Dietary Neuroketotherapeutics for Alzheimer’s Disease: An Evidence Update and the Potential Role for Diet Quality


Abstract
1. Introduction
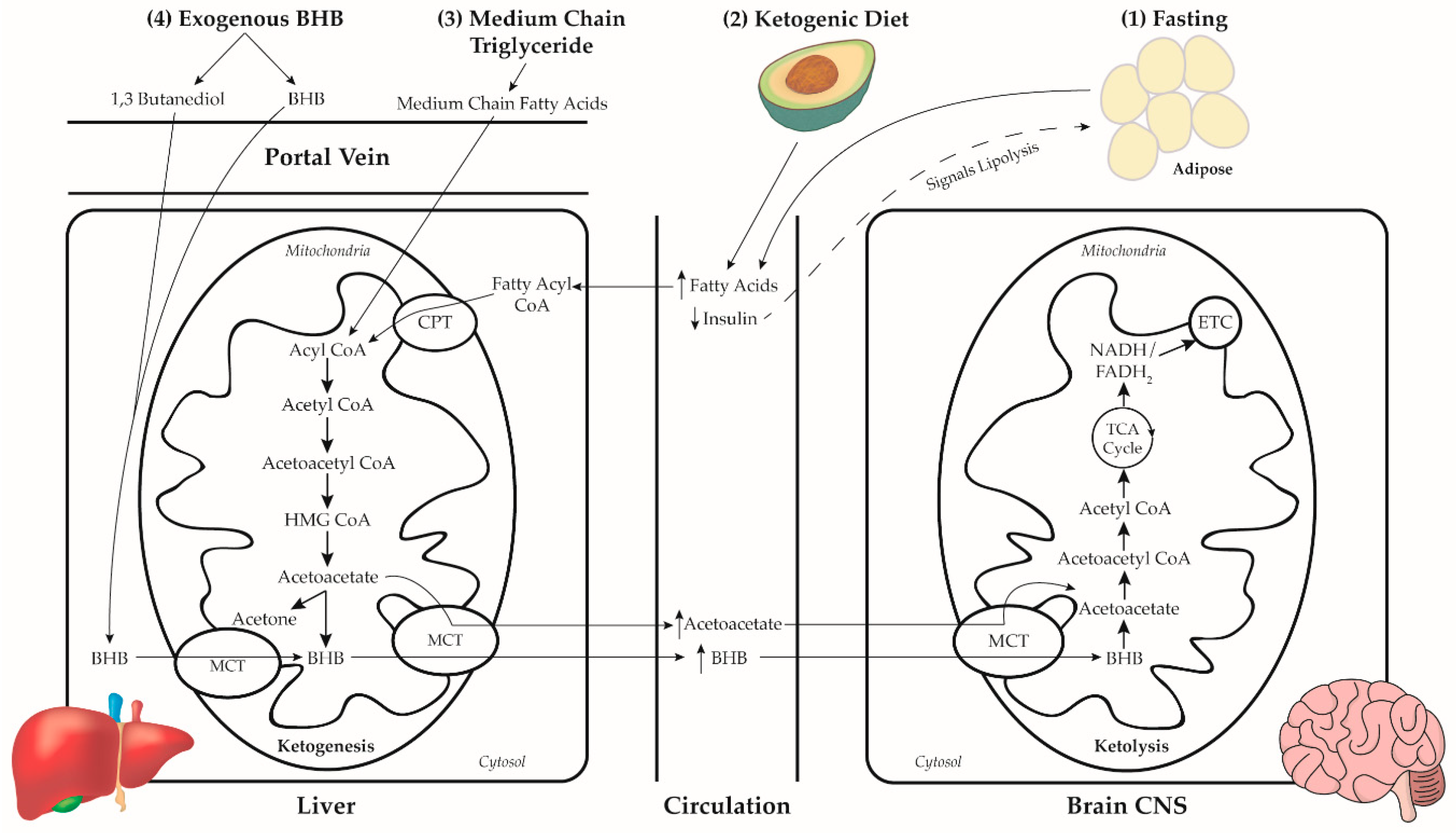
2. Dietary Neuroketotherapeutic Approaches
2.1. Prolonged Fasting
2.2. Ketogenic Diet
2.3. Medium Chain Triglycerides
- (1)
- MCT is hydrolyzed in the gut by pancreatic lipase to their constituent fatty acids (FA) much more rapidly and completely than LCT [75]. Due to this hydrolysis action, MCT is primarily absorbed in the gut as MCFA while LCT is primarily absorbed as monoglyceride and, to a lesser extent, diglyceride and LCFA [76].
- (2)
- MCFA is absorbed across the enterocyte and enters the portal vein for direct hepatic access [61]. In contrast, LCFA is emulsified by bile acids and packaged in micelles for absorption across the enterocyte where they are incorporated into chylomicrons and enter the lymphatic system before entering circulation [77].
- (3)
- Once in the liver, MCFA freely enters the mitochondria for rapid β-oxidation to acetyl CoA [61]. LCFA in the cytoplasm of the hepatocytes is converted to long-chain fatty acyl CoA (LCFAcyl-CoA) [76]. The entry of LCFAcyl-CoA into the mitochondria is facilitated by binding to carnitine, where carnitine is unbound and the LCFAcyl-CoA undergoes β-oxidation. Because MCFA is absorbed at the same rate as glucose [78] and has a rapid rate of β-oxidation, the ingestion of MCT is effective at inducing ketosis.
2.4. Exogenous Ketones
3. Bioenergetic Deficit in AD
4. Putative Ketotherapeutic Benefits are Multi-Mechanistic
5. Evidence in MCI and AD from Animal and Human Studies
5.1. Animal Models
5.2. Humans
5.2.1. MCT Treatment
5.2.2. Coconut Oil
5.2.3. Exogenous Ketone Treatment
5.2.4. Ketogenic Diet
5.2.5. Limitations
5.2.6. Ongoing Clinical Trials
6. Role of Diet Quality in NKTs
6.1. Components of a High-Quality KD
6.2. Considerations of Diet Quality in Non-KD NKTs
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Colby, S.L.; Ortman, J.M. Projections of the Size and Composition of the U.S. Population: 2014 to 2060. Available online: https://www.census.gov/content/dam/Census/library/publications/2015/demo/p25-1143.pdf (accessed on 23 April 2019).
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frolich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimer’s Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Honea, R.A.; Vidoni, E.D.; Swerdlow, R.H.; Burns, J.M. Is Alzheimer’s disease a systemic disease? Biochim. Biophys. Acta 2014, 1842, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, N.; Leon, J.; Sheng, Z.; Oka, S.; Hamasaki, H.; Iwaki, T.; Nakabeppu, Y. Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer’s disease brain. Mech. Ageing Dev. 2017, 161, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Holliday, M.A. Metabolic rate and organ size during growth from infancy to maturity and during late gastation and early infancy. Pediatrics 1971, 47, 169. [Google Scholar]
- Sokoloff, L. Energetics of functional activation in neural tissues. Neurochem. Res. 1999, 24, 321–329. [Google Scholar] [CrossRef]
- De Leon, M.J.; Convit, A.; Wolf, O.T.; Tarshish, C.Y.; DeSanti, S.; Rusinek, H.; Tsui, W.; Kandil, E.; Scherer, A.J.; Roche, A.; et al. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET). Proc. Natl. Acad. Sci. USA 2001, 98, 10966–10971. [Google Scholar] [CrossRef]
- Ishii, K.; Sasaki, M.; Kitagaki, H.; Yamaji, S.; Sakamoto, S.; Matsuda, K.; Mori, E. Reduction of cerebellar glucose metabolism in advanced Alzheimer’s disease. J. Nucl. Med. 1997, 38, 925–928. [Google Scholar]
- Mosconi, L.; De Santi, S.; Brys, M.; Tsui, W.H.; Pirraglia, E.; Glodzik-Sobanska, L.; Rich, K.E.; Switalski, R.; Mehta, P.D.; Pratico, D.; et al. Hypometabolism and altered cerebrospinal fluid markers in normal apolipoprotein E E4 carriers with subjective memory complaints. Biol. Psychiatry 2008, 63, 609–618. [Google Scholar] [CrossRef]
- Mosconi, L.; De Santi, S.; Li, J.; Tsui, W.H.; Li, Y.; Boppana, M.; Laska, E.; Rusinek, H.; de Leon, M.J. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 2008, 29, 676–692. [Google Scholar] [CrossRef]
- Mosconi, L.; Mistur, R.; Switalski, R.; Brys, M.; Glodzik, L.; Rich, K.; Pirraglia, E.; Tsui, W.; De Santi, S.; de Leon, M.J. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology 2009, 72, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, M.H.; Alkalay, A.; Agarwal, N.; Baker, S.L.; O’Neil, J.P.; Janabi, M.; Yen, I.V.; Growdon, M.; Jang, J.; Madison, C.; et al. Distinct clinical and metabolic deficits in PCA and AD are not related to amyloid distribution. Neurology 2011, 76, 1789–1796. [Google Scholar] [CrossRef]
- Castellano, C.A.; Nugent, S.; Paquet, N.; Tremblay, S.; Bocti, C.; Lacombe, G.; Imbeault, H.; Turcotte, E.; Fulop, T.; Cunnane, S.C. Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. J. Alzheimer’ Dis. 2015, 43, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Ferris, S.H.; de Leon, M.J.; Wolf, A.P.; Farkas, T.; Christman, D.R.; Reisberg, B.; Fowler, J.S.; Macgregor, R.; Goldman, A.; George, A.E.; et al. Positron emission tomography in the study of aging and senile dementia. Neurobiol. Aging 1980, 1, 127–131. [Google Scholar] [CrossRef]
- Foster, N.L.; Chase, T.N.; Fedio, P.; Patronas, N.J.; Brooks, R.A.; Di Chiro, G. Alzheimer’s disease: Focal cortical changes shown by positron emission tomography. Neurology 1983, 33, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Friedland, R.P.; Budinger, T.F.; Ganz, E.; Yano, Y.; Mathis, C.A.; Koss, B.; Ober, B.A.; Huesman, R.H.; Derenzo, S.E. Regional cerebral metabolic alterations in dementia of the Alzheimer type: Positron emission tomography with [18F] fluorodeoxyglucose. J. Comput. Assist. Tomogr. 1983, 7, 590–598. [Google Scholar] [CrossRef]
- De Leon, M.J.; Ferris, S.H.; George, A.E.; Christman, D.R.; Fowler, J.S.; Gentes, C.; Reisberg, B.; Gee, B.; Emmerich, M.; Yonekura, Y.; et al. Positron emission tomographic studies of aging and Alzheimer disease. AJNR Am. J. Neuroradiol. 1983, 4, 568–571. [Google Scholar]
- Mullins, R.; Reiter, D.; Kapogiannis, D. Magnetic resonance spectroscopy reveals abnormalities of glucose metabolism in the Alzheimer’s brain. Ann. Clin. Transl. Neurol. 2018, 5, 262–272. [Google Scholar] [CrossRef]
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 811–822. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimer’s Dis. 2010, 20, S265–S279. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Berti, V.; Quinn, C.; McHugh, P.; Petrongolo, G.; Osorio, R.S.; Connaughty, C.; Pupi, A.; Vallabhajosula, S.; Isaacson, R.S.; et al. Correction: Perimenopause and emergence of an Alzheimer’s bioenergetic phenotype in brain and periphery. PLoS ONE 2018, 13, e0193314. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; E., L.; Roy, N.; Hutfles, L.; Burns, J.M.; Michaelis, E.K.; Yan, S.; Cardoso, S.M.; et al. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum. Mol. Genet. 2013, 22, 3931–3946. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef]
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F., Jr. Brain metabolism during fasting. J. Clin. Investig. 1967, 46, 1589–1595. [Google Scholar] [CrossRef]
- Stafstrom, C.E.; Rho, J.M. Epilepsy and the Ketogenic Diet; Humana Press: Totowa, NJ, USA, 2004; p. 352. [Google Scholar]
- Walshe, T.M. Neurological Concepts in Ancient Greek Medicine; Oxford University Press: Oxford, NY, USA, 2016; p. 204. [Google Scholar]
- Zondervan Publishing House (Grand Rapids Mich.). Today’s Parallel Bible: New International Version, New American Standard Bible; updated edition King James Version; Zondervan Pub. House: Grand Rapids, MI, USA, 2000; p. 2861. [Google Scholar]
- Guelpa, G. La lutte contre l’épilepsie par la désintoxication et par la rééducation alimentaire. Rev. Ther. Med. Chir. 1911, 78, 8–13. [Google Scholar]
- Freeman, J.M.; Kelly, M.T.; Freeman, J.B. The Epilepsy Diet Treatment: An Introduction to the Ketogenic Diet; Demos Publications: New York, NY, USA, 1994; p. 180. [Google Scholar]
- Geyelin, H.R. Fasting as a method for treating epilepsy. Med. Rec. 1921, 99, 1037–1039. [Google Scholar]
- Lennox, W.G.; Cobb, S. Studies in epilepsy VIII. The clinical effect of fasting. Arch. Neuro Psychiatr. 1928, 20, 771–779. [Google Scholar] [CrossRef]
- Talbot, F.B.; Metcalf, K.M.; Moriarty, M.E. The ketogenic diet in the treatment of idiopathic epilepsy. Am. J. Dis. Child 1926, 32, 316–318. [Google Scholar]
- Weeks, D.F.; Renner, D.S.; Allen, F.M.; Wishart, M.B. Observations on fasting and diets in the treatment of epilepsy. J. Metab. Res. 1923, 3, 317–364. [Google Scholar]
- Patterson, R.E.; Sears, D.D. Metabolic Effects of Intermittent Fasting. Annu. Rev. Nutr. 2017, 37, 371–393. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Vela, M.E.; Torres, N.; Tovar, A.R. White adipose tissue as endocrine organ and its role in obesity. Arch. Med. Res. 2008, 39, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.J.; Menzel, P.H.; Boden, G.; Owen, O.E. Hepatic ketogenesis and gluconeogenesis in humans. J. Clin. Investig. 1974, 54, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, G.R.; Lardy, H.A. Fatty acid oxidation by a soluble enzyme system from mitochondria. J. Biol. Chem. 1953, 202, 119–136. [Google Scholar] [PubMed]
- Hegardt, F.G. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: A control enzyme in ketogenesis. Biochem. J. 1999, 338, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Lehninger, A.L.; Sudduth, H.C.; Wise, J.B. D-beta-Hydroxybutyric dehydrogenase of muitochondria. J. Biol. Chem. 1960, 235, 2450–2455. [Google Scholar] [PubMed]
- Lin, A.L.; Zhang, W.; Gao, X.; Watts, L. Caloric restriction increases ketone bodies metabolism and preserves blood flow in aging brain. Neurobiol. Aging 2015, 36, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Woodyatt, R.T. Objects and method of diet adjustment in diabetes. Arch. Intern. Med. 1921, 28, 125–141. [Google Scholar] [CrossRef]
- Wilder, R.M. The effect of ketonemia on the course of epilepsy. Mayo Clin. Bull. 1921, 2, 307. [Google Scholar]
- Wilder, R.M. High fat diets in epilepsy. Mayo Clin. Bull. 1921, 2, 308. [Google Scholar]
- Brodie, M.J. Antiepileptic drug therapy the story so far. Seizure 2010, 19, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Weber, D.D.; Aminazdeh-Gohari, S.; Kofler, B. Ketogenic diet in cancer therapy. Aging 2018, 10, 164–165. [Google Scholar] [CrossRef] [PubMed]
- McSwiney, F.T.; Wardrop, B.; Hyde, P.N.; Lafountain, R.A.; Volek, J.S.; Doyle, L. Keto-adaptation enhances exercise performance and body composition responses to training in endurance athletes. Metabolism 2018, 81, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.J.; Villamena, F.A.; Volek, J.S. Nutritional Ketosis and Mitohormesis: Potential Implications for Mitochondrial Function and Human Health. J. Nutr. Metab. 2018, 2018, 5157645. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.K.; Swerdlow, R.H.; Burns, J.M.; Sullivan, D.K. An Experimental Ketogenic Diet for Alzheimer Disease Was Nutritionally Dense and Rich in Vegetables and Avocado. Curr. Dev. Nutr. 2019, 3, nzz003. [Google Scholar] [CrossRef] [PubMed]
- Harkins, R.W.; Sarett, H.P. Medium-chain triglycerides. JAMA 1968, 203, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Hilditch, T.P.; Meara, M.L. Human-milk fat: 1. Component fatty acids. Biochem. J. 1944, 38, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.D.; Limketkai, B.N. The Use of Medium-Chain Triglycerides in Gastrointestinal Disorders. Pract. Gastroenterol. 2017, 41, 20–28. [Google Scholar]
- Mazzocchi, A.; D’Oria, V.; De Cosmi, V.; Bettocchi, S.; Milani, G.P.; Silano, M.; Agostoni, C. The Role of Lipids in Human Milk and Infant Formulae. Nutrients 2018, 10, 567. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, T.; Yoshida, I.; Kaneko, A.; Tashiro, K.; Tashiro, S.; Jogo, M.; Aoki, K.; Tanaka, M. Neonatal ketosis is not rare: Experience of neonatal screening using gas chromatography-mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 2001, 758, 57–60. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Crawford, M.A. Energetic and nutritional constraints on infant brain development: Implications for brain expansion during human evolution. J. Hum. Evol. 2014, 77, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Adam, P.A.; Raiha, N.; Rahiala, E.L.; Kekomaki, M. Oxidation of glucose and D-B-OH-butyrate by the early human fetal brain. Acta Paediatr. Scand. 1975, 64, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.C.; Babayan, V.K. Medium-chain triglycerides: An update. Am. J. Clin. Nutr. 1982, 36, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Traitler, H.; Dieffenbacher, A. Palm Oil and Palm Kernel Oil in Food-Products. J. Am. Oil Chem. Soc. 1985, 62, 417–421. [Google Scholar] [CrossRef]
- Beveridge, J.M.R.; Connell, W.F.; Haust, H.L.; Mayer, G.A. Dietary Cholesterol and Plasma Cholesterol Levels in Man. Can. J. Biochem. Phys. 1959, 37, 575–582. [Google Scholar] [CrossRef]
- Hashim, S.A.; Arteaga, A.; Vanitallie, T.B. Effect of a Saturated Medium-Chain Triglyceride on Serum-Lipids in Man. Lancet 1960, 1, 1105–1108. [Google Scholar] [CrossRef]
- Pinter, K.G.; Lamar, C.; Goldsmith, G.A.; Mccracken, B.H. Fat Absorption Studies in Various Forms of Steatorrhea. Am. J. Clin. Nutr. 1964, 15, 293–298. [Google Scholar] [CrossRef]
- Holt, P.R.; Hashim, S.A.; Vanitallie, T.B. Treatment of Malabsorption Syndrome and Exudative Enteropathy with Synthetic Medium Chain Triglycerides. Am. J. Gastroenterol. 1965, 43, 549. [Google Scholar] [PubMed]
- Bergen, S.S., Jr.; Hashim, S.A.; Van Itallie, T.B. Hyperketonemia induced in man by medium-chain triglyceride. Diabetes 1966, 15, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Freund, G.; Weinsier, R.L. Standardized ketosis in man following medium chain triglyceride ingestion. Metabolism 1966, 15, 980–991. [Google Scholar] [CrossRef]
- Huttenlocher, P.R.; Wilbourn, A.J.; Signore, J.M. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology 1971, 21, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.H.; Eaton, J.; Bower, B.D.; Aynsley-Green, A. Ketogenic diets in the treatment of epilepsy: Short-term clinical effects. Dev. Med. Child. Neurol. 1989, 31, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.M.; Boyes, S.; Aynsley-Green, A. Metabolic effects of three ketogenic diets in the treatment of severe epilepsy. Dev. Med. Child. Neurol. 1989, 31, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Wang, H.S. Medium-chain triglyceride ketogenic diet, an effective treatment for drug-resistant epilepsy and a comparison with other ketogenic diets. Biomed. J. 2013, 36, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Bloom, B.; Chaikoff, I.L. Reinhardt. Intestinal lymph as pathway for transport of absorbed fatty acids of different chain lengths. Am. J. Physiol. 1951, 166, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, F.X.; Hashim, S.A.; Van Itallie, T.B. Insulin and ketone responses to ingestion of medium and long-chain triglycerides in man. Diabetes 1969, 18, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Desnuelle, P.; Savary, P. Specificities of Lipases. J. Lipid Res. 1963, 4, 369–384. [Google Scholar]
- Iqbal, J.; Hussain, M.M. Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1183–E1194. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Amate, L.; Gil, A. Absorption and distribution of dietary fatty acids from different sources. Early Hum. Dev. 2001, 65, S95–S101. [Google Scholar] [CrossRef]
- Iber, F.L. Relative rates of metabolism MCT, LCT and ethanol in man. Z. Ernahrungswiss. 1974, 17, 9–16. [Google Scholar]
- Lieben, F.; Ehrlich, G. The behaviour of Aldol in animal bodies and in fresh organ paps. Biochem. Z. 1928, 198, 317–327. [Google Scholar]
- Mehlman, M.A.; Tobin, R.B.; Johnston, J.B. Metabolic Control of Enzymes in Normal, Diabetic, and Diabetic Insulin-Treated Rats Utilizing 1,3 Butanediol. Metab. Clin. Exp. 1971, 20, 149–167. [Google Scholar] [CrossRef]
- Tate, R.L.; Mehlman, M.A.; Tobin, R.B. Metabolic Fate of 1,3-Butanediol in Rat—Conversion to Beta-Hydroxybutyrate. J. Nutr. 1971, 101, 1719–1726. [Google Scholar] [CrossRef]
- Brunengraber, H. Potential of ketone body esters for parenteral and oral nutrition. Nutrition 1997, 13, 233–235. [Google Scholar] [CrossRef]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Santer, P.; Miller, J.J.; Faull, O.K.; Magor-Elliott, S.; Hiyama, S.; Stirling, M.; Clarke, K. On the Metabolism of Exogenous Ketones in Humans. Front. Physiol. 2017, 8, 848. [Google Scholar] [CrossRef]
- Stubbs, B.J.; Cox, P.J.; Kirk, T.; Evans, R.D.; Clarke, K. Gastrointestinal Effects of Exogenous Ketone Drinks are Infrequent, Mild and Vary According to Ketone Compound and Dose. Int. J. Sport Nutr. Exerc. Metab. 2019, 1, 1–8. [Google Scholar] [CrossRef]
- Veech, R.L. Ketone ester effects on metabolism and transcription. J. Lipid Res. 2014, 55, 2004–2006. [Google Scholar] [CrossRef]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; Todd King, M.; Musa-Veloso, K.; Ho, M.; Roberts, A.; Robertson, J.; Vanitallie, T.B.; et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul. Toxicol. Pharm. 2012, 63, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Shivva, V.; Cox, P.J.; Clarke, K.; Veech, R.L.; Tucker, I.G.; Duffull, S.B. The Population Pharmacokinetics of D-beta-hydroxybutyrate Following Administration of (R)-3-Hydroxybutyl (R)-3-Hydroxybutyrate. AAPS J. 2016, 18, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.J.; Kirk, T.; Ashmore, T.; Willerton, K.; Evans, R.; Smith, A.; Murray, A.J.; Stubbs, B.; West, J.; McLure, S.W.; et al. Nutritional Ketosis Alters Fuel Preference and Thereby Endurance Performance in Athletes. Cell Metab. 2016, 24, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth, D.A.; Cox, P.J.; Kirk, T.; Stradling, H.; Impey, S.G.; Clarke, K. A Ketone Ester Drink Increases Postexercise Muscle Glycogen Synthesis in Humans. Med. Sci. Sports Exerc. 2017, 49, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Leckey, J.J.; Ross, M.L.; Quod, M.; Hawley, J.A.; Burke, L.M. Ketone Diester Ingestion Impairs Time-Trial Performance in Professional Cyclists. Front. Physiol. 2017, 8, 806. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, B.J.; Koutnik, A.P.; Poff, A.M.; Ford, K.M.; D’Agostino, D.P. Commentary: Ketone Diester Ingestion Impairs Time-Trial Performance in Professional Cyclists. Front. Physiol. 2018, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Mehlman, M.A.; Hanson, R.W. Energy metabolism and the regulation of metabolic processes in mitochondria. In Proceedings of the A Symposium Held at the University of Nebraska Medical School, Omaha, NE, USA, 3–4 May 1971; Academic Press: New York, NY, USA, 1972; p. 296. [Google Scholar]
- Alzheimer, A. Über einen eigenartigen schweren Erkrankungsprozeβ der Hirnrincle. Neurol. Cent. 1906, 25, 1129–1136. [Google Scholar]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef]
- Johnson, A.B.; Blum, N.R. Nucleoside phosphatase activities associated with the tangles and plaques of alzheimer’s disease: A histochemical study of natural and experimental neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 1970, 29, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.; Terry, R.D.; Hirano, A. Neurofibrillary pathology. J. Neuropathol. Exp. Neurol. 1970, 29, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Lying-Tunell, U.; Lindblad, B.S.; Malmlund, H.O.; Persson, B. Cerebral blood flow and metabolic rate of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids. Acta Neurol. Scand. 1981, 63, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Dastur, D.K. Cerebral blood flow and metabolism in normal human aging, pathological aging, and senile dementia. J. Cereb. Blood Flow Metab. 1985, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Scholl, M.; Almkvist, O.; Bogdanovic, N.; Wall, A.; Langstrom, B.; Viitanen, M.; Nordberg, A. Time course of glucose metabolism in relation to cognitive performance and postmortem neuropathology in Met146Val PSEN1 mutation carriers. J. Alzheimer’s Dis. 2011, 24, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Velliquette, R.A.; O’Connor, T.; Vassar, R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: Possible early events in Alzheimer’s disease pathogenesis. J. Neurosci. 2005, 25, 10874–10883. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Blass, J.P.; Zemcov, A. Alzheimer’s disease. A metabolic systems degeneration? Neurochem. Pathol. 1984, 2, 103–114. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Courchesne-Loyer, A.; Vandenberghe, C.; St-Pierre, V.; Fortier, M.; Hennebelle, M.; Croteau, E.; Bocti, C.; Fulop, T.; Castellano, C.A. Can Ketones Help Rescue Brain Fuel Supply in Later Life? Implications for Cognitive Health during Aging and the Treatment of Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 53. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Marcus, D.M.; Landman, J.; Harooni, M.; Freedman, M.L. Brain glucose and ketone body metabolism in patients with Alzheimer’s disease. Clin. Res. 1989, 37, 461A. [Google Scholar]
- Ogawa, M.; Fukuyama, H.; Ouchi, Y.; Yamauchi, H.; Kimura, J. Altered energy metabolism in Alzheimer’s disease. J. Neurol. Sci. 1996, 139, 78–82. [Google Scholar] [CrossRef]
- Courchesne-Loyer, A.; Croteau, E.; Castellano, C.A.; St-Pierre, V.; Hennebelle, M.; Cunnane, S.C. Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: A dual tracer quantitative positron emission tomography study. J. Cereb. Blood Flow Metab. 2017, 37, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Croteau, E.; Castellano, C.A.; Richard, M.A.; Fortier, M.; Nugent, S.; Lepage, M.; Duchesne, S.; Whittingstall, K.; Turcotte, E.E.; Bocti, C.; et al. Ketogenic Medium Chain Triglycerides Increase Brain Energy Metabolism in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Fortier, M.; Castellano, C.A.; Croteau, E.; Langlois, F.; Bocti, C.; St-Pierre, V.; Vandenberghe, C.; Bernier, M.; Roy, M.; Descoteaux, M.; et al. A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimer’s Dement. 2019, 15, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.L. Cerebral metabolic adaptation and ketone metabolism after brain injury. J. Cereb. Blood Flow Metab. 2008, 28, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Achanta, L.B.; Rae, C.D. beta-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Kashiwaya, Y.; Pawlosky, R.; Markis, W.; King, M.T.; Bergman, C.; Srivastava, S.; Murray, A.; Clarke, K.; Veech, R.L. A ketone ester diet increases brain malonyl-CoA and Uncoupling proteins 4 and 5 while decreasing food intake in the normal Wistar Rat. J. Biol. Chem. 2010, 285, 25950–25956. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chan, S.L.; de Souza-Pinto, N.C.; Slevin, J.R.; Wersto, R.P.; Zhan, M.; Mustafa, K.; de Cabo, R.; Mattson, M.P. Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromolecular. Med. 2006, 8, 389–414. [Google Scholar] [CrossRef]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar] [CrossRef]
- Srivastava, S.; Kashiwaya, Y.; King, M.T.; Baxa, U.; Tam, J.; Niu, G.; Chen, X.; Clarke, K.; Veech, R.L. Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB J. 2012, 26, 2351–2362. [Google Scholar] [CrossRef]
- Chu, A.C.; Ho, P.W.; Kwok, K.H.; Ho, J.W.; Chan, K.H.; Liu, H.F.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Mitochondrial UCP4 attenuates MPP+− and dopamine-induced oxidative stress, mitochondrial depolarization, and ATP deficiency in neurons and is interlinked with UCP2 expression. Free Radic. Biol. Med. 2009, 46, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.W.; Chu, A.C.; Kwok, K.H.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Knockdown of uncoupling protein-5 in neuronal SH-SY5Y cells: Effects on MPP+− induced mitochondrial membrane depolarization, ATP deficiency, and oxidative cytotoxicity. J. Neurosci. Res. 2006, 84, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Kwok, K.H.; Ho, P.W.; Chu, A.C.; Ho, J.W.; Liu, H.F.; Yiu, D.C.; Chan, K.H.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Mitochondrial UCP5 is neuroprotective by preserving mitochondrial membrane potential, ATP levels, and reducing oxidative stress in MPP+ and dopamine toxicity. Free Radic. Biol. Med. 2010, 49, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, D.B.; Ho, P.W.; Ho, J.W.; Liu, H.F.; So, D.H.; Tse, H.M.; Chan, K.H.; Ho, S.L. Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): Structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav. 2012, 2, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Chigurupati, S.; Bagsiyao, P.; Henriquez, A.; Chan, S.L. The brain uncoupling protein UCP4 attenuates mitochondrial toxin-induced cell death: Role of extracellular signal-regulated kinases in bioenergetics adaptation and cell survival. Neurotox. Res. 2009, 16, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.W.; Ho, P.W.; Liu, H.F.; So, D.H.; Chan, K.H.; Tse, Z.H.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. UCP4 is a target effector of the NF-kappaB c-Rel prosurvival pathway against oxidative stress. Free Radic. Biol. Med. 2012, 53, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.W.; Ho, J.W.; Tse, H.M.; So, D.H.; Yiu, D.C.; Liu, H.F.; Chan, K.H.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Uncoupling protein-4 (UCP4) increases ATP supply by interacting with mitochondrial Complex II in neuroblastoma cells. PLoS ONE 2012, 7, e32810. [Google Scholar] [CrossRef]
- Juge, N.; Gray, J.A.; Omote, H.; Miyaji, T.; Inoue, T.; Hara, C.; Uneyama, H.; Edwards, R.H.; Nicoll, R.A.; Moriyama, Y. Metabolic control of vesicular glutamate transport and release. Neuron 2010, 68, 99–112. [Google Scholar] [CrossRef]
- Simeone, T.A.; Simeone, K.A.; Rho, J.M. Ketone Bodies as Anti-Seizure Agents. Neurochem. Res. 2017, 42, 2011–2018. [Google Scholar] [CrossRef]
- McNally, M.A.; Hartman, A.L. Ketone bodies in epilepsy. J. Neurochem. 2012, 121, 28–35. [Google Scholar] [CrossRef]
- Hernandez, A.R.; Hernandez, C.M.; Campos, K.; Truckenbrod, L.; Federico, Q.; Moon, B.; McQuail, J.A.; Maurer, A.P.; Bizon, J.L.; Burke, S.N. A Ketogenic Diet Improves Cognition and Has Biochemical Effects in Prefrontal Cortex That Are Dissociable From Hippocampus. Front. Aging Neurosci. 2018, 10, 391. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S. Mitochondrial uncoupling proteins--what is their physiological role? Free Radic. Biol. Med. 2007, 43, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Hansford, R.G.; Hogue, B.A.; Mildaziene, V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J. Bioenerg. Biomembr. 1997, 29, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Votyakova, T.V.; Reynolds, I.J. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2001, 79, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Milder, J.B.; Liang, L.P.; Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 2008, 106, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Milder, J.B.; Liang, L.P.; Patel, M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol. Dis. 2010, 40, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yang, Y.Y.; Zhou, M.W.; Liu, N.; Xing, H.Y.; Liu, X.X.; Li, F. Ketogenic diet attenuates oxidative stress and inflammation after spinal cord injury by activating Nrf2 and suppressing the NF-kappaB signaling pathways. Neurosci. Lett. 2018, 683, 13–18. [Google Scholar] [CrossRef]
- Maalouf, M.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 293–315. [Google Scholar] [CrossRef]
- Emerit, J.; Edeas, M.; Bricaire, F. Neurodegenerative diseases and oxidative stress. Biomed. Pharm. 2004, 58, 39–46. [Google Scholar] [CrossRef]
- Benzi, G.; Moretti, A. Are reactive oxygen species involved in Alzheimer’s disease? Neurobiol. Aging 1995, 16, 661–674. [Google Scholar] [CrossRef]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimer 2018, 4, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.; Buchholz, A.; Henry-Barron, B.; Vizthum, D.; Avramopoulos, D.; Cervenka, M.C. Preliminary Report on the Feasibility and Efficacy of the Modified Atkins Diet for Treatment of Mild Cognitive Impairment and Early Alzheimer’s Disease. J. Alzheimer 2019, 68, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Muller-Fielitz, H.; Pokorna, B.; Vollbrandt, T.; Stolting, I.; Nadrowitz, R.; et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef] [PubMed]
- Selfridge, J.E.; Wilkins, H.M.; Lezi, E.; Carl, S.M.; Koppel, S.; Funk, E.; Fields, T.; Lu, J.; Tang, E.P.; Slawson, C.; et al. Effect of one month duration ketogenic and non-ketogenic high fat diets on mouse brain bioenergetic infrastructure. J. Bioenerg. Biomembr. 2015, 47, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zandi-Nejad, K.; Takakura, A.; Jurewicz, M.; Chandraker, A.K.; Offermanns, S.; Mount, D.; Abdi, R. The role of HCA2 (GPR109A) in regulating macrophage function. FASEB J. 2013, 27, 4366–4374. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, L.; Abel, T. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology 2013, 38, 62–76. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, S.; Yu, L.; Jin, J.; Ye, X.; Liu, Y.; Xu, Y. HDAC3 negatively regulates spatial memory in a mouse model of Alzheimer’s disease. Aging Cell 2017, 16, 1073–1082. [Google Scholar] [CrossRef]
- Koppel, I.; Timmusk, T. Differential regulation of Bdnf expression in cortical neurons by class-selective histone deacetylase inhibitors. Neuropharmacology 2013, 75, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Marosi, K.; Kim, S.W.; Moehl, K.; Scheibye-Knudsen, M.; Cheng, A.; Cutler, R.; Camandola, S.; Mattson, M.P. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem. 2016, 139, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body beta-hydroxybutyrate. Elife 2016, 5. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Nabeshima, T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J. Pharm. Sci. 2003, 91, 267–270. [Google Scholar] [CrossRef]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 174, 497. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wang, A.C.; Parikh, I.; Green, S.J.; Hoffman, J.D.; Chlipala, G.; Murphy, M.P.; Sokola, B.S.; Bauer, B.; Hartz, A.M.S.; et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci. Rep. 2018, 8, 6670. [Google Scholar] [CrossRef]
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2005, 2, 28. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef]
- Yin, J.X.; Maalouf, M.; Han, P.; Zhao, M.; Gao, M.; Dharshaun, T.; Ryan, C.; Whitelegge, J.; Wu, J.; Eisenberg, D.; et al. Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiol. Aging 2016, 39, 25–37. [Google Scholar] [CrossRef]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS ONE 2013, 8, e75713. [Google Scholar] [CrossRef] [PubMed]
- Beckett, T.L.; Studzinski, C.M.; Keller, J.N.; Paul Murphy, M.; Niedowicz, D.M. A ketogenic diet improves motor performance but does not affect beta-amyloid levels in a mouse model of Alzheimer’s disease. Brain Res. 2013, 1505, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Wang, D.D.; Sun, Y.X.; Zhao, D.J.; Ni, H. Neuro-Behavioral Status and the Hippocampal Expression of Metabolic Associated Genes in Wild-Type Rat Following a Ketogenic Diet. Front. Neurol. 2019, 10, 65. [Google Scholar] [CrossRef]
- Droogsma, E.; van Asselt, D.Z.; Scholzel-Dorenbos, C.J.; van Steijn, J.H.; van Walderveen, P.E.; van der Hooft, C.S. Nutritional status of community-dwelling elderly with newly diagnosed Alzheimer’s disease: Prevalence of malnutrition and the relation of various factors to nutritional status. J. Nutr. Health Aging 2013, 17, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Roman, M.W. Axona (Accera, Inc): A new medical food therapy for persons with Alzheimer’s disease. Issues Ment. Health Nurs. 2010, 31, 435–436. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.D.; Gelblum, J. Retrospective cohort study of the efficacy of caprylic triglyceride in patients with mild-to-moderate alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2013, 9, 1619–1627. [Google Scholar] [CrossRef]
- Farah, B.A. Effects of caprylic triglyceride on cognitive performance and cerebral glucose metabolism in mild Alzheimer’s disease: A single-case observation. Front. Aging Neurosci. 2014, 6, 133. [Google Scholar] [CrossRef]
- Rebello, C.J.; Keller, J.N.; Liu, A.G.; Johnson, W.D.; Greenway, F.L. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: A randomized controlled trial. BBA Clin. 2015, 3, 123–125. [Google Scholar] [CrossRef]
- Riederer, I.; Bohn, K.P.; Preibisch, C.; Wiedemann, E.; Zimmer, C.; Alexopoulos, P.; Forster, S. Alzheimer Disease and Mild Cognitive Impairment: Integrated Pulsed Arterial Spin-Labeling MRI and (18) F-FDG PET. Radiology 2018, 288, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Torosyan, N.; Sethanandha, C.; Grill, J.D.; Dilley, M.L.; Lee, J.; Cummings, J.L.; Ossinalde, C.; Silverman, D.H. Changes in regional cerebral blood flow associated with a 45day course of the ketogenic agent, caprylidene, in patients with mild to moderate Alzheimer’s disease: Results of a randomized, double-blinded, pilot study. Exp. Gerontol. 2018, 111, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Eyres, L.; Eyres, M.F.; Chisholm, A.; Brown, R.C. Coconut oil consumption and cardiovascular risk factors in humans. Nutr. Rev. 2016, 74, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Swift, L.L.; Hill, J.O.; Peters, J.C.; Greene, H.L. Medium-chain fatty acids: Evidence for incorporation into chylomicron triglycerides in humans. Am. J. Clin. Nutr. 1990, 52, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Vandenberghe, C.; St-Pierre, V.; Pierotti, T.; Fortier, M.; Castellano, C.A.; Cunnane, S.C. Tricaprylin Alone Increases Plasma Ketone Response More Than Coconut Oil or Other Medium-Chain Triglycerides: An Acute Crossover Study in Healthy Adults. Curr. Dev. Nutr. 2017, 1, e000257. [Google Scholar] [CrossRef]
- Hu Yang, I.; De la Rubia Orti, J.E.; Selvi Sabater, P.; Sancho Castillo, S.; Rochina, M.J.; Manresa Ramon, N.; Montoya-Castilla, I. Coconut Oil: Non-Alternative Drug Treatment against Alzheimer S Disease. Nutr. Hosp. 2015, 32, 2822–2827. [Google Scholar] [CrossRef]
- Chan, S.C.; Esther, G.E.; Yip, H.L.; Sugathan, S.; Chin, P.S. Effect of cold pressed coconut oil on cognition and behavior among patients with Alzheimer’s disease—A pilot intervention study. Natl. J. Physiol. Pharm. Pharmacol. 2017, 7, 1432–1435. [Google Scholar]
- De la Rubia Orti, J.E.; Garcia-Pardo, M.P.; Drehmer, E.; Sancho Cantus, D.; Julian Rochina, M.; Aguilar, M.A.; Hu Yang, I. Improvement of Main Cognitive Functions in Patients with Alzheimer’s Disease after Treatment with Coconut Oil Enriched Mediterranean Diet: A Pilot Study. J. Alzheimer 2018, 65, 577–587. [Google Scholar] [CrossRef]
- Newport, M.T.; VanItallie, T.B.; Kashiwaya, Y.; King, M.T.; Veech, R.L. A new way to produce hyperketonemia: Use of ketone ester in a case of Alzheimer’s disease. Alzheimer 2015, 11, 99–103. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, e419–e427. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; McGrogan, J.R.; Bluml, R.M.; Pillas, D.J.; Rubenstein, J.E.; Vining, E.P. A modified Atkins diet is effective for the treatment of intractable pediatric epilepsy. Epilepsia 2006, 47, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Ernst, N.D.; Cleeman, J.I. National cholesterol education program keeps a priority on lifestyle modification to decrease cardiovascular disease risk. Curr. Opin. Lipidol. 2002, 13, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.M.; Reedy, J.; Krebs-Smith, S.M. American Diet Quality: Where It Is, Where It Is Heading, and What It Could Be. J. Acad. Nutr. Diet. 2016, 116, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.K.; Graves, D.E.; Craven, T.E.; Lipkin, E.W.; Austin, M.; Margolis, K.L. Restricted-carbohydrate diets in patients with type 2 diabetes: A meta-analysis. J. Am. Diet. Assoc. 2008, 108, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Vining, E.P. Long-term health consequences of epilepsy diet treatments. Epilepsia 2008, 49, 27–29. [Google Scholar] [CrossRef]
- Davies, M.J.; D’Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2018, 41, 2669–2701. [Google Scholar] [CrossRef]
- Hallberg, S.J.; McKenzie, A.L.; Williams, P.T.; Bhanpuri, N.H.; Peters, A.L.; Campbell, W.W.; Hazbun, T.L.; Volk, B.M.; McCarter, J.P.; Phinney, S.D.; et al. Effectiveness and Safety of a Novel Care Model for the Management of Type 2 Diabetes at 1 Year: An Open-Label, Non-Randomized, Controlled Study. Diabetes Ther. 2018, 9, 583–612. [Google Scholar] [CrossRef]
- Athinarayanan, S.J.; Adams, R.N.; Hallberg, S.J.; McKenzie, A.L.; Bhanpuri, N.H.; Campbell, W.W.; Volek, J.S.; Phinney, S.D.; McCarter, J.P. Long-Term Effects of a Novel Continuous Remote Care Intervention Including Nutritional Ketosis for the Management of Type 2 Diabetes: A 2-Year Non-randomized Clinical Trial. Front. Endocrinol. 2019, 10, 348. [Google Scholar] [CrossRef]
- de Souza, R.J.; Mente, A.; Maroleanu, A.; Cozma, A.I.; Ha, V.; Kishibe, T.; Uleryk, E.; Budylowski, P.; Schunemann, H.; Beyene, J.; et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: Systematic review and meta-analysis of observational studies. BMJ 2015, 351, h3978. [Google Scholar] [CrossRef] [PubMed]
- Clifton, P.M.; Keogh, J.B. A systematic review of the effect of dietary saturated and polyunsaturated fat on heart disease. Nutr. Metab. Cardiovasc Dis. 2017, 27, 1060–1080. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Lichtenstein, A.H.; Wu, J.H.Y.; Appel, L.J.; Creager, M.A.; Kris-Etherton, P.M.; Miller, M.; Rimm, E.B.; Rudel, L.L.; Robinson, J.G.; et al. Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association. Circulation 2017, 136, e1–e23. [Google Scholar] [CrossRef] [PubMed]
- Fuehrlein, B.S.; Rutenberg, M.S.; Silver, J.N.; Warren, M.W.; Theriaque, D.W.; Duncan, G.E.; Stacpoole, P.W.; Brantly, M.L. Differential metabolic effects of saturated versus polyunsaturated fats in ketogenic diets. J. Clin. Endocrinol. Metab. 2004, 89, 1641–1645. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, C.E.; Phinney, S.D.; Feinman, R.D.; Volk, B.M.; Freidenreich, D.; Quann, E.; Ballard, K.; Puglisi, M.J.; Maresh, C.M.; Kraemer, W.J.; et al. Limited effect of dietary saturated fat on plasma saturated fat in the context of a low carbohydrate diet. Lipids 2010, 45, 947–962. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.W.; Hauser, M.E.; Aronica, L.; Rigdon, J.; Gardner, C.D. Changes in blood lipid concentrations associated with changes in intake of dietary saturated fat in the context of a healthy low-carbohydrate weight-loss diet: A secondary analysis of the Diet Intervention Examining The Factors Interacting with Treatment Success (DIETFITS) trial. Am. J. Clin. Nutr. 2019, 109, 433–441. [Google Scholar] [CrossRef]
- Volek, J.S.; Fernandez, M.L.; Feinman, R.D.; Phinney, S.D. Dietary carbohydrate restriction induces a unique metabolic state positively affecting atherogenic dyslipidemia, fatty acid partitioning, and metabolic syndrome. Prog. Lipid Res. 2008, 47, 307–318. [Google Scholar] [CrossRef]
- Mashek, D.G.; Wu, C. MUFAs. Adv. Nutr. 2015, 6, 276–277. [Google Scholar] [CrossRef]
- Zong, G.; Li, Y.; Sampson, L.; Dougherty, L.W.; Willett, W.C.; Wanders, A.J.; Alssema, M.; Zock, P.L.; Hu, F.B.; Sun, Q. Monounsaturated fats from plant and animal sources in relation to risk of coronary heart disease among US men and women. Am. J. Clin. Nutr. 2018, 107, 445–453. [Google Scholar] [CrossRef]
- Calder, P.C. Omega-3 polyunsaturated fatty acids and inflammatory processes: Nutrition or pharmacology? Br. J. Clin. Pharmacol. 2013, 75, 645–662. [Google Scholar] [CrossRef]
- Calder, P.C. Polyunsaturated fatty acids and inflammatory processes: New twists in an old tale. Biochimie 2009, 91, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, F.; Ortiz, M.; Valenzuela, R.; Videla, L.A. Long-chain polyunsaturated fatty acids regulation of PPARs, signaling: Relationship to tissue development and aging. Prostaglandins Leukot. Essent. Fat. Acids 2016, 114, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Yuen, A.W.C.; Walcutt, I.A.; Sander, J.W. An acidosis-sparing ketogenic (ASK) diet to improve efficacy and reduce adverse effects in the treatment of refractory epilepsy. Epilepsy Behav. 2017, 74, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Slavin, J.L.; Lloyd, B. Health benefits of fruits and vegetables. Adv. Nutr. 2012, 3, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.C.; Koh, W.P.; Yuan, J.M.; Qin, L.Q.; van Dam, R.M. Green leafy and cruciferous vegetable consumption and risk of type 2 diabetes: Results from the Singapore Chinese Health Study and meta-analysis. Br. J. Nutr. 2018, 119, 1057–1067. [Google Scholar] [CrossRef]
- Khoo, H.E.; Prasad, K.N.; Kong, K.W.; Jiang, Y.; Ismail, A. Carotenoids and their isomers: Color pigments in fruits and vegetables. Molecules 2011, 16, 1710. [Google Scholar] [CrossRef] [PubMed]
- Jardim, F.R.; de Rossi, F.T.; Nascimento, M.X.; da Silva Barros, R.G.; Borges, P.A.; Prescilio, I.C.; de Oliveira, M.R. Resveratrol and Brain Mitochondria: A Review. Mol. Neurobiol. 2018, 55, 2085–2101. [Google Scholar] [CrossRef]
- Tan, S.; Wong, E. Mitophagy Transcriptome: Mechanistic Insights into Polyphenol-Mediated Mitophagy. Oxid. Med. Cell Longev. 2017, 2017, 9028435. [Google Scholar] [CrossRef]
- Teixeira, J.; Deus, C.M.; Borges, F.; Oliveira, P.J. Mitochondria: Targeting mitochondrial reactive oxygen species with mitochondriotropic polyphenolic-based antioxidants. Int. J. Biochem. Cell Biol. 2018, 97, 98–103. [Google Scholar] [CrossRef]
- Crawford, P.A.; Crowley, J.R.; Sambandam, N.; Muegge, B.D.; Costello, E.K.; Hamady, M.; Knight, R.; Gordon, J.I. Regulation of myocardial ketone body metabolism by the gut microbiota during nutrient deprivation. Proc. Natl. Acad. Sci. USA 2009, 106, 11276–11281. [Google Scholar] [CrossRef]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Mahmassani, H.A.; Avendano, E.E.; Raman, G.; Johnson, E.J. Avocado consumption and risk factors for heart disease: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2018, 107, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Istas, G.; Wood, E.; Le Sayec, M.; Rawlings, C.; Yoon, J.; Dandavate, V.; Cera, D.; Rampelli, S.; Costabile, A.; Fromentin, E.; et al. Effects of aronia berry (poly)phenols on vascular function and gut microbiota: A double-blind randomized controlled trial in adult men. Am. J. Clin. Nutr. 2019. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.B.; Erlbaum, A.I. Role of ketogenesis in urinary sodium excretion: Elucidation by nicotinic acid administration during fasting. J. Clin. Endocrinol. Metab. 1979, 49, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Briet, M.; Schiffrin, E.L. Aldosterone: Effects on the kidney and cardiovascular system. Nat. Rev. Nephrol. 2010, 6, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.; Spencer, H.; Welsh, J.J. Magnesium absorption in human subjects from leafy vegetables, intrinsically labeled with stable 26Mg. Am. J. Clin. Nutr. 1984, 39, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.J.; Lee, C.W.; Tsai, W.C.; Chien, Y.C. Essential and toxic metals in animal bone broths. Food Nutr. Res. 2017, 61, 1347478. [Google Scholar] [CrossRef] [PubMed]
- Harasym, J.; Oledzki, R. Effect of fruit and vegetable antioxidants on total antioxidant capacity of blood plasma. Nutrition 2014, 30, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Renzi-Hammond, L.M.; Bovier, E.R.; Fletcher, L.M.; Miller, L.S.; Mewborn, C.M.; Lindbergh, C.A.; Baxter, J.H.; Hammond, B.R. Effects of a Lutein and Zeaxanthin Intervention on Cognitive Function: A Randomized, Double-Masked, Placebo-Controlled Trial of Younger Healthy Adults. Nutrients 2017, 9, 1246. [Google Scholar] [CrossRef] [PubMed]
- Scott, T.M.; Rasmussen, H.M.; Chen, O.; Johnson, E.J. Avocado Consumption Increases Macular Pigment Density in Older Adults: A Randomized, Controlled Trial. Nutrients 2017, 9, 919. [Google Scholar] [CrossRef] [PubMed]
- Omar, S.H. Mediterranean and MIND Diets Containing Olive Biophenols Reduces the Prevalence of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 2797. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Diet Formulation | Fat % (g) | Carbohydrate % (g) | Protein % (g) |
|---|---|---|---|
| 4:1 Ketogenic Diet | 90% (200) | 2% (10) | 8% (40) |
| 3:1 Ketogenic Diet | 87% (193) | 4% (20) | 9% (45) |
| 2:1 Ketogenic Diet | 82% (182) | 8% (40) | 10% (50) |
| 1:1 Ketogenic Diet | 70% (156) | 10% (50) | 20% (100) |
| Modifed Atkins Diet | 70% (156) | 5% (25) | 25% (125) |
| MCT Diet 2 | 71% (158) 3 | 19% (95) | 10% (50) |
| MUFA 1 | Omega-3 PUFA 2 | Omega-6 PUFA 2 | SFA 3 |
|---|---|---|---|
| Avocado | Chia Seeds | Nuts & Seeds | Butter |
| Lard | Fatty Fish | Dark Poultry | Coconut Oil |
| Nuts & Seeds | Flaxseeds | Red Meat | Eggs |
| Olive Oil | Walnuts | Non-Starchy Vegetables | MCT Oil |
| Olives | Dark Poultry | ||
| Red Meat |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, M.K.; Swerdlow, R.H.; Sullivan, D.K. Dietary Neuroketotherapeutics for Alzheimer’s Disease: An Evidence Update and the Potential Role for Diet Quality. Nutrients 2019, 11, 1910. https://doi.org/10.3390/nu11081910
Taylor MK, Swerdlow RH, Sullivan DK. Dietary Neuroketotherapeutics for Alzheimer’s Disease: An Evidence Update and the Potential Role for Diet Quality. Nutrients. 2019; 11(8):1910. https://doi.org/10.3390/nu11081910
Chicago/Turabian StyleTaylor, Matthew K., Russell H. Swerdlow, and Debra K. Sullivan. 2019. "Dietary Neuroketotherapeutics for Alzheimer’s Disease: An Evidence Update and the Potential Role for Diet Quality" Nutrients 11, no. 8: 1910. https://doi.org/10.3390/nu11081910
APA StyleTaylor, M. K., Swerdlow, R. H., & Sullivan, D. K. (2019). Dietary Neuroketotherapeutics for Alzheimer’s Disease: An Evidence Update and the Potential Role for Diet Quality. Nutrients, 11(8), 1910. https://doi.org/10.3390/nu11081910