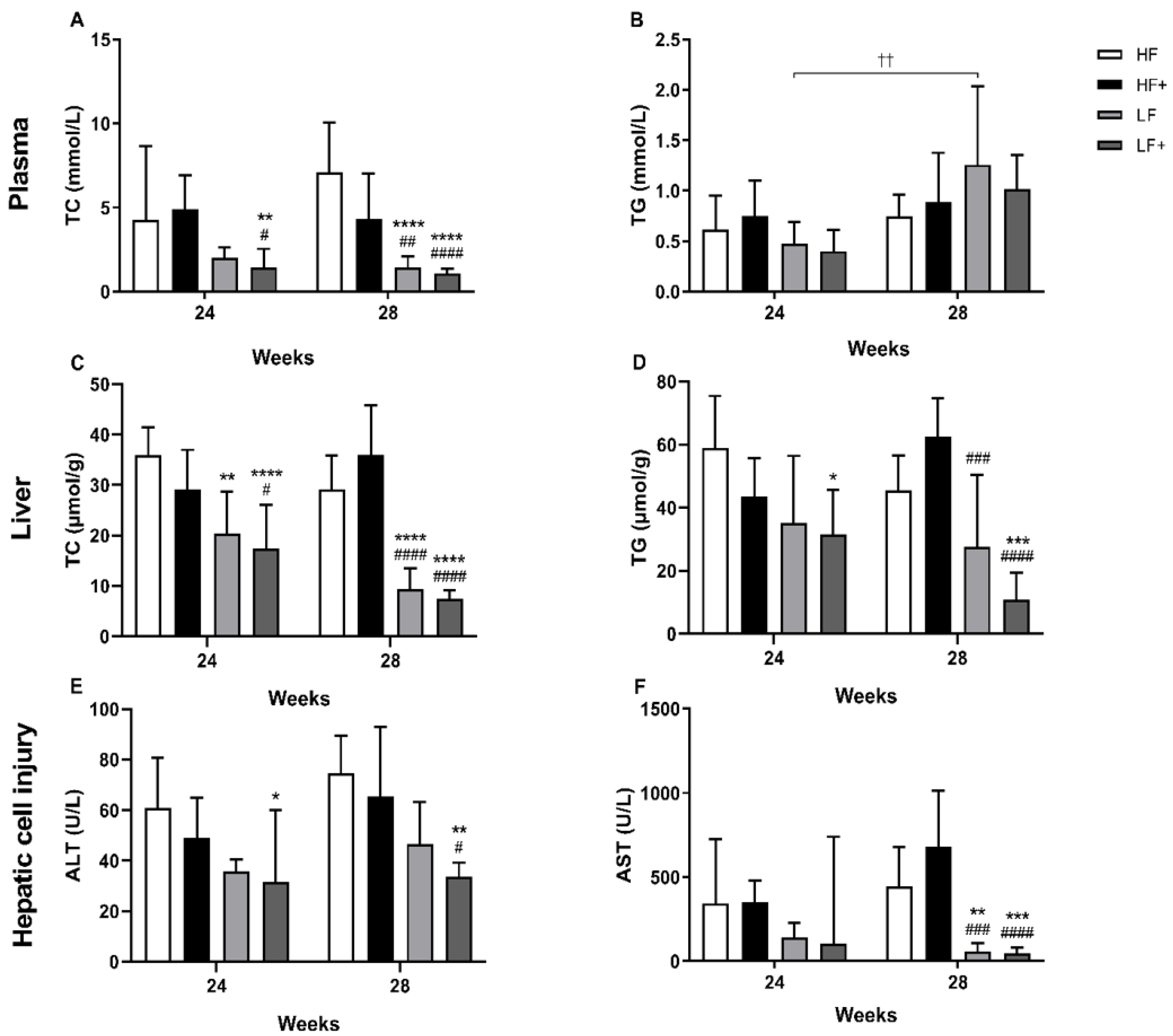
3.2. Dyslipidemia
3.3. Biochemical Markers
3.4. Liver Status
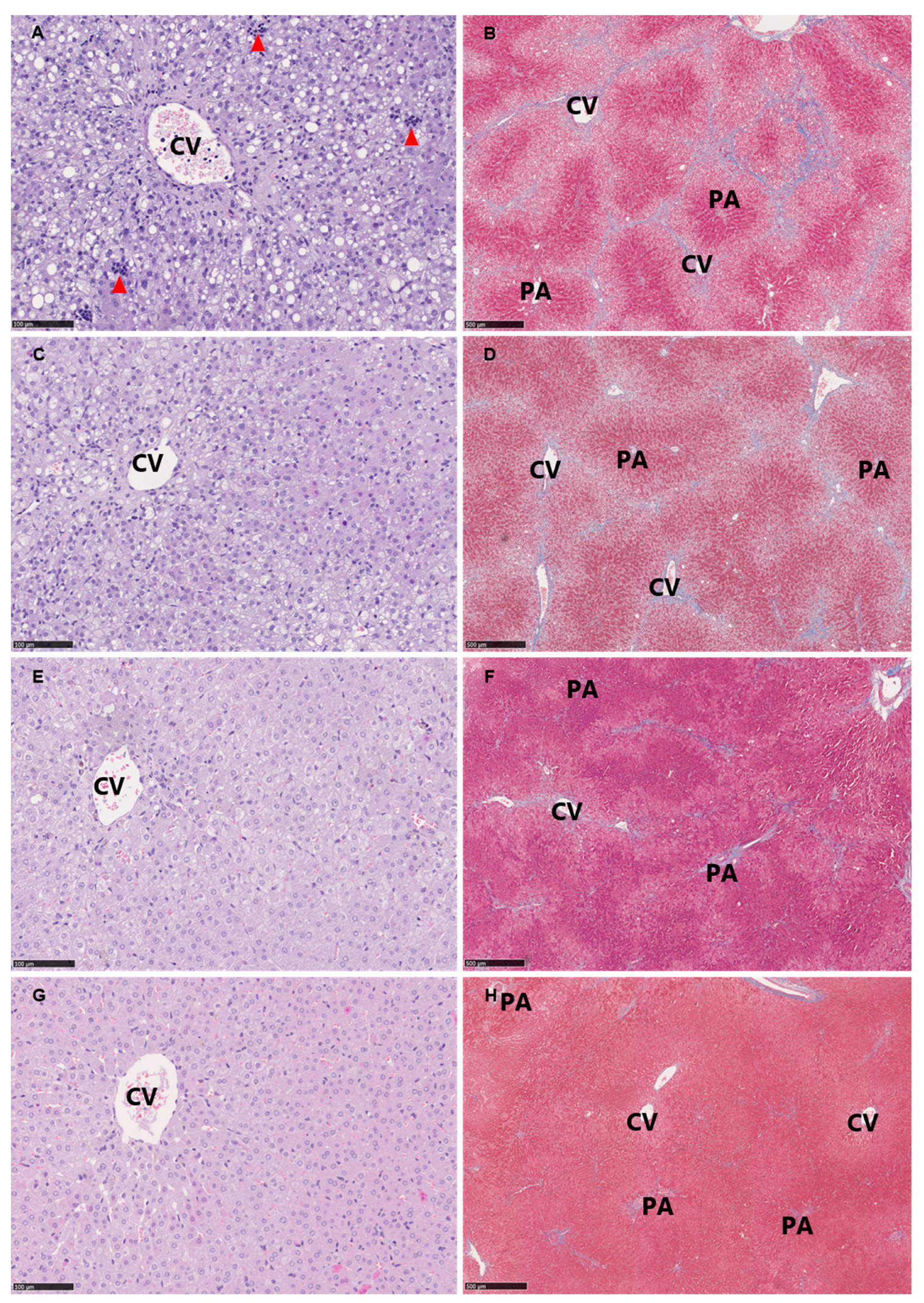
3.5. Histopathological Evaluation
3.6. Expression of Target Genes/qPCR
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.; Arrese, T.M.F.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.; Yilmaz, Y.; George, J.; Fan, J.; Vos, M.B. Global Perspectives on Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis. Hepatology 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (Nafld). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Birck, M.M.; Ipsen, D.H.; Thiessen, T.; Feldmann, L.B.; Lindblad, M.M.; Jensen, H.E.; Lykkesfeldt, J. Diet-Induced Dyslipidemia Leads to Nonalcoholic Fatty Liver Disease and Oxidative Stress in Guinea Pigs. Transl. Res. 2016, 168, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of Nash. Int. J. Mol. Sci. 2016, 17, 1575. [Google Scholar] [CrossRef]
- Ekstedt, M.; Hagstrom, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis Stage Is the Strongest Predictor for Disease-Specific Mortality in Nafld after up to 33 Years of Follow-Up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Rafiq, N.; Makhlouf, H.; Younoszai, Z.; Agrawal, R.; Goodman, Z. Pathologic Criteria for Nonalcoholic Steatohepatitis: Interprotocol Agreement and Ability to Predict Liver-Related Mortality. Hepatology 2011, 53, 1874–1882. [Google Scholar] [CrossRef]
- Kim, D.; Kim, W.R.; Kim, H.J.; Therneau, T.M. Association between Noninvasive Fibrosis Markers and Mortality among Adults with Nonalcoholic Fatty Liver Disease in the United States. Hepatology 2013, 57, 1357–1365. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. Diseases American Association for the Study of Liver, Gastroenterology American College of, and Association American Gastroenterological. The Diagnosis and Management of Non-Alcoholic Fatty Liver Disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Am. J. Gastroenterol. 2012, 107, 811–826. [Google Scholar] [CrossRef]
- Catapano, A.L.; Graham, I.; De Backer, G.; Wiklund, O.; Chapman, M.J.; Drexel, H.; Hoes, A.W.; Jennings, C.S.; Landmesser, U.; Pedersen, T.R.; et al. 2016 Esc/Eas Guidelines for the Management of Dyslipidaemias. Rev. Esp. Cardiol. 2017, 70, 115. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). Easl-Easd-Easo Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Banini, B.A.; Sanyal, A.J. Nonalcoholic Fatty Liver Disease: Epidemiology, Pathogenesis, Natural History, Diagnosis, and Current Treatment Options. Clin. Med. Insights Ther. 2016, 8, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Hyogo, H.; Tazuma, S.; Arihiro, K.; Iwamoto, K.; Nabeshima, Y.; Inoue, M.; Ishitobi, T.; Nonaka, M.; Chayama, K. Efficacy of Atorvastatin for the Treatment of Nonalcoholic Steatohepatitis with Dyslipidemia. Metabolism 2008, 57, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Athyros, V.G.; Boutari, C.; Stavropoulos, K.; Anagnostis, P.; Imprialos, K.P.; Doumas, M.; Karagiannis, A. Statins: An under-Appreciated Asset for the Prevention and the Treatment of Nafld or Nash and the Related Cardiovascular Risk. Curr. Vasc. Pharmacol. 2018, 16, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Nagashimada, M.; Ota, T. Role of Vitamin E in Nonalcoholic Fatty Liver Disease. IUBMB Life 2019, 71, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Gosho, M.; Yamamoto, T.; Kobayashi, Y.; Ishii, N.; Ohashi, T.; Nakade, Y.; Ito, K.; Fukuzawa, Y.; Yoneda, M. Vitamin E Has a Beneficial Effect on Nonalcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. Nutrition 2015, 31, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Tao, A.; Zhang, S.; Deng, Y.; Chen, G. Association between Vitamin E and Non-Alcoholic Steatohepatitis: A Meta-Analysis. Int. J. Clin. Exp. Med. 2015, 8, 3924–3934. [Google Scholar]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Role of Vitamin E in the Treatment of Nafld. Diseases 2018, 6, 86. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Tveden-Nyborg, P.; Rolin, B.; Rakipovski, G.; Beck, M.; Mortensen, L.W.; Faerk, L.; Heegaard, P.M.; Moller, P.; Lykkesfeldt, J. High-Fat but Not Sucrose Intake Is Essential for Induction of Dyslipidemia and Non-Alcoholic Steatohepatitis in Guinea Pigs. Nutr. Metab. 2016, 13, 51. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Skat-Rordam, J.; Tsamouri, M.M.; Latta, M.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Drivers of Non-Alcoholic Steatohepatitis Are Sustained in Mild-to-Late Fibrosis Progression in a Guinea Pig Model. Mol. Genet Genom. 2019, 294, 649–661. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Ascorbate and Dehydroascorbic Acid as Reliable Biomarkers of Oxidative Stress: Analytical Reproducibility and Long-Term Stability of Plasma Samples Subjected to Acidic Deproteinization. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2513–2516. [Google Scholar] [CrossRef]
- Reimann, M.J.; Haggstrom, J.; Mortensen, A.; Lykkesfeldt, J.; Moller, J.E.; Falk, T.; Olsen, L.H. Biopterin Status in Dogs with Myxomatous Mitral Valve Disease Is Associated with Disease Severity and Cardiovascular Risk Factors. J. Vet. Intern. Med. 2014, 28, 1520–1526. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, A.; Hasselholt, S.; Tveden-Nyborg, P.; Lykkesfeldt, J. Guinea Pig Ascorbate Status Predicts Tetrahydrobiopterin Plasma Concentration and Oxidation Ratio in Vivo. Nutr. Res. 2013, 33, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Ekeloef, S.; Larsen, M.H.; Schou-Pedersen, A.M.; Lykkesfeldt, J.; Rosenberg, J.; Gogenur, I. Endothelial Dysfunction in the Early Postoperative Period after Major Colon Cancer Surgery. Br. J. Anaesth. 2017, 118, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Hissin, P.J.; Hilf, R. A Fluorometric Method for Determination of Oxidized and Reduced Glutathione in Tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Determination of Malondialdehyde as Dithiobarbituric Acid Adduct in Biological Samples by Hplc with Fluorescence Detection: Comparison with Ultraviolet-Visible Spectrophotometry. Clin. Chem. 2001, 47, 1725–1727. [Google Scholar]
- Burton, G.W.; Webb, A.; Ingold, K.U. A Mild, Rapid, and Efficient Method of Lipid Extraction for Use in Determining Vitamin E/Lipid Ratios. Lipids 1985, 20, 29–39. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. And Network Nonalcoholic Steatohepatitis Clinical Research. Design and Validation of a Histological Scoring System for Nonalcoholic Fatty Liver Disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Hasselholt, S.; Miyashita, N.; Moos, T.; Poulsen, H.E.; Lykkesfeldt, J. Chronic Vitamin C Deficiency Does Not Accelerate Oxidative Stress in Ageing Brains of Guinea Pigs. Basic Clin. Pharmacol. Toxicol. 2012, 110, 524–529. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of Nafld Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and Future Pharmacological Therapies for Nafld/Nash. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Hadzi-Petrushev, N.; Dimovska, K.; Jankulovski, N.; Mitrov, D.; Mladenov, M. Supplementation with Alpha-Tocopherol and Ascorbic Acid to Nonalcoholic Fatty Liver Disease’s Statin Therapy in Men. Adv. Pharmacol. Sci. 2018, 2018, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Costache, I.I.; Garleanu, I.; Aursulesei, V.; Namat, R.A.; Ion, A.; Miftode, R.S.; Tesloianu, D.; Iliescu, D.; Petris, A.O.; Costache, A.D.; et al. Atorvastatin in the Treatment of Dyslipidemic Patients with Very High Cardiovascular Risk and Nonalcoholic Fatty Liver Disease. Rev. Chim. 2019, 70, 2159–2163. [Google Scholar]
- Sigler, M.A.; Congdon, L.; Edwards, K.L. An Evidence-Based Review of Statin Use in Patients with Nonalcoholic Fatty Liver Disease. Clin. Med. Insights Gastroenterol. 2018, 11, 1179552218787502. [Google Scholar] [CrossRef]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. And Network Nonalcoholic Steatohepatitis Clinical Research. Effect of Vitamin E or Metformin for Treatment of Nonalcoholic Fatty Liver Disease in Children and Adolescents: The Tonic Randomized Controlled Trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef]
- Athyros, V.G.; Tziomalos, K.; Gossios, T.D.; Griva, T.; Anagnostis, P.; Kargiotis, K.; Pagourelias, E.D.; Theocharidou, E.; Karagiannis, A.; Mikhailidis, D.P. Greace Study Collaborative Group. Safety and Efficacy of Long-Term Statin Treatment for Cardiovascular Events in Patients with Coronary Heart Disease and Abnormal Liver Tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (Greace) Study: A Post-Hoc Analysis. Lancet 2010, 376, 1916–1922. [Google Scholar] [CrossRef]
- Oshakbayev, K.; Bimbetov, B.; Manekenova, K.; Bedelbayeva, G.; Mustafin, K.; Dukenbayeva, B. Severe Nonalcoholic Steatohepatitis and Type 2 Diabetes: Liver Histology after Weight Loss Therapy in a Randomized Clinical Trial. Curr. Med. Res. Opin. 2019, 35, 157–165. [Google Scholar] [CrossRef]
- Chalasani, N.P.; Sanyal, A.J.; Kowdley, K.V.; Robuck, P.R.; Hoofnagle, J.; Kleiner, D.E.; Unalp, A.; Tonascia, J. And Nash Crn Research Group. Pioglitazone Versus Vitamin E Versus Placebo for the Treatment of Non-Diabetic Patients with Non-Alcoholic Steatohepatitis: Pivens Trial Design. Contemp. Clin. Trials 2009, 30, 88–96. [Google Scholar] [CrossRef]
- Connolly, J.J.; Ooka, K.; Lim, J.K. Future Pharmacotherapy for Non-Alcoholic Steatohepatitis (Nash): Review of Phase 2 and 3 Trials. J. Clin. Transl. Hepatol. 2018, 6, 264–275. [Google Scholar] [CrossRef]
- Okada, Y.; Yamaguchi, K.; Nakajima, T.; Nishikawa, T.; Jo, M.; Mitsumoto, Y.; Kimura, H.; Nishimura, T.; Tochiki, N.; Yasui, K.; et al. Rosuvastatin Ameliorates High-Fat and High-Cholesterol Diet-Induced Nonalcoholic Steatohepatitis in Rats. Liver Int. 2013, 33, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Yoneda, M.; Nakamura, K.; Makino, I.; Terano, A. Plasma Transforming Growth Factor-Beta1 Level and Efficacy of Alpha-Tocopherol in Patients with Non-Alcoholic Steatohepatitis: A Pilot Study. Aliment. Pharmacol. Ther. 2001, 15, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. Tgf-Beta in Progression of Liver Disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Nemes, K.; Aberg, F. Interpreting Lipoproteins in Nonalcoholic Fatty Liver Disease. Curr. Opin. Lipidol. 2017, 28, 355–360. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, K.L.; Ruan, X.Z.; Liu, B.C. Dysregulation of the Low-Density Lipoprotein Receptor Pathway Is Involved in Lipid Disorder-Mediated Organ Injury. Int. J. Biol. Sci. 2016, 12, 569–579. [Google Scholar] [CrossRef]
- Kayden, H.J.; Traber, M.G. Lipoprotein Transport, and Regulation of Plasma Concentrations of Vitamin E in Humans. J. Lipid Res. 1993, 34, 343–358. [Google Scholar]
- Jeanes, Y.M.; Hall, W.L.; Ellard, S.; Lee, E.; Lodge, J.K. The Absorption of Vitamin E Is Influenced by the Amount of Fat in a Meal and the Food Matrix. Br. J. Nutr. 2004, 92, 575–579. [Google Scholar] [CrossRef]
- Traber, M.G.; Leonard, S.W.; Ebenuwa, I.; Violet, P.C.; Wang, Y.; Niyyati, M.; Padayatty, S.; Tu, H.; Courville, A.; Bernstein, S.; et al. Vitamin E Absorption and Kinetics in Healthy Women, as Modulated by Food and by Fat, Studied Using 2 Deuterium-Labeled Alpha-Tocopherols in a 3-Phase Crossover Design. Am. J. Clin. Nutr. 2019, 110, 1148–1167. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nutrient | HF | HF+ | LF | LF+ |
|---|---|---|---|---|
| Protein (%) | 16.7 | 16.7 | 16.8 | 16.8 |
| Carbohydrates (%) | 37.9 | 37.9 | 47.1 | 47.1 |
| Fat (%) | 20 | 20 | 4 | 4 |
| Cholesterol (%) | 0.35 | 0.35 | - | - |
| Sucrose (%) | 15 | 15 | - | - |
| Vitamin E (all-rac-alpha-tocopheryl acetate (mg/kg feed)) | 125 | 375 | 125 | 375 |
| Atorvastatin (mg/kg feed) | - | 20 | - | 20 |
| Gene | Accession no. | Forward (5’-3’) | Reverse (3’-5’) | Product (bp) |
|---|---|---|---|---|
| HMGR | XM_003461336.3 | TACAGACATGGGCATTGG GT | GGCAGGGAAAGTGTTGAGTG | 184 |
| LDLR | XM_013149927.1 | GACGTGTCCCAGAGG AAGAT | CGAGTCGGTCCAGTAGATGTT | 144 |
| SREBP2 | XM_003470391.3 | GGGGCTCAAAGGTCTTCTCT | AGGACCCCATCAAAGTGAGG | 189 |
| ABCG5 | XM_003472925.3 | ATCCTGAGGCTGCTCGATTT | CCAGATCCAATCAGCAACCC | 163 |
| ABCG8 | XM_003472926.2 | GTCTCAACTCCCACCCTCTC | CTGAAGGGTCTTCTCCGAGG | 170 |
| CYP7a1 | GQ507494.1 | CTGGAGAAGGCAGGTCAACA | CTCCTTAGCTGTCCGGATGT | 150 |
| IL-8 | NM_001173399.2 | GGCAGCCTTCCTGCTCTCT | CAGCTCCGAGACCAACTTTGT | 67 |
| MCP-1 | NM_001172926.1 | TGCCAAACTGGACCAGAGAA | CGAATGTTCAAAGGCTTTGAAGT | 75 |
| TNF-α | NM_001173025.1 | GCCGTCTCCTACCCGGAAAA | TAGATCTGCCCGGAATCGGC | 203 |
| TGF-β | NM_001173023.1 | AACCCGAGCCGGACTACTATG | TGCTTTTATAGATATTGTGGCTGT TGT | 78 |
| Col1a1 | XM_003466865.2 | CTGGACAGCGTGGTGTAGTC | TCCAGAAGGACCTTGTTTGC | 104 |
| α-sma | ENSCPOT00000011693.2 | GACATCAAGGAGAAGCTGTG | GCTGTTGTAGGTGGTTTCAT | 273 |
| Pre-intervention | Week 24 | Week 28 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HF | HF+ | LF | LF+ | HF | HF+ | LF | LF+ | ||
| FFA (mmol/L) | 0.41 ± 0.04 | 0.64 ± 0.17 | 0.40 ± 0.10 | 0.63 ± 0.20 | 0.61 ±0.16 | 0.50 ± 0.21 | 0.42 ± 0.14 | 0.58 ± 0.20 | 0.42 ± 0.17 |
| ALP (U/L) | 46.13 ± 9.73 | 37.38 ± 7.76 | 40.0 ± 7.60 | 48.0 ± 9.32 | 47.25 ± 12.61 | 41.90 ± 8.36 | 38.5 ± 7.35 | 51.88 ± 10.66 | 50.13 ± 13.81 |
| LW (%) j | 4.15 (3.44–4.86) | 5.15 (4.58−5.72) | 4.82 (4.10−5.54) | 3.34 (2.91−3.77) f, h | 3.18 (2.73−3.63) f, i | 4.60 (3.64−5.56) | 5.96 (4.76−7.16) | 3.15 (2.81−3.50) e, i | 2.85 (2.53−3.16) b, f, i |
| BH2/BH4 | 0.05 ± 0.02 | 0.13 ± 0.037 c | 0.11 ± 0.03 a | 0.13 ± 0.03 c | 0.12 ± 0.04 b | 0.08 ± 0.04 | 0.10 ± 0.04 | 0.08 ± 0.03 | 0.07 ± 0.03 |
| TAA (µM) | 40.56 ± 17.09 | 37.39 ± 14.40 | 32.28 ± 12.93 | 70.73 ± 27.94 a, e, i | 57.64 ± 19.34 | 28.69 ± 6.92 | 33.85 ± 15.51 | 55.05 ± 11.11 d | 56.51 ± 10.56 d |
| αToc (µM) j | 4.52 (2.84−6.20) | 3.10 (1.91−4.29) | 6.70 (3.37−10.03) d | 1.35 (0.89−1.82) b, i | 1.89 (1.32−2.46) i | 2.70 (1.73−3.68) | 6.63 (2.88−10.38) d | 1.59 (0.94−2.23) a, h | 2.69 (1.62−3.77) g |
| Pre-intervention | HF | HF+ | LF | LF+ | |
|---|---|---|---|---|---|
| AA (nmol/g) | 1283.5 ± 379.51 | 1343 ± 533.44 | 1230.88 ± 474.33 | 1381.5 ± 518.21 | 1663.13 ± 598.48 |
| BH2/BH4 | 0.21 ± 0.05 | 0.15 ± 0.10 | 0.23 ± 0.11 | 0.20 ± 0.12 | 0.23 ± 0.12 |
| L-arg (nmol/g) | 32.96 ± 8.73 | 30.47 ± 11.83 | 27.54 ± 7.10 | 29.14 ± 13.40 | 21.60 ± 6.54 |
| ADMA (nmol/g) | 0.73 ± 0.92 | 0.40 ± 0.53 | 0.92 ± 1.20 | 0.50 ± 0.53 | 0.17 ± 0.50 |
| GSH (nmol/g) | 3160.63 ± 503.63 | 3131.5 ± 316.22 | 2967.75 ± 336.71 | 3305.90 ± 498.19 | 3118.00 ± 424.33 |
| GSSG (nmol/g) | 317.72 ± 49.40 | 331.40 ± 51.99 | 293.81 ± 83.70 | 315.01 ± 61.90 | 307.70 ± 49.51 |
| SOD (U/mg) | 2489.5 ± 432.10 | 2621.40 ± 343.50 | 2495.00 ± 782.92 | 2449.40 ± 405.90 | 3107.80 ± 952.75 |
| αToc (nmol/g) j | 1.71 (1.22–2.19) | 3.47 (0.92–6.03) | 5.14 (2.02–8.26) a | 3.39 (1.88–4.91) | 3.30 (1.53–5.06) |
| Pre-intervention | HF | HF+ | LF | LF+ | |
|---|---|---|---|---|---|
| AA (nmol/g) | 1283.5 ± 379.51 | 1404.5 ± 497.99 | 1357 ± 457.91 | 1287.75 ± 573.73 | 1770 ± 380.30 |
| BH2/BH4 | 0.21 ± 0.05 | 0.20 ± 0.12 | 0.30 ± 0.16 | 0.23 ± 0.10 | 0.23 ± 0.20 |
| L-arg (nmol/g) | 32.96 ± 8.73 | 22.10 ± 9.56 | 24.95 ± 6.47 | 23.60 ± 6.50 | 36.74 ± 10.80 |
| ADMA (nmol/g) | 0.73 ± 0.92 | 0.99 ± 0.80 | 0.70 ± 0.60 | 0.50 ± 0.60 | 0.91 ± 1.10 |
| GSH (nmol/g) | 3160.63 ± 503.63 | 3440.40 ± 487.40 | 3138.63 ± 301.32 | 3072.25 ± 305.80 | 3303.25 ± 170.97 |
| GSSG (nmol/g) | 317.72 ± 49.40 | 300.52 ± 69.90 | 295.40 ± 68.50 | 322.70 ± 32.30 | 324.80 ± 61.96 |
| SOD (U/mg) | 2489.5 ± 432.10 | 2783.90 ± 786.60 | 2512.00 ± 642.80 | 3206.40 ± 1059.80 | 2899.13 ± 502.82 |
| αToc (nmol/g) j | 1.71 (1.22–2.19) | 2.87 (0.90–4.84) | 9.43 (3.68–15.18) a, b, c | 3.49 (1.95–5.02) | 8.61 (4.98–12.24) a, b, c |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klaebel, J.H.; Skjødt, M.; Skat-Rørdam, J.; Rakipovski, G.; Ipsen, D.H.; Schou-Pedersen, A.M.V.; Lykkesfeldt, J.; Tveden-Nyborg, P. Atorvastatin and Vitamin E Accelerates NASH Resolution by Dietary Intervention in a Preclinical Guinea Pig Model. Nutrients 2019, 11, 2834. https://doi.org/10.3390/nu11112834
Klaebel JH, Skjødt M, Skat-Rørdam J, Rakipovski G, Ipsen DH, Schou-Pedersen AMV, Lykkesfeldt J, Tveden-Nyborg P. Atorvastatin and Vitamin E Accelerates NASH Resolution by Dietary Intervention in a Preclinical Guinea Pig Model. Nutrients. 2019; 11(11):2834. https://doi.org/10.3390/nu11112834
Chicago/Turabian StyleKlaebel, Julie Hviid, Mia Skjødt, Josephine Skat-Rørdam, Günaj Rakipovski, David H. Ipsen, Anne Marie V. Schou-Pedersen, Jens Lykkesfeldt, and Pernille Tveden-Nyborg. 2019. "Atorvastatin and Vitamin E Accelerates NASH Resolution by Dietary Intervention in a Preclinical Guinea Pig Model" Nutrients 11, no. 11: 2834. https://doi.org/10.3390/nu11112834
APA StyleKlaebel, J. H., Skjødt, M., Skat-Rørdam, J., Rakipovski, G., Ipsen, D. H., Schou-Pedersen, A. M. V., Lykkesfeldt, J., & Tveden-Nyborg, P. (2019). Atorvastatin and Vitamin E Accelerates NASH Resolution by Dietary Intervention in a Preclinical Guinea Pig Model. Nutrients, 11(11), 2834. https://doi.org/10.3390/nu11112834