The Potential for Renal Injury Elicited by Physical Work in the Heat

 , ,
, ,  and
and
Abstract
:1. Background
2. Acute Kidney Injury
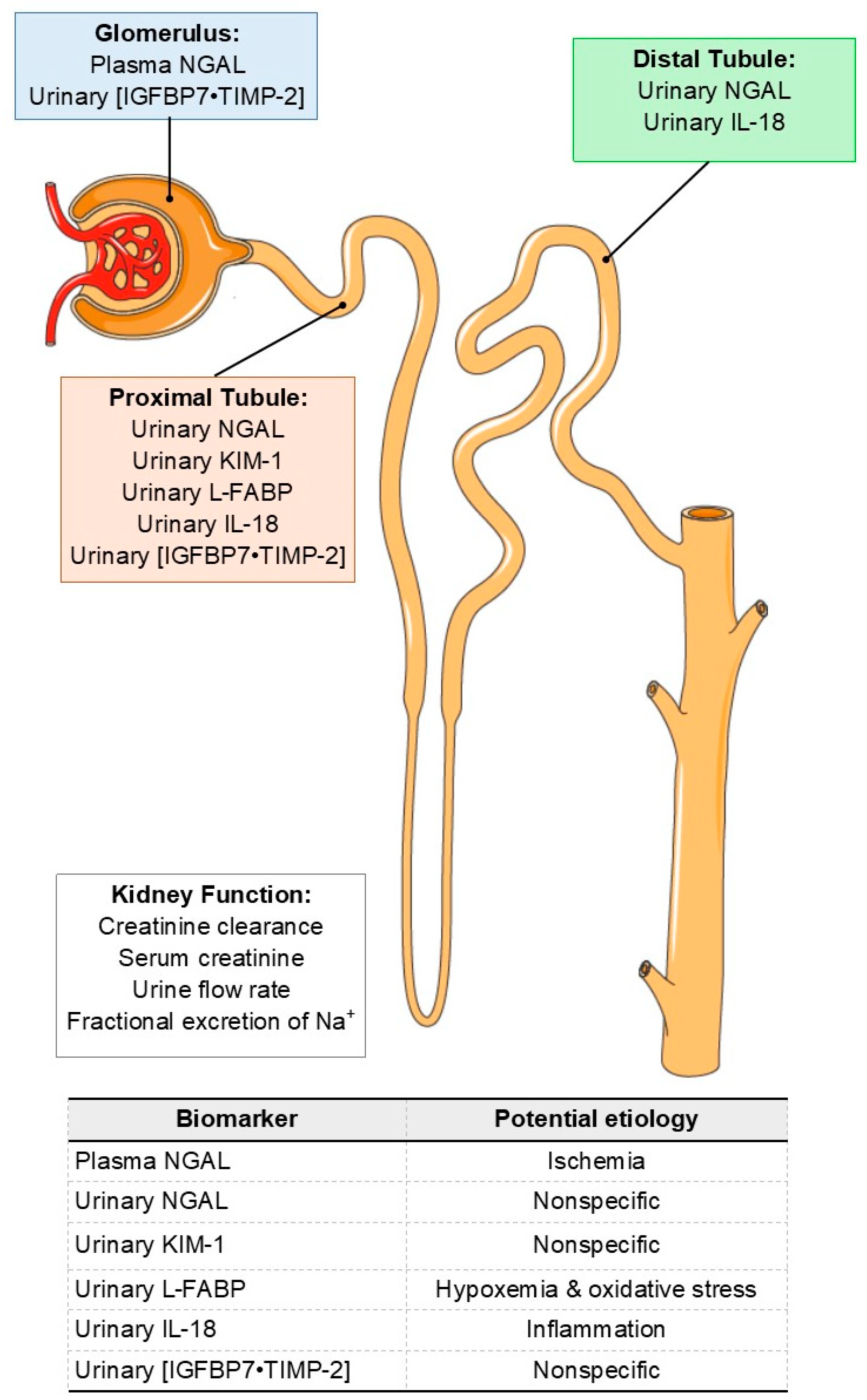
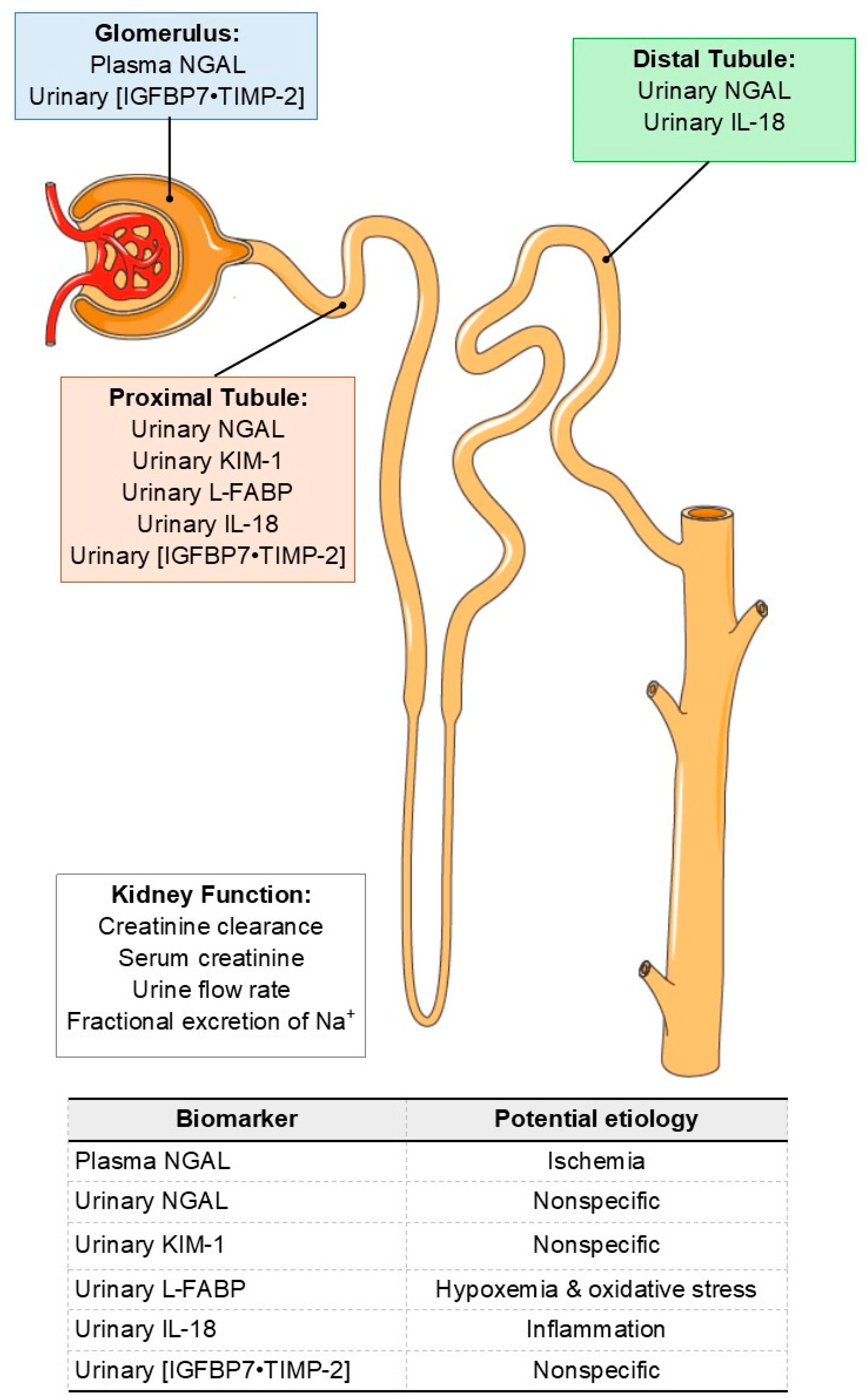
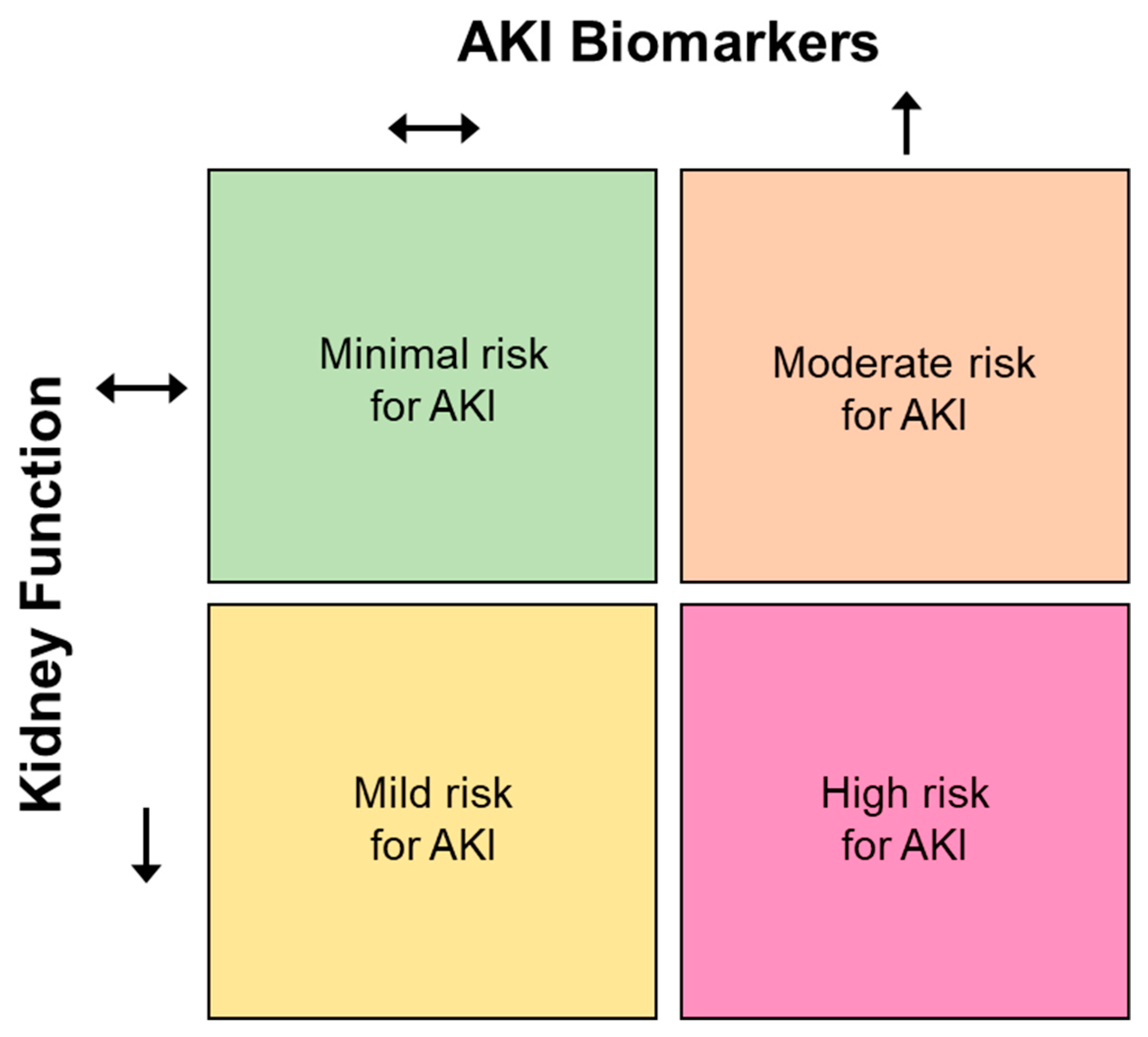
AKI Biomarkers
3. Interpretation of AKI Biomarkers in Non-Clinical Settings
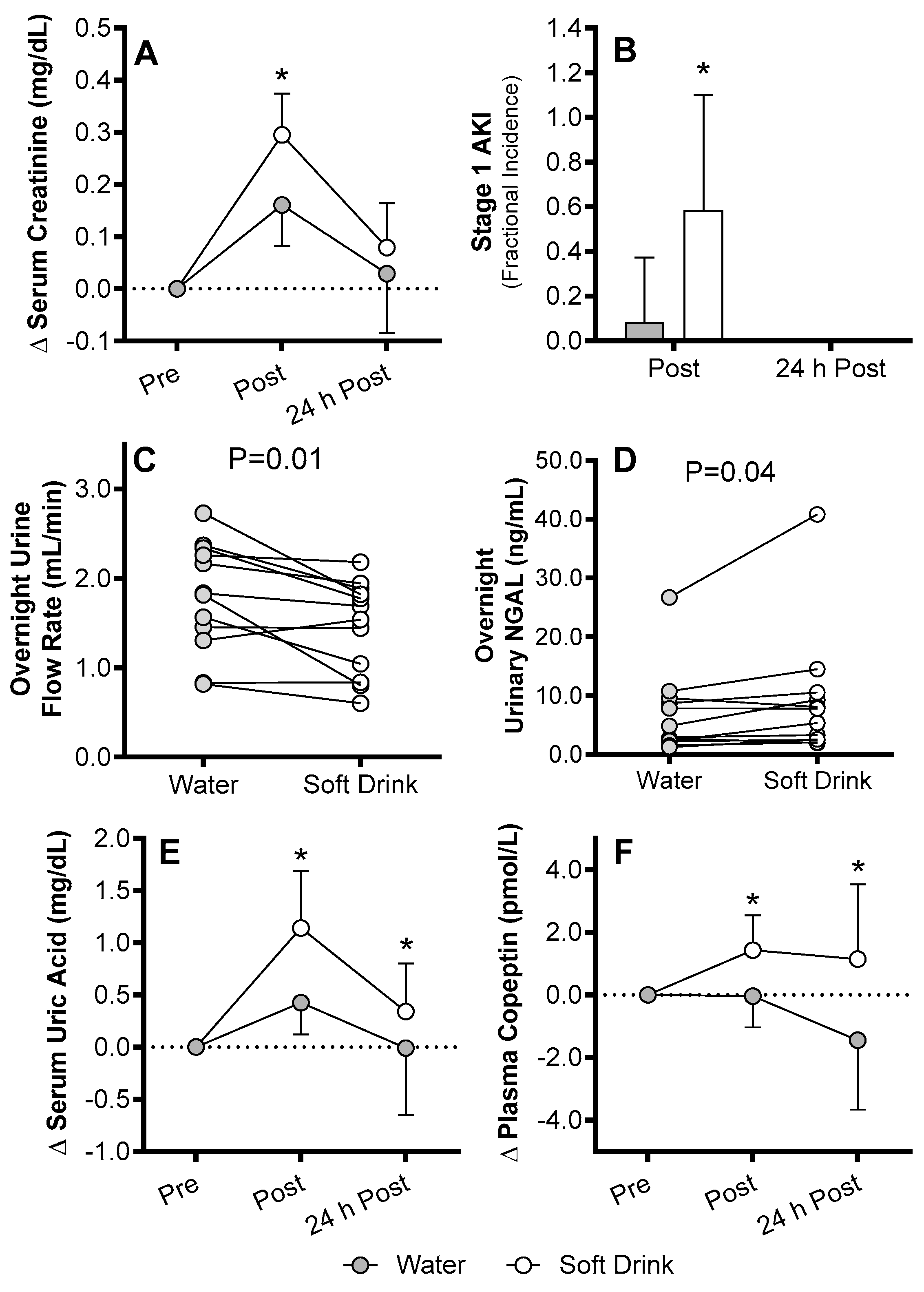
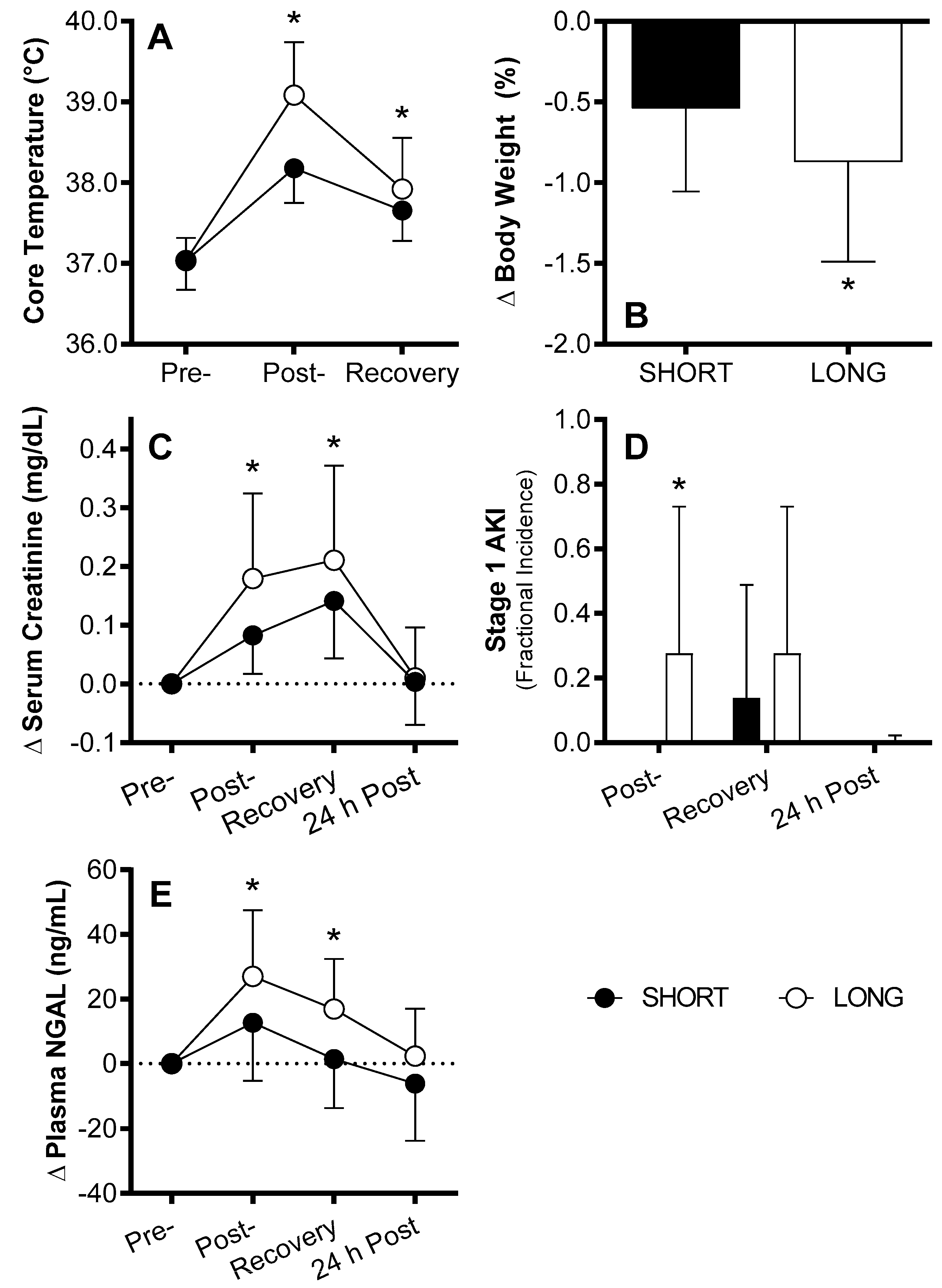
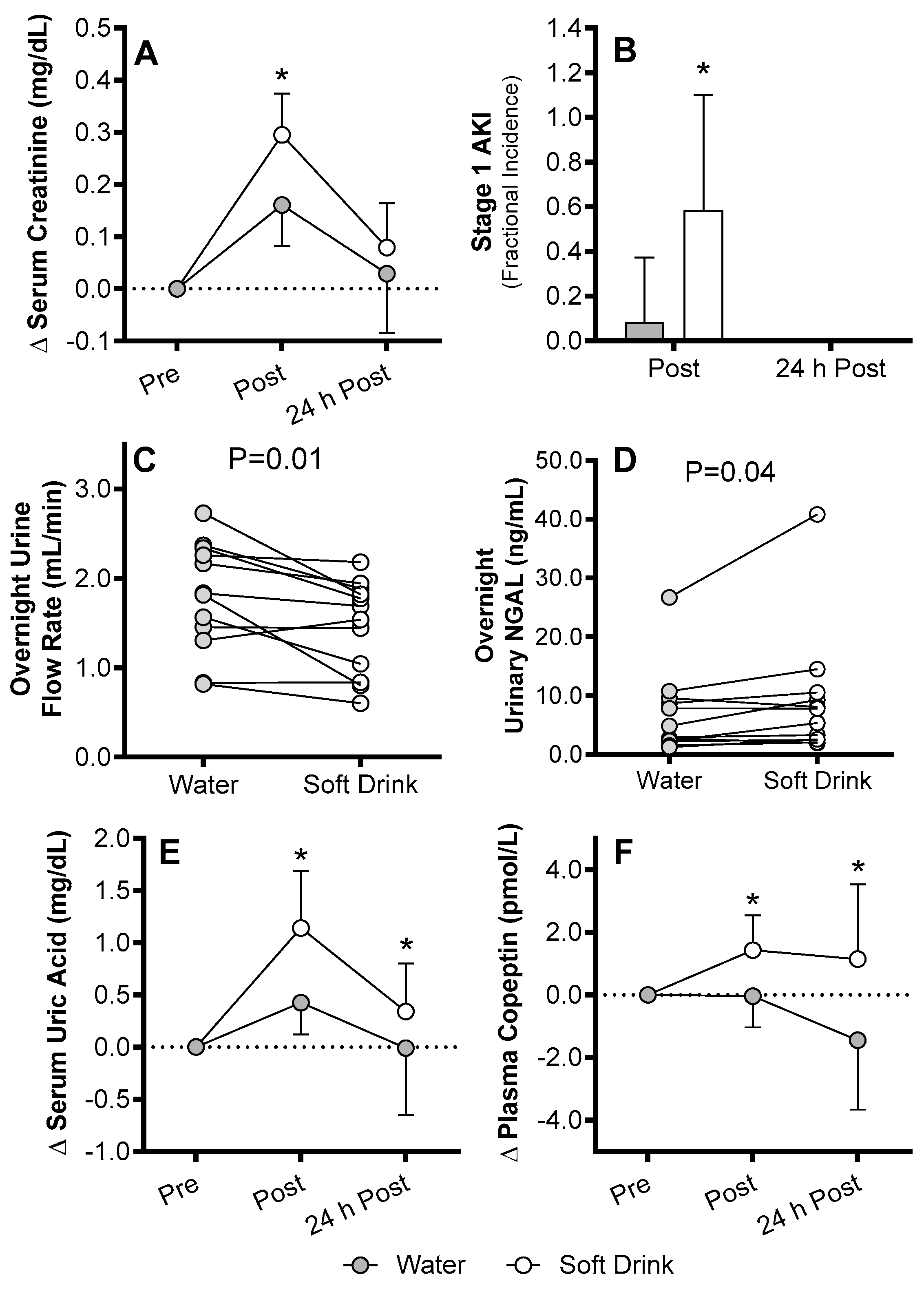
4. AKI Susceptibility Evoked by Exercise in the Heat in Humans
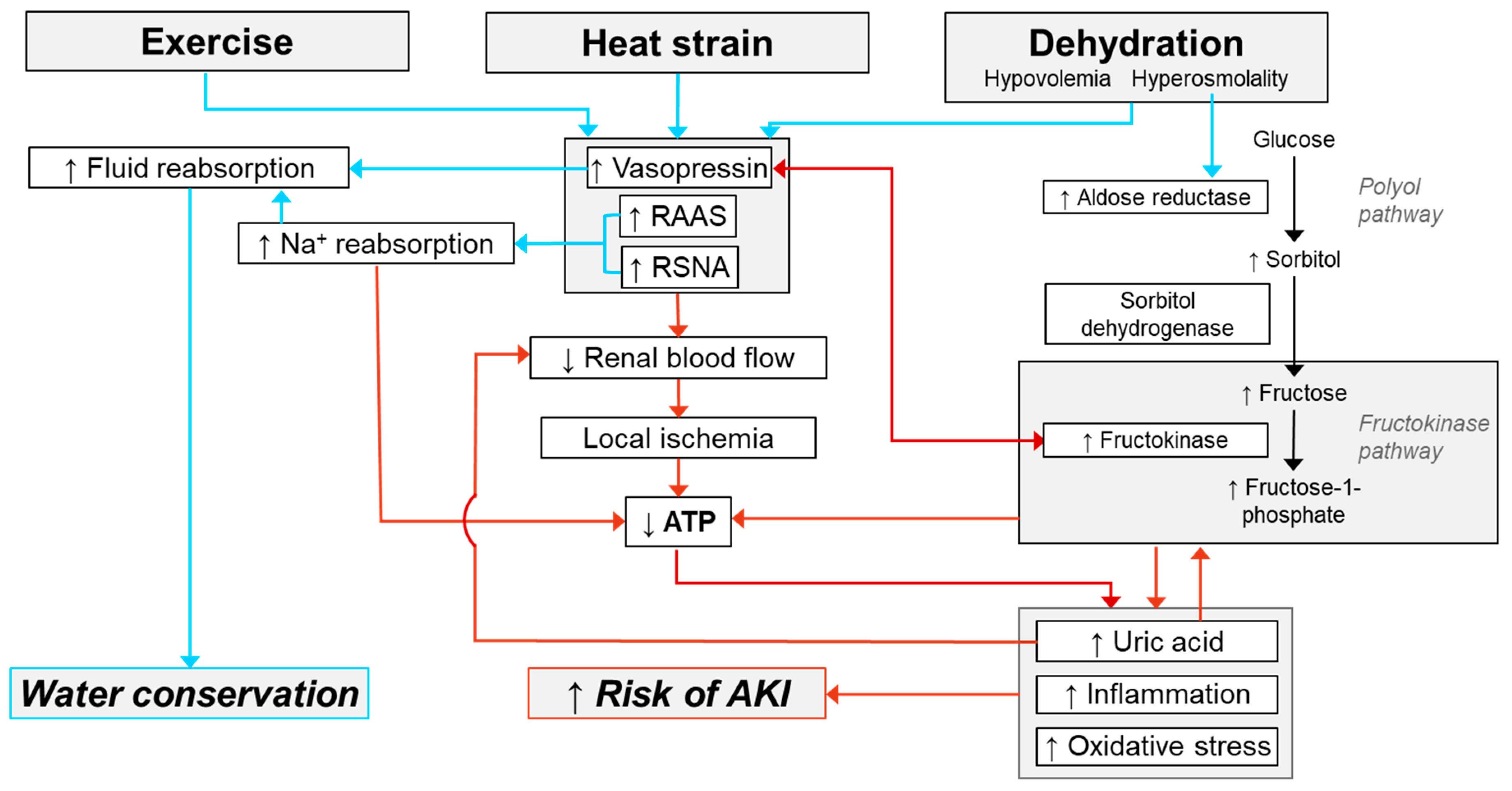
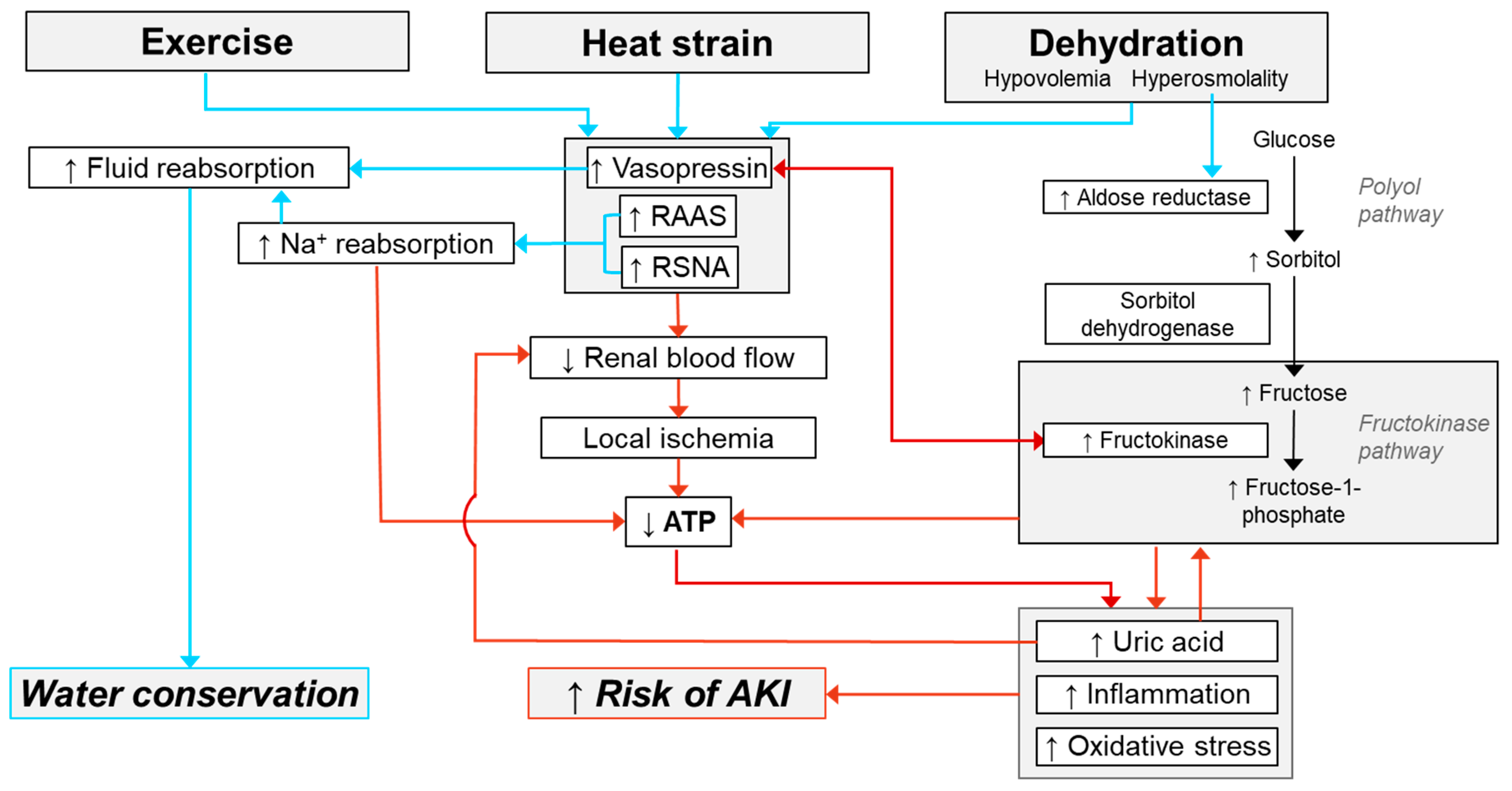
Mechanisms of AKI Susceptibility Evoked by Exercise in the Heat
5. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chatterjee, R. Occupational hazard. Science 2016, 352, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Wesseling, C.; Newman, L.S. Chronic kidney disease of unknown cause in agricultural communities. New Engl. J. Med. 2019, 380, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Trabanino, R.G.; Aguilar, R.; Silva, C.R.; Mercado, M.O.; Merino, R.L. Nefropatía terminal en pacientes de un hospital de referencia en El Salvador. Revista Panamericana de Salud Pública 2002, 12, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.; Aragón, A.; González, M.; Jakobsson, K.; Elinder, C.-G.; Lundberg, I.; Wesseling, C. Decreased kidney function of unknown cause in Nicaragua: A community-based survey. Am. J. Kidney Dis. 2010, 55, 485–496. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.K.; Tobey, M.; Weiner, D.E.; Stevens, L.A.; Johnson, S.; Stringham, P.; Cohen, B.; Brooks, D.R. Prevalence of and risk factors for chronic kidney disease in rural Nicaragua. Nephrol. Dial. Transplant. 2011, 26, 2798–2805. [Google Scholar] [CrossRef] [PubMed]
- Peraza, S.; Wesseling, C.; Aragon, A.; Leiva, R.; García-Trabanino, R.A.; Torres, C.; Jakobsson, K.; Elinder, C.G.; Hogstedt, C. Decreased kidney function among agricultural workers in El Salvador. Am. J. Kidney Dis. 2012, 59, 531–540. [Google Scholar] [CrossRef]
- Glaser, J.; Lemery, J.; Rajagopalan, B.; Diaz, H.F.; García-Trabanino, R.; Taduri, G.; Madero, M.; Amarasinghe, M.; Abraham, G.; Anutrakulchai, S. Climate Change and the Emergent Epidemic of CKD from Heat Stress in Rural Communities: The Case for Heat Stress Nephropathy. Clin. J. Am. Soc. Nephrol. 2016, 11, 1472–1483. [Google Scholar] [CrossRef] [Green Version]
- Flores, S.; Rider, A.C.; Alter, H.J. Mesoamerican nephropathy: A novel case of kidney failure in a US ED. Am. J. Emerg Med. 2015, 34, 1323.e5–1323.e6. [Google Scholar] [CrossRef]
- Organization, P.A.H. Epidemic of Chronic Kidney Disease in Agricultural Communities in Central America Case Definitions, Methodological Basis and Approaches for Public Health Surveillance; Pan American Health Organization: Washington, DC, USA, 2017. [Google Scholar]
- Elledge, M.F.; Redmon, J.H.; Levine, K.E.; Wickremasinghe, R.J.; Wanigasariya, K.P.; Peiris-John, R.J. Chronic Kidney Disease of Unknown Etiology in Sri Lanka: Quest for Understanding and Global Implications; RTI Research Brief. Research; RTI Press: Triangle Park, NC, USA, 2014. [Google Scholar]
- Flouris, A.D.; Dinas, P.C.; Ioannou, L.G.; Nybo, L.; Havenith, G.; Kenny, G.P.; Kjellstrom, T. Workers’ health and productivity under occupational heat strain: A systematic review and meta-analysis. Lancet Planet. Health 2018, 2, e521–e531. [Google Scholar] [CrossRef]
- Pearce, N.; Caplin, B. Let’s Take the Heat OUT of the CKDu Debate: More Evidence Is Needed; BMJ Publishing Group Ltd.: London, UK, 2019. [Google Scholar]
- Wijkström, J.; Leiva, R.; Elinder, C.-G.; Leiva, S.; Trujillo, Z.; Trujillo, L.; Söderberg, M.; Hultenby, K.; Wernerson, A. Clinical and pathological characterization of Mesoamerican nephropathy: A new kidney disease in Central America. Am. J. Kidney Dis. 2013, 62, 908–918. [Google Scholar] [CrossRef]
- Correa-Rotter, R.; Wesseling, C.; Johnson, R.J. CKD of unknown origin in Central America: The case for a Mesoamerican nephropathy. Am. J. Kidney Dis. 2014, 63, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Roncal-Jimenez, C.; Lanaspa, M.; Jensen, T.; Sanchez-Lozada, L.; Johnson, R. Mechanisms by which dehydration may lead to chronic kidney disease. Ann. Nutr. Metab. 2015, 66, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Roncal-Jimenez, C.A.; García-Trabanino, R.; Wesseling, C.; Johnson, R.J. Mesoamerican Nephropathy or Global Warming Nephropathy? Blood Purif. 2016, 41, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Orantes, C.M.; Herrera, R.; Almaguer, M.; Brizuela, E.G.; Hernández, C.E.; Bayarre, H.; Amaya, J.C.; Calero, D.J.; Orellana, P.; Colindres, R.M. Chronic kidney disease and associated risk factors in the Bajo Lempa region of El Salvador: Nefrolempa study, 2009. Med. Rev. 2011, 13, 14–22. [Google Scholar]
- Herrera, R.; Orantes, C.M.; Almaguer, M.; Alfonso, P.; Bayarre, H.D.; Leiva, I.M.; Smith, M.J.; Cubias, R.A.; Almendárez, W.O.; Cubias, F.R. Clinical characteristics of chronic kidney disease of nontraditional causes in Salvadoran farming communities. Med. Rev. 2014, 16, 39–48. [Google Scholar]
- Butler-Dawson, J.; Krisher, L.; Asensio, C.; Cruz, A.; Tenney, L.; Weitzenkamp, D.; Dally, M.; Asturias, E.J.; Newman, L.S. Risk factors for declines in kidney function in sugarcane workers in Guatemala. J. Occup. Environ. Med. 2018, 60, 548. [Google Scholar] [CrossRef] [PubMed]
- González-Quiroz, M.; Pearce, N.; Caplin, B.; Nitsch, D. What do epidemiological studies tell us about chronic kidney disease of undetermined cause in Meso-America? A systematic review and meta-analysis. Clin. Kidney J. 2017, 11, 496–506. [Google Scholar] [CrossRef] [Green Version]
- McClean, M.; Amador, J.J.; Laws, R.; Kaufman, J.S.; Weiner, D.E.; Sanchez-Rodriguez, J.; Brooks, D. Biological Sampling Report: Investigating Biomarkers of Kidney Injury and Chronic Kidney Disease among Workers in Western Nicaragua; University School of Public Health; Compliance Advisor Ombudsman: Boston, MA, USA, 2012. [Google Scholar]
- Gifford, F.J.; Gifford, R.M.; Eddleston, M.; Dhaun, N. Endemic Nephropathy Across the World. Kidney Int. 2016, 2, 282–292. [Google Scholar] [CrossRef]
- Johnson, R.J.; Glaser, J.; Sánchez-Lozada, L.G. Chronic kidney disease of unknown etiology: A disease related to global warming? Med. Rev. 2014, 16, 79. [Google Scholar]
- Chapman, E.; Haby, M.M.; Illanes, E.; Sanchez-Viamonte, J.; Elias, V.; Reveiz, L. Risk factors for chronic kidney disease of non-traditional causes: A systematic review. Pan Am. J. Pub. Health 2019, 43, e35. [Google Scholar] [CrossRef]
- Valcke, M.; Levasseur, M.-E.; da Silva, A.S.; Wesseling, C. Pesticide exposures and chronic kidney disease of unknown etiology: An epidemiologic review. Environ. Health 2017, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.J.; Butler-Dawson, J.; Dally, M.; Krisher, L.; Griffin, B.R.; Johnson, R.J.; Lemery, J.; Asensio, C.; Tenney, L.; Newman, L.S. Risk Factors and Mechanisms Underlying Cross-Shift Decline in Kidney Function in Guatemalan Sugarcane Workers. J. Occup. Environ. Med. 2019, 61, 239. [Google Scholar] [CrossRef] [PubMed]
- Roncal-Jimenez, C.; García-Trabanino, R.; Barregard, L.; Lanaspa, M.A.; Wesseling, C.; Harra, T.; Aragón, A.; Grases, F.; Jarquin, E.R.; González, M.A. Heat stress nephropathy from exercise-induced uric acid crystalluria: A perspective on Mesoamerican nephropathy. Am. J. Kidney Dis. 2016, 67, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Madero, M.; García-Arroyo, F.E.; Sánchez-Lozada, L.-G. Pathophysiologic insight into MesoAmerican nephropathy. Curr. Opin. Nephrol. Hypertens. 2017, 26, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Schlader, Z.J.; Chapman, C.L.; Sarkar, S.; Russo, L.; Rideout, T.C.; Parker, M.D.; Johnson, B.D.; Hostler, D. Firefighter work duration influences the extent of acute kidney injury. Med. Sci. Sport Exerc. 2017, 49, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.L.; Johnson, B.D.; Sackett, J.R.; Parker, M.D.; Schlader, Z.J. Soft drink consumption during and following exercise in the heat elevates biomarkers of acute kidney injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R189–R198. [Google Scholar] [CrossRef]
- Laws, R.L.; Brooks, D.R.; Amador, J.J.; Weiner, D.E.; Kaufman, J.S.; Ramírez-Rubio, O.; Riefkohl, A.; Scammell, M.K.; López-Pilarte, D.; Sánchez, J.M. Biomarkers of kidney injury among Nicaraguan sugarcane workers. Am. J. Kidney Dis. 2016, 67, 209–217. [Google Scholar] [CrossRef]
- Butler-Dawson, J.; Krisher, L.; Yoder, H.; Dally, M.; Sorensen, C.; Johnson, R.J.; Asensio, C.; Cruz, A.; Johnson, E.C.; Carlton, E.J. Evaluation of heat stress and cumulative incidence of acute kidney injury in sugarcane workers in Guatemala. Int. Arch. Occup. Environ. Health 2019, 1–14. [Google Scholar] [CrossRef]
- Au, V.; Feit, J.; Barasch, J.; Sladen, R.N.; Wagener, G. Urinary neutrophil gelatinase–associated lipocalin (NGAL) distinguishes sustained from transient acute kidney injury after general surgery. Kidney Int. Rep. 2016, 1, 3–9. [Google Scholar] [CrossRef]
- Rawson, E.S.; Clarkson, P.M.; Tarnopolsky, M.A. Perspectives on exertional rhabdomyolysis. Sports Med. 2017, 47, 33–49. [Google Scholar] [CrossRef]
- Correa-Rotter, R.; García-Trabanino, R. Mesoamerican nephropathy. Semin. Nephrol. 2019, 39, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Junglee, N.A.; Di Felice, U.; Dolci, A.; Fortes, M.B.; Jibani, M.M.; Lemmey, A.B.; Walsh, N.P.; Macdonald, J.H. Exercising in a hot environment with muscle damage: Effects on acute kidney injury biomarkers and kidney function. Am. J. Physiol. Ren. Physiol. 2013, 305, F813–F820. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Lozada, L.-G.; García-Arroyo, F.E.; Gonzaga, G.; Silverio, O.; Blas-Marron, M.G.; Muñoz-Jimenez, I.; Tapia, E.; Osorio-Alonso, H.; Madero, M.; Roncal-Jiménez, C.A. Kidney Injury from Recurrent Heat Stress and Rhabdomyolysis: Protective Role of Allopurinol and Sodium Bicarbonate. Am. J. Nephrol. 2018, 48, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.J.; Child, R.B.; Day, S.H.; Donnelly, A.E. Exercise-induced skeletal muscle damage and adaptation following repeated bouts of eccentric muscle contractions. J. Sports Sci. 1997, 15, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Herath, C.; Jayasumana, C.; De Silva, P.M.C.; De Silva, P.C.; Siribaddana, S.; De Broe, M.E. Kidney diseases in agricultural communities: A case against heat-stress nephropathy. Kidney Int. Rep. 2018, 3, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Bonventre, J.V.; Mehta, R.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Progression after AKI: Understanding maladaptive repair processes to predict and identify therapeutic treatments. J. Am. Soc. Nephrol. 2015, 27, 687–697. [Google Scholar] [CrossRef]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2011, 2, 1303–1353. [Google Scholar]
- Basile, D.P.; Donohoe, D.; Roethe, K.; Osborn, J.L. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am. J. Physiol. Ren. Physiol. 2001, 281, F887–F899. [Google Scholar] [CrossRef]
- Tanaka, S.; Tanaka, T.; Nangaku, M. Hypoxia as a key player in the AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 2014, 307, F1187–F1195. [Google Scholar] [CrossRef] [Green Version]
- Ferenbach, D.A.; Bonventre, J.V. Acute kidney injury and chronic kidney disease: From the laboratory to the clinic. Nephrol. Ther. 2016, 12, S41–S48. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D. Fibrotic Changes Mediating Acute Kidney Injury to Chronic Kidney Disease Transition. Nephron 2017, 137, 264–267. [Google Scholar]
- Ko, G.J.; Grigoryev, D.N.; Linfert, D.; Jang, H.R.; Watkins, T.; Cheadle, C.; Racusen, L.; Rabb, H. Transcriptional analysis of kidneys during repair from AKI reveals possible roles for NGAL and KIM-1 as biomarkers of AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 2010, 298, F1472–F1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roncal-Jimenez, C.A.; Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Nakagawa, T.; Ejaz, A.A.; Cicerchi, C.; Inaba, S.; Le, M.; Miyazaki, M. Fructokinase activity mediates dehydration-induced renal injury. Kidney Int. 2014, 86, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roncal-Jimenez, C.A.; Milagres, T.; Andres-Hernando, A.; Kuwabara, M.; Jensen, T.; Song, Z.; Bjornstad, P.; Garcia, G.E.; Sato, Y.; Sanchez-Lozada, L.G. Effects of exogenous desmopressin on a model of heat stress nephropathy in mice. Am. J. Physiol. Ren. Physiol. 2017, 312, F418–F426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roncal-Jimenez, C.A.; Sato, Y.; Milagres, T.; Andres-Hernando, A.; Garcia, G.E.; Bjornstad, P.; Butler-Dawson, J.; Sorensen, C.; Newman, L.; Krisher, L. Experimental Heat Stress Nephropathy and Liver Injury are Improved by Allopurinol. Am. J. Physiol. Ren. Physiol. 2018, 315, F726–F733. [Google Scholar] [CrossRef]
- Moyce, S.; Joseph, J.; Tancredi, D.; Mitchell, D.; Schenker, M. Cumulative Incidence of Acute Kidney Injury in California’s Agricultural Workers. J. Occup. Environ. Med. 2016, 58, 391–397. [Google Scholar] [CrossRef] [PubMed]
- García-Trabanino, R.; Jarquín, E.; Wesseling, C.; Johnson, R.J.; González-Quiroz, M.; Weiss, I.; Glaser, J.; Vindell, J.J.; Stockfelt, L.; Roncal, C. Heat stress, dehydration, and kidney function in sugarcane cutters in El Salvador—A cross-shift study of workers at risk of Mesoamerican nephropathy. Environ. Res. 2015, 142, 746–755. [Google Scholar] [CrossRef]
- Laws, R.L.; Brooks, D.R.; Amador, J.J.; Weiner, D.E.; Kaufman, J.S.; Ramírez-Rubio, O.; Riefkohl, A.; Scammell, M.K.; López-Pilarte, D.; Sánchez, J.M. Changes in kidney function among Nicaraguan sugarcane workers. Int. J. Occup. Environ. Health 2015, 21, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Santos, U.P.; Zanetta, D.M.T.; Terra-Filho, M.; Burdmann, E.A. Burnt sugarcane harvesting is associated with acute renal dysfunction. Kidney Int. 2015, 87, 792–799. [Google Scholar] [CrossRef] [Green Version]
- Wesseling, C.; Aragón, A.; González, M.; Weiss, I.; Glaser, J.; Bobadilla, N.A.; Roncal-Jiménez, C.; Correa-Rotter, R.; Johnson, R.J.; Barregard, L. Kidney function in sugarcane cutters in Nicaragua–A longitudinal study of workers at risk of Mesoamerican nephropathy. Environ. Res. 2016, 147, 125–132. [Google Scholar] [CrossRef]
- Moyce, S.; Mitchell, D.; Armitage, T.; Tancredi, D.; Joseph, J.; Schenker, M. Heat strain, volume depletion and kidney function in California agricultural workers. Occup. Environ. Med. 2017, 74, 402–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesseling, C.; García-Trabanino, R.; Wegman, D.H. Mesoamerican Nephropathy: Do Novel Biomarkers of Kidney Damage Have a Role to Play? Am. J. Kidney Dis. 2016, 67, 173–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heung, M.; Steffick, D.E.; Zivin, K.; Gillespie, B.W.; Banerjee, T.; Hsu, C.-Y.; Powe, N.R.; Pavkov, M.E.; Williams, D.E.; Saran, R. Acute kidney injury recovery pattern and subsequent risk of CKD: An analysis of veterans health administration data. Am. J. Kidney Dis. 2016, 67, 742–752. [Google Scholar] [CrossRef]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef]
- Arias-Cabrales, C.; Rodríguez, E.; Bermejo, S.; Sierra, A.; Burballa, C.; Barrios, C.; Soler, M.J.; Pascual, J. Short-and long-term outcomes after non-severe acute kidney injury. Clin. Exp. Nephrol. 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ishani, A.; Xue, J.L.; Himmelfarb, J.; Eggers, P.W.; Kimmel, P.L.; Molitoris, B.A.; Collins, A.J. Acute kidney injury increases risk of ESRD among elderly. J. Am. Soc. Nephrol. 2009, 20, 223–228. [Google Scholar] [CrossRef]
- Schiffl, H.; Fischer, R. Five-year outcomes of severe acute kidney injury requiring renal replacement therapy. Nephrol. Dial. Transplant. 2008, 23, 2235–2241. [Google Scholar] [CrossRef] [Green Version]
- Uchino, S.; Bellomo, R.; Bagshaw, S.M.; Goldsmith, D. Transient azotaemia is associated with a high risk of death in hospitalized patients. Nephrol. Dial. Transplant. 2010, 25, 1833–1839. [Google Scholar] [CrossRef] [Green Version]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. New Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef]
- Chawla, L.S.; Kimmel, P.L. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 2012, 82, 516–524. [Google Scholar] [CrossRef]
- Belayev, L.Y.; Palevsky, P.M. The link between AKI and CKD. Curr. Opin. Nephrol. Hypertens. 2014, 23, 149. [Google Scholar] [CrossRef] [PubMed]
- Roncal, C.A.; Mu, W.; Croker, B.; Reungjui, S.; Ouyang, X.; Tabah-Fisch, I.; Johnson, R.J.; Ejaz, A.A. Effect of elevated serum uric acid on cisplatin-induced acute renal failure. Am. J. Physiol. Ren. Physiol. 2007, 292, F116–F122. [Google Scholar] [CrossRef] [PubMed]
- Ryu, E.-S.; Kim, M.J.; Shin, H.-S.; Jang, Y.-H.; Choi, H.S.; Jo, I.; Johnson, R.J.; Kang, D.-H. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2013, 304, F471–F480. [Google Scholar] [CrossRef] [PubMed]
- Suga, S.-I.; Phillips, M.I.; Ray, P.E.; Raleigh, J.A.; Vio, C.P.; Kim, Y.-G.; Mazzali, M.; Gordon, K.L.; Hughes, J.; Johnson, R.J. Hypokalemia induces renal injury and alterations in vasoactive mediators that favor salt sensitivity. Am. J. Physiol. Ren. Physiol. 2001, 281, F620–F629. [Google Scholar] [CrossRef] [PubMed]
- Perner, A.; Prowle, J.; Joannidis, M.; Young, P.; Hjortrup, P.B.; Pettilä, V. Fluid management in acute kidney injury. Intensive Care Med. 2017, 43, 807–815. [Google Scholar] [CrossRef] [PubMed]
- McDermott, B.P.; Smith, C.R.; Butts, C.L.; Caldwell, A.R.; Lee, E.C.; Vingren, J.L.; Munoz, C.X.; Kunces, L.J.; Williamson, K.; Ganio, M.S. Renal stress and kidney injury biomarkers in response to endurance cycling in the heat with and without ibuprofen. J. Sci. Med. Sport 2018, 21, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Lameire, N.; Aspelin, P.; Barsoum, R.S.; Burdmann, E.A.; Goldstein, S.L.; Herzog, C.A.; Joannidis, M.; Kribben, A.; Levey, A.S. Kidney disease: Improving global outcomes (KDIGO) acute kidney injury work group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int. Suppl. 2012, 2, 1–138. [Google Scholar]
- Levey, A.S.; Eckardt, K.-U.; Tsukamoto, Y.; Levin, A.; Coresh, J.; Rossert, J.; Zeeuw, D.D.; Hostetter, T.H.; Lameire, N.; Eknoyan, G. Definition and classification of chronic kidney disease: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2005, 67, 2089–2100. [Google Scholar] [CrossRef] [Green Version]
- Mehta, R.L.; Kellum, J.A.; Shah, S.V.; Molitoris, B.A.; Ronco, C.; Warnock, D.G.; Levin, A. Acute Kidney Injury Network: Report of an initiative to improve outcomes in acute kidney injury. Crit. Care 2007, 11, R31. [Google Scholar] [CrossRef]
- Bellomo, R.; Ronco, C.; Kellum, J.A.; Mehta, R.L.; Palevsky, P. Acute renal failure–definition, outcome measures, animal models, fluid therapy and information technology needs: The Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit. Care 2004, 8, R204. [Google Scholar] [CrossRef]
- Kellum, J.A.; Lameire, N. Diagnosis, evaluation, and management of acute kidney injury: A KDIGO summary (Part 1). Crit. Care 2013, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Beierwaltes, W.H.; Harrison-Bernard, L.M.; Sullivan, J.C.; Mattson, D.L. Assessment of renal function; clearance, the renal microcirculation, renal blood flow, and metabolic balance. Compr. Physiol. 2013, 3, 165–200. [Google Scholar] [PubMed]
- Traynor, J.; Mactier, R.; Geddes, C.C.; Fox, J.G. How to measure renal function in clinical practice. BMJ 2006, 333, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Yong, Z.; Pei, X.; Zhu, B.; Yuan, H.; Zhao, W. Predictive value of serum cystatin C for acute kidney injury in adults: A meta-analysis of prospective cohort trials. Sci. Rep. 2017, 7, 41012. [Google Scholar] [CrossRef] [PubMed]
- Odutayo, A.; Cherney, D. Cystatin C and acute changes in glomerular filtration rate. Clin. Nephrol. 2012, 78, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Coca, S.G. Acute kidney injury: Defining prerenal azotemia in clinical practice and research. Nat. Rev. Nephrol. 2010, 6, 641. [Google Scholar] [CrossRef]
- McCance, R.; Widdowson, E. The secretion of urine in man during experimental salt deficiency. J. Physiol. 1937, 91, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Nadal, J.W.; Pedersen, S.; Maddock, W.G. A comparison between dehydration from salt loss and from water deprivation. J. Clin. Invest. 1941, 20, 691. [Google Scholar] [CrossRef]
- Kirkebø, A.; Tyssebotn, I. Effect of dehydration on renal blood flow in dog. Acta Physiol. Scand. 1977, 101, 257–263. [Google Scholar] [CrossRef]
- Hope, A.; Tyssebotn, I. The effect of water deprivation on local renal blood flow and filtration in the laboratory rat. Circ. Shock 1982, 11, 175–186. [Google Scholar]
- Stocker, S.D.; Hunwick, K.J.; Toney, G.M. Hypothalamic paraventricular nucleus differentially supports lumbar and renal sympathetic outflow in water-deprived rats. J. Physiol. 2005, 563, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Bardgett, M.E.; Chen, Q.-H.; Guo, Q.; Calderon, A.S.; Andrade, M.A.; Toney, G.M. Coping with dehydration: Sympathetic activation and regulation of glutamatergic transmission in the hypothalamic PVN. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R804–R813. [Google Scholar] [CrossRef] [PubMed]
- Bie, P. Osmoreceptors, vasopressin, and control of renal water excretion. Physiol. Rev. 1980, 60, 961–1048. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, R.; Mendelsohn, F. Plasma renin and angiotensin in dehydrated and rehydrated rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1986, 250, R898–R901. [Google Scholar] [CrossRef] [PubMed]
- Mack, G.W.; Nadel, E.R. Body fluid balance during heat stress in humans. In Comprehensive Physiology; Supplement 14: Handbook of Physiology, Environmental Physiology; Wiley: Hoboken, NJ, USA, 2011; pp. 187–214. [Google Scholar]
- Cheuvront, S.N.; Ely, B.R.; Kenefick, R.W.; Sawka, M.N. Biological variation and diagnostic accuracy of dehydration assessment markers. Am. J. Clin. Nutr. 2010, 92, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Griffin, B.R.; Gist, K.M.; Faubel, S. Current status of novel biomarkers for the diagnosis of acute kidney injury: A historical perspective. J. Intensive Care Med. 2019, 0885066618824531. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Siew, E.D. Biomarkers for the early detection and prognosis of acute kidney injury. Clin. J. Am. Soc. Nephrol. 2017, 12, 149–173. [Google Scholar] [CrossRef]
- Koyner, J.L.; Parikh, C.R. Clinical utility of biomarkers of AKI in cardiac surgery and critical illness. Clin. J. Am. Soc. Nephrol. 2013, 8, 1034–1042. [Google Scholar] [CrossRef]
- Schrezenmeier, E.; Barasch, J.; Budde, K.; Westhoff, T.; Schmidt-Ott, K. Biomarkers in acute kidney injury–pathophysiological basis and clinical performance. Acta Physiol. 2017, 219, 556–574. [Google Scholar] [CrossRef]
- Schaub, J.A.; Parikh, C.R. Biomarkers of acute kidney injury and associations with short-and long-term outcomes. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Kashani, K.; Kellum, J.A. Novel biomarkers indicating repair or progression after acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2015, 24, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Haase, M.; Kellum, J.A.; Ronco, C. Subclinical AKI—An emerging syndrome with important consequences. Nat. Rev. Nephrol. 2012, 8, 735. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Kaur, S.; Guha, S.; Batra, S.K. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim. Biophys. Acta Rev. Cancer 2012, 1826, 129–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Mishra, J.; Mori, K.; Ma, Q.; Kelly, C.; Barasch, J.; Devarajan, P. Neutrophil gelatinase-associated lipocalin: A novel early urinary biomarker for cisplatin nephrotoxicity. Am. J. Nephrol. 2004, 24, 307–315. [Google Scholar] [CrossRef]
- Mishra, J.; Dent, C.; Tarabishi, R.; Mitsnefes, M.M.; Ma, Q.; Kelly, C.; Ruff, S.M.; Zahedi, K.; Shao, M.; Bean, J. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 2005, 365, 1231–1238. [Google Scholar] [CrossRef]
- Mishra, J.; Ma, Q.; Prada, A.; Mitsnefes, M.; Zahedi, K.; Yang, J.; Barasch, J.; Devarajan, P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J. Am. Soc. Nephrol. 2003, 14, 2534–2543. [Google Scholar] [CrossRef]
- Supavekin, S.; Zhang, W.; Kucherlapati, R.; Kaskel, F.J.; Moore, L.C.; Devarajan, P. Differential gene expression following early renal ischemia/reperfusion. Kidney Int. 2003, 63, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Ott, K.M.; Mori, K.; Li, J.Y.; Kalandadze, A.; Cohen, D.J.; Devarajan, P.; Barasch, J. Dual action of neutrophil gelatinase–associated lipocalin. J. Am. Soc. Nephrol. 2007, 18, 407–413. [Google Scholar] [CrossRef]
- Hvidberg, V.; Jacobsen, C.; Strong, R.K.; Cowland, J.B.; Moestrup, S.K.; Borregaard, N. The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett. 2005, 579, 773–777. [Google Scholar] [CrossRef]
- Singer, E.; Elger, A.; Elitok, S.; Kettritz, R.; Nickolas, T.L.; Barasch, J.; Luft, F.C.; Schmidt-Ott, K.M. Urinary neutrophil gelatinase-associated lipocalin distinguishes pre-renal from intrinsic renal failure and predicts outcomes. Kidney Int. 2011, 80, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nejat, M.; Pickering, J.W.; Devarajan, P.; Bonventre, J.V.; Edelstein, C.L.; Walker, R.J.; Endre, Z.H. Some biomarkers of acute kidney injury are increased in pre-renal acute injury. Kidney Int. 2012, 81, 1254–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, T.; Bonventre, J.V.; Bailly, V.; Wei, H.; Hession, C.A.; Cate, R.L.; Sanicola, M. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J. Biol. Chem. 1998, 273, 4135–4142. [Google Scholar] [CrossRef] [PubMed]
- Prozialeck, W.; Vaidya, V.; Liu, J.; Waalkes, M.; Edwards, J.; Lamar, P.; Bernard, A.; Dumont, X.; Bonventre, J. Kidney injury molecule-1 is an early biomarker of cadmium nephrotoxicity. Kidney Int. 2007, 72, 985–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, T.; Hung, C.C.; Yang, S.A.; Stevens, J.L.; Bonventre, J.V. Kidney injury molecule-1: A tissue and urinary biomarker for nephrotoxicant-induced renal injury. Am. J. Physiol. Ren. Physiol. 2004, 286, F552–F563. [Google Scholar] [CrossRef]
- Han, W.K.; Bailly, V.; Abichandani, R.; Thadhani, R.; Bonventre, J.V. Kidney Injury Molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 2002, 62, 237–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Gong, Y.; Anderson, J.; Sun, D.; Minuk, G.; Roberts, M.S.; Burczynski, F.J. Antioxidative function of L-FABP in L-FABP stably transfected Chang liver cells. Hepatology 2005, 42, 871–879. [Google Scholar] [CrossRef]
- Yamamoto, T.; Noiri, E.; Ono, Y.; Doi, K.; Negishi, K.; Kamijo, A.; Kimura, K.; Fujita, T.; Kinukawa, T.; Taniguchi, H. Renal L-type fatty acid–binding protein in acute ischemic injury. J. Am. Soc. Nephrol. 2007, 18, 2894–2902. [Google Scholar] [CrossRef]
- Doi, K.; Negishi, K.; Ishizu, T.; Katagiri, D.; Fujita, T.; Matsubara, T.; Yahagi, N.; Sugaya, T.; Noiri, E. Evaluation of new acute kidney injury biomarkers in a mixed intensive care unit. Crit. Care Med. 2011, 39, 2464–2469. [Google Scholar] [CrossRef]
- Gauer, S.; Sichler, O.; Obermüller, N.; Holzmann, Y.; Kiss, E.; Sobkowiak, E.; Pfeilschifter, J.; Geiger, H.; Mühl, H.; Hauser, I. IL-18 is expressed in the intercalated cell of human kidney. Kidney Int. 2007, 72, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Franke, E.I.; Vanderbrink, B.A.; Hile, K.L.; Zhang, H.; Cain, A.; Matsui, F.; Meldrum, K.K. Renal IL-18 production is macrophage independent during obstructive injury. PLoS ONE 2012, 7, e47417. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yuan, J.; Zhao, Y.; Zha, Y. Urine interleukin-18 in prediction of acute kidney injury: A systemic review and meta-analysis. J. Nephrol. 2015, 28, 7–16. [Google Scholar] [CrossRef]
- Kellum, J.A.; Chawla, L.S. Cell-cycle arrest and acute kidney injury: The light and the dark sides. Nephrol. Dial. Transplant. 2015, 31, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J.; Bhaumik, D. Two faces of p53: Aging and tumor suppression. Nucleic Acids Res. 2007, 35, 7475–7484. [Google Scholar] [CrossRef] [PubMed]
- Shankland, S.J. Cell cycle regulatory proteins in glomerular disease. Kidney Int. 1999, 56, 1208–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witzgall, R.; Brown, D.; Schwarz, C.; Bonventre, J.V. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J. Clin. Investig. 1994, 93, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535. [Google Scholar] [CrossRef]
- Kashani, K.; Al-Khafaji, A.; Ardiles, T.; Artigas, A.; Bagshaw, S.M.; Bell, M.; Bihorac, A.; Birkhahn, R.; Cely, C.M.; Chawla, L.S. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit. Care 2013, 17, R25. [Google Scholar] [CrossRef]
- Endre, Z.H.; Pickering, J.W. Acute kidney injury: Cell cycle arrest biomarkers win race for AKI diagnosis. Nat. Rev. Nephrol. 2014, 10, 683. [Google Scholar] [CrossRef]
- Price, P.M.; Safirstein, R.L.; Megyesi, J. The cell cycle and acute kidney injury. Kidney Int. 2009, 76, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.C.; Zager, R.A. Mechanisms Underlying Increased TIMP2 and IGFBP7 Urinary Excretion in Experimental AKI. J. Am. Soc. Nephrol. 2018, 76, 604–613. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A.; Chinnaiyan, K.M.; Gallagher, M.J.; Colar, J.M.; Geddes, T.; Gold, J.M.; Trivax, J.E. Changes in renal markers and acute kidney injury after marathon running. Nephrology 2011, 16, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.D.; Stuempfle, K.J.; Fogard, K.; Hew-Butler, T.; Winger, J.; Weiss, R.H. Urine dipstick analysis for identification of runners susceptible to acute kidney injury following an ultramarathon. J. Sport Sci. 2013, 31, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.-K.; Chiu, Y.-H.; Tsai, Y.-F.; Tai, L.-C.; Hou, P.C.; How, C.-K.; Yang, C.-C.; Kao, W.-F. Clinical impact of speed variability to identify ultramarathon runners at risk for acute kidney injury. PLoS ONE 2015, 10, e0133146. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.G.; Verma, G.; Pata, R.W.; Martin, T.G.; Perazella, M.A.; Parikh, C.R. Kidney injury and repair biomarkers in marathon runners. Am. J. Kidney Dis. 2017, 70, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Bongers, C.C.; Alsady, M.; Nijenhuis, T.; Hartman, Y.A.; Eijsvogels, T.M.; Deen, P.M.; Hopman, M.T. Impact of acute versus repetitive moderate intensity endurance exercise on kidney injury markers. Physiol. Rep. 2017, 5, e13544. [Google Scholar] [CrossRef] [PubMed]
- Bongers, C.C.; Alsady, M.; Nijenhuis, T.; Tulp, A.D.; Eijsvogels, T.M.; Deen, P.M.; Hopman, M.T. Impact of acute versus prolonged exercise and dehydration on kidney function and injury. Physiol. Rep. 2018, 6, e13734. [Google Scholar] [CrossRef] [PubMed]
- Eichner, E.R. Is Heat Stress Nephropathy a Concern for Endurance Athletes? Curr. Sport Med. Rep. 2017, 16, 299–300. [Google Scholar] [CrossRef]
- Latham, A. The history of a habit: Jogging as a palliative to sedentariness in 1960s America. Cult. Geogr. 2015, 22, 103–126. [Google Scholar] [CrossRef]
- Rewa, O.; Bagshaw, S.M. Acute kidney injury—Epidemiology, outcomes and economics. Nat. Rev. Nephrol. 2014, 10, 193. [Google Scholar] [CrossRef]
- Stump, C.S. Physical activity in the prevention of chronic kidney disease. Cardiorenal Med. 2011, 1, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.T.; Mehta, R.L.; Shaw, A.; Ronco, C.; Endre, Z.; Kellum, J.A.; Chawla, L.S.; Cruz, D.; Ince, C.; Okusa, M.D. Potential use of biomarkers in acute kidney injury: Report and summary of recommendations from the 10th Acute Dialysis Quality Initiative consensus conference. Kidney Int. 2014, 85, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Katz, N.; Ronco, C. Acute kidney stress—A useful term based on evolution in the understanding of acute kidney injury. Crit. Care 2015, 20, 23. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A. Diagnostic criteria for acute kidney injury: Present and future. Crit. Care Clin. 2015, 31, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Waikar, S.S.; Sabbisetti, V.S.; Bonventre, J.V. Normalization of urinary biomarkers to creatinine during changes in glomerular filtration rate. Kidney Int. 2010, 78, 486–494. [Google Scholar] [CrossRef] [Green Version]
- Junglee, N.A.; Lemmey, A.B.; Burton, M.; Searell, C.; Jones, D.; Lawley, J.S.; Jibani, M.M.; Macdonald, J.H. Does proteinuria-inducing physical activity increase biomarkers of acute kidney injury? Kidney Blood Press. Res. 2012, 36, 278–289. [Google Scholar] [CrossRef]
- Schrier, R.W.; Henderson, H.S.; Tisher, C.C.; Tannen, R.L. Nephropathy associated with heat stress and exercise. Ann. Intern. Med. 1967, 67, 356–376. [Google Scholar] [CrossRef]
- Schrier, R.W.; Hano, J.; Keller, H.I.; Finkel, R.M.; Gilliland, P.F.; Cirksena, W.J.; Teschan, P.E. Renal, metabolic, and circulatory responses to heat and exercise. Studies in military recruits during summer training, with implications for acute renal failure. Ann. Intern. Med. 1970, 73, 213–223. [Google Scholar] [CrossRef]
- Montain, S.J.; Coyle, E.F. Influence of graded dehydration on hyperthermia and cardiovascular drift during exercise. J. Appl. Physiol. 1992, 73, 1340–1350. [Google Scholar] [CrossRef]
- Farquhar, W.; Morgan, A.; Zambraski, E.; Kenney, W. Effects of acetaminophen and ibuprofen on renal function in the stressed kidney. J. Appl. Physiol. 1999, 86, 598–604. [Google Scholar] [CrossRef]
- Radigan, L.R.; Robinson, S. Effects of environmental heat stress and exercise on renal blood flow and filtration rate. J. Appl. Physiol. 1949, 2, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Barclay, J.; Cooke, W.; Kenney, R.; Nutt, M.E. The effects of water diuresis and exercise on the volume and composition of the urine. Am. J. Physiol. 1947, 148, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Kenney, W.; Zappe, D. Effect of age on renal blood flow during exercise. Aging Clin. Exp. Res. 1994, 6, 293–302. [Google Scholar] [CrossRef]
- Kawakami, S.; Yasuno, T.; Matsuda, T.; Fujimi, K.; Ito, A.; Yoshimura, S.; Uehara, Y.; Tanaka, H.; Saito, T.; Higaki, Y. Association between exercise intensity and renal blood flow evaluated using ultrasound echo. Clin. Exp. Nephrol. 2018, 22, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Robinson, S.; Pearcy, M. Renal responses to exercise, heat and dehydration. J. Appl. Physiol. 1952, 4, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.E. Renal sympathetic nerve, blood flow, and epithelial transport responses to thermal stress. Auton. Neurosci. Basic Clin. 2017, 204, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Azzawi, S.; Shirley, D. The effect of vasopressin on renal blood flow and its distribution in the rat. J. Physiol. 1983, 341, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R.H.; Davis, J.O.; Vitale, S.J.; Johnson, J.A. Intrarenal role of angiotensin II: Homeostatic regulation of renal blood flow in the dog. Circ. Res. 1973, 32, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Mittleman, K.D. Influence of angiotensin II blockade during exercise in the heat. Eur. J. Appl. Physiol. Occup. Physiol. 1996, 72, 542–547. [Google Scholar] [CrossRef]
- Miyamoto, M. Renal cortical and medullary tissue blood flow during experimental hyperthermia in dogs. Therm. Med 1994, 10, 78–89. [Google Scholar] [CrossRef]
- Devarajan, P. Update on mechanisms of ischemic acute kidney injury. J. Am. Soc. Nephrol. 2006, 17, 1503–1520. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.L.; Li, X.C. Proximal nephron. Compr. Physiol. 2013, 3, 1079–1123. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.G.; Schnermann, J. Integrated control of Na transport along the nephron. Clin. J. Am. Soc. Nephrol. 2015, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- El Mernissi, G.; Doucet, A. Short-term effect of aldosterone on renal sodium transport and tubular Na−K-ATPase in the rat. Pflügers Arch. 1983, 399, 139–146. [Google Scholar] [CrossRef]
- Johnson, M.D.; Malvin, R.L. Stimulation of renal sodium reabsorption by angiotensin II. Am. J. Physiol. Ren. Physiol. 1977, 232, F298–F306. [Google Scholar] [CrossRef]
- Bell-Reuss, E.; Trevino, D.; Gottschalk, C. Effect of renal sympathetic nerve stimulation on proximal water and sodium reabsorption. J. Clin. Investig. 1976, 57, 1104–1107. [Google Scholar] [CrossRef]
- Montain, S.J.; Laird, J.E.; Latzka, W.A.; Sawka, M.N. Aldosterone and vasopressin responses in the heat: Hydration level and exercise intensity effects. Med. Sci. Sports Exerc. 1997, 29, 661–668. [Google Scholar] [CrossRef]
- Kenney, M.J.; Barney, C.C.; Hirai, T.; Gisolfi, C.V. Sympathetic nerve responses to hyperthermia in the anesthetized rat. J. Appl. Physiol. 1995, 78, 881–889. [Google Scholar] [CrossRef]
- Melin, B.; Koulmann, N.; Jimenez, C.; Savourey, G.; Launay, J.-C.; Cottet-Emard, J.-M.; Pequignot, J.-M.; Allevard, A.-M.; Gharib, C. Comparison of passive heat or exercise-induced dehydration on renal water and electrolyte excretion: The hormonal involvement. Eur. J. Appl. Physiol. 2001, 85, 250–258. [Google Scholar] [CrossRef]
- Doucet, A. Function and control of Na-K-ATPase in single nephron segments of the mammalian kidney. Kidney Int. 1988, 34, 749–760. [Google Scholar] [CrossRef] [Green Version]
- Barrera-Chimal, J.; Pérez-Villalva, R.; Ortega, J.A.; Sánchez, A.; Rodríguez-Romo, R.; Durand, M.; Jaisser, F.; Bobadilla, N.A. Mild ischemic injury leads to long-term alterations in the kidney: Amelioration by spironolactone administration. Int. J. Biol. Sci. 2015, 11, 892. [Google Scholar] [CrossRef] [PubMed]
- Lattenist, L.; Lechner, S.M.; Messaoudi, S.; Le Mercier, A.; El Moghrabi, S.; Prince, S.; Bobadilla, N.A.; Kolkhof, P.; Jaisser, F.; Barrera-Chimal, J. Nonsteroidal Mineralocorticoid Receptor Antagonist Finerenone Protects Against Acute Kidney Injury–Mediated Chronic Kidney Disease: Role of Oxidative Stress. Hypertension 2017, 69, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Otani, H.; Kaya, M.; Tsujita, J. Effect of the volume of fluid ingested on urine concentrating ability during prolonged heavy exercise in a hot environment. J. Sports Sci. Med. 2013, 12, 197. [Google Scholar] [PubMed]
- Evans, R.G.; Ince, C.; Joles, J.A.; Smith, D.W.; May, C.N.; O’Connor, P.M.; Gardiner, B.S. Haemodynamic influences on kidney oxygenation: Clinical implications of integrative physiology. Clin. Exp. Pharmacol. Physiol. 2013, 40, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Chayoth, R.; Kleinman, D.; Kaplanski, J.; Sod Moriah, U. Renal clearance of urea, inulin, and p-aminohippurate in heat-acclimated rats. J. Appl. Physiol. 1984, 57, 731–732. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.A. Human heat adaptation. Compr. Physiol. 2014, 4, 325–365. [Google Scholar] [PubMed]
- Omassoli, J.; Hill, N.E.; Woods, D.R.; Delves, S.K.; Fallowfield, J.L.; Brett, S.J.; Wilson, D.; Corbett, R.W.; Allsopp, A.J.; Stacey, M.J. Variation in renal responses to exercise in the heat with progressive acclimatisation. J. Sci. Med. Sport 2019, 22, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.W.; Beard, J.L.; Farrell, P.A.; Minson, C.T.; Kenney, W.L. Age, fitness, and regional blood flow during exercise in the heat. J. Appl. Physiol. 1997, 82, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Minson, C.T.; Wladkowski, S.L.; Cardell, A.F.; Pawelczyk, J.A.; Kenney, W.L. Age alters the cardiovascular response to direct passive heating. J. Appl. Physiol. 1998, 84, 1323–1332. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.-H.; So, R.; Lee, C.; Hong, Y.-C.; Park, M.; Kim, L.; Yoon, H.-J. Ambient temperature and hospital admissions for acute kidney injury: A time-series analysis. Sci. Total Environ. 2018, 616, 1134–1138. [Google Scholar] [CrossRef]
- McTavish, R.K.; Richard, L.; McArthur, E.; Shariff, S.Z.; Acedillo, R.; Parikh, C.R.; Wald, R.; Wilk, P.; Garg, A.X. Association between high environmental heat and risk of acute kidney injury among older adults in a northern climate: A matched case-control study. Am. J. Kidney Dis. 2018, 71, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, H.; Kim, Y.C.; Lee, J.P. Association between extreme temperature and kidney disease in South Korea, 2003–2013: Stratified by sex and age groups. Sci. Total Environ. 2018, 642, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Bobb, J.F.; Obermeyer, Z.; Wang, Y.; Dominici, F. Cause-specific risk of hospital admission related to extreme heat in older adults. JAMA 2014, 312, 2659–2667. [Google Scholar] [CrossRef] [PubMed]
- Roncal-Jimenez, C.A.; Ishimoto, T.; Lanaspa, M.A.; Milagres, T.; Hernando, A.A.; Jensen, T.; Miyazaki, M.; Doke, T.; Hayasaki, T.; Nakagawa, T. Aging-associated renal disease in mice is fructokinase dependent. Am. J. Physiol. Ren. Physiol. 2016, 311, F722–F730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachenfeld, N.S.; Mack, G.W.; Takamata, A.; DiPietro, L.; Nadel, E.R. Thirst and fluid regulatory responses to hypertonicity in older adults. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1996, 271, R757–R765. [Google Scholar] [CrossRef]
- Phillips, P.A.; Rolls, B.J.; Ledingham, J.G.; Forsling, M.L.; Morton, J.J.; Crowe, M.J.; Wollner, L. Reduced thirst after water deprivation in healthy elderly men. New Engl. J. Med. 1984, 311, 753–759. [Google Scholar] [CrossRef]
- Garcia-Arroyo, F.E.; Cristóbal, M.; Arellano-Buendía, A.S.; Osorio, H.; Tapia, E.; Soto, V.; Madero, M.; Lanaspa, M.A.; Roncal-Jimenez, C.A.; Bankir, L. Rehydration with Soft Drink-like Beverages Exacerbates Dehydration and Worsens Dehydration-associated Renal Injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 111, R57–R65. [Google Scholar] [CrossRef]
- García-Arroyo, F.E.; Tapia, E.; Blas-Marron, M.G.; Gonzaga, G.; Silverio, O.; Cristóbal, M.; Osorio, H.; Arellano-Buendía, A.S.; Zazueta, C.; Aparicio-Trejo, O.E. Vasopressin Mediates the Renal Damage Induced by Limited Fructose Rehydration in Recurrently Dehydrated Rats. Int. J. Biol. Sci. 2017, 13, 961–975. [Google Scholar] [CrossRef] [Green Version]
- Burg, M.B. Molecular basis of osmotic regulation. Am. J. Physiol. Ren. Physiol. 1995, 268, F983–F996. [Google Scholar] [CrossRef]
- Johnson, R.J.; Rodriguez-Iturbe, B.; Roncal-Jimenez, C.; Lanaspa, M.A.; Ishimoto, T.; Nakagawa, T.; Correa-Rotter, R.; Wesseling, C.; Bankir, L.; Sanchez-Lozada, L.G. Hyperosmolarity drives hypertension and CKD--water and salt revisited. Nat. Rev. Nephrol. 2014, 10, 415–420. [Google Scholar] [CrossRef]
- Cirillo, P.; Gersch, M.S.; Mu, W.; Scherer, P.M.; Kim, K.M.; Gesualdo, L.; Henderson, G.N.; Johnson, R.J.; Sautin, Y.Y. Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. J. Am. Soc. Nephrol. 2009, 20, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Ishimoto, T.; Cicerchi, C.; Tamura, Y.; Roncal-Jimenez, C.A.; Chen, W.; Tanabe, K.; Andres-Hernando, A.; Orlicky, D.J.; Finol, E. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 2526–2538. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Tapia, E.; Santamaria, J.; Avila-Casado, C.; Soto, V.; Nepomuceno, T.; Rodriguez-Iturbe, B.; Johnson, R.J.; Herrera-Acosta, J. Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney Int. 2005, 67, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Hong, Q.; Zhang, X.; Xiao, W.; Wang, L.; Cui, S.; Feng, Z.; Lv, Y.; Cai, G.; Chen, X. Aldose reductase mediates endothelial cell dysfunction induced by high uric acid concentrations. Cell Commun. Signal. 2017, 15, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Roncal-Jimenez, C.A.; Lanaspa-Garcia, M.A.; Oppelt, S.A.; Kuwabara, M.; Jensen, T.; Milagres, T.; Andres-Hernando, A.; Ishimoto, T.; Garcia, G.E. Role of fructose and fructokinase in acute dehydration-induced vasopressin gene expression and secretion in mice. J. Neurophysiol. 2016, 117, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.; Berl, T.; Anderson, R. Osmotic and nonosmotic control of vasopressin release. Am. J. Physiol. Ren. Physiol. 1979, 236, F321–F332. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criteria | ||
|---|---|---|
| Serum Creatinine | Urine Output | |
| Acute Kidney Injury Network (AKIN) Classification | ||
| Stage 1 | Increase ≥0.3 mg/dL (≥26.5 µmol/L) OR increase ≥1.5–2.0-fold from baseline | <0.5 mL/kg/h for 6 h |
| Stage 2 | Increase >2.0–3.0-fold from baseline | <0.5 mL/kg/h for 12 h |
| Stage 3 | Increase >3.0-fold from baseline OR serum creatinine ≥4.0 mg/dL (≥354 µmol/L) with an acute increase of ≥0.5 mg/dL (44 µmol/L) OR need for renal replacement therapy | <0.3 mL/kg/h for 24 h OR anuria for 12 h OR need for renal replacement therapy |
| Kidney Disease: Improving Global Outcomes (KDIGO) Classification | ||
| Stage 1 | Increase ≥0.3 mg/dL (≥26.5 µmol/L) OR 1.5–1.9 times baseline | <0.5 mL/kg/h for 6-12 h |
| Stage 2 | 2.0–2.9 times baseline | <0.5 mL/kg/h for 12 h |
| Stage 3 | 3.0 times baseline OR increase in serum creatinine to ≥4.0 mg/dL (≥354 µmol/L) OR need for renal replacement therapy OR in patients <18 years old a decrease in eGFR to <35 mL/min/1.73 m2 | <0.3 mL/kg/h for 24 h OR anuria for 12 h |
| Acute Dialysis Quality Initiative (ADQI): Risk, Injury, Failure, Loss of Kidney Function and End-Stage Kidney Disease (RIFLE) Classification | ||
| Risk | 1.5-fold increase OR GFR decrease >25% from baseline | <0.5 mL/kg/h for 6-12 h |
| Injury | 2.0-fold increase OR GFR decrease >50% from baseline | <0.5 mL/kg/h for 12 h |
| Failure | 3.0-fold increase OR GFR decrease >75% from baseline OR serum creatinine ≥4.0 mg/dL (≥354 µmol/L) with an acute increase of ≥0.5 mg/dL (44 µmol/L) | <0.3 mL/gk/h for 24 h OR anuria for 12 h |
| Loss of kidney function | Complete loss of kidney function >4 weeks | |
| End-stage kidney disease | Complete loss of kidney function >3 months | |
| • Acute Kidney Injury and the Development of Chronic kidney Disease: |
| • Can exercise in the heat induce intrinsic renal damage? |
| • How do we interpret increases in AKI biomarkers associated with exercise in the heat? |
| • Can repeated exposures to AKI caused by exercise in the heat lead to CKDu? How does the frequency and severity of this AKI relate to the development and severity of CKDu? |
| • Does heat acclimation alleviate the risk of AKI (and CKDu) associated with exercise in the heat? |
| Mechanisms by Which Exercise in the Heat May Increase the Risk Of Acute Kidney Injury: |
| • What is the relative importance of heat strain versus dehydration on the magnitude of the risk of AKI evoked by exercise in the heat? |
| • Do NSAIDs (or other common substances with nephrotoxic side effects) modulate the risk of AKI evoked by exercise in the heat? |
| • To what extent does exercise in the heat invoke a heterogenous distribution of blood flow in the kidneys? What are the contributions of heat strain and/or dehydration? |
| • Do reductions in renal blood flow during exercise in the heat cause localized ischemia, reductions in oxygenation, and/or decreases in ATP availability? Where do these changes occur within the kidneys? |
| • Does exercise in the heat promote inflammation and oxidative stress within the kidneys? What are the contributions of heat strain and/or dehydration? |
| • What is the extent by which activation of the Na+/K+ pump contributes to the risk of AKI during exercise in the heat? |
| • Does the polyol-fructokinase pathway directly contribute to the risk of AKI during exercise in the heat? |
| • What are the roles of vasopressin and hyperuricemia in the risk of AKI during exercise in the heat? |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlader, Z.J.; Hostler, D.; Parker, M.D.; Pryor, R.R.; Lohr, J.W.; Johnson, B.D.; Chapman, C.L. The Potential for Renal Injury Elicited by Physical Work in the Heat. Nutrients 2019, 11, 2087. https://doi.org/10.3390/nu11092087
Schlader ZJ, Hostler D, Parker MD, Pryor RR, Lohr JW, Johnson BD, Chapman CL. The Potential for Renal Injury Elicited by Physical Work in the Heat. Nutrients. 2019; 11(9):2087. https://doi.org/10.3390/nu11092087
Chicago/Turabian StyleSchlader, Zachary J., David Hostler, Mark D. Parker, Riana R. Pryor, James W. Lohr, Blair D. Johnson, and Christopher L. Chapman. 2019. "The Potential for Renal Injury Elicited by Physical Work in the Heat" Nutrients 11, no. 9: 2087. https://doi.org/10.3390/nu11092087
APA StyleSchlader, Z. J., Hostler, D., Parker, M. D., Pryor, R. R., Lohr, J. W., Johnson, B. D., & Chapman, C. L. (2019). The Potential for Renal Injury Elicited by Physical Work in the Heat. Nutrients, 11(9), 2087. https://doi.org/10.3390/nu11092087