Selenium, Selenoproteins and Viral Infection

 and
and
Abstract

1. Introduction
2. Reactive Oxygen Species (ROS) in Immunity and Viral Infection
2.1. ROS and Oxidative Stress
2.2. ROS Function in Immunity and Cell Signaling
2.3. ROS and Viral Infection
3. Selenium, Selenoproteins and ROS
3.1. Selenium Insertion in Selenoproteins
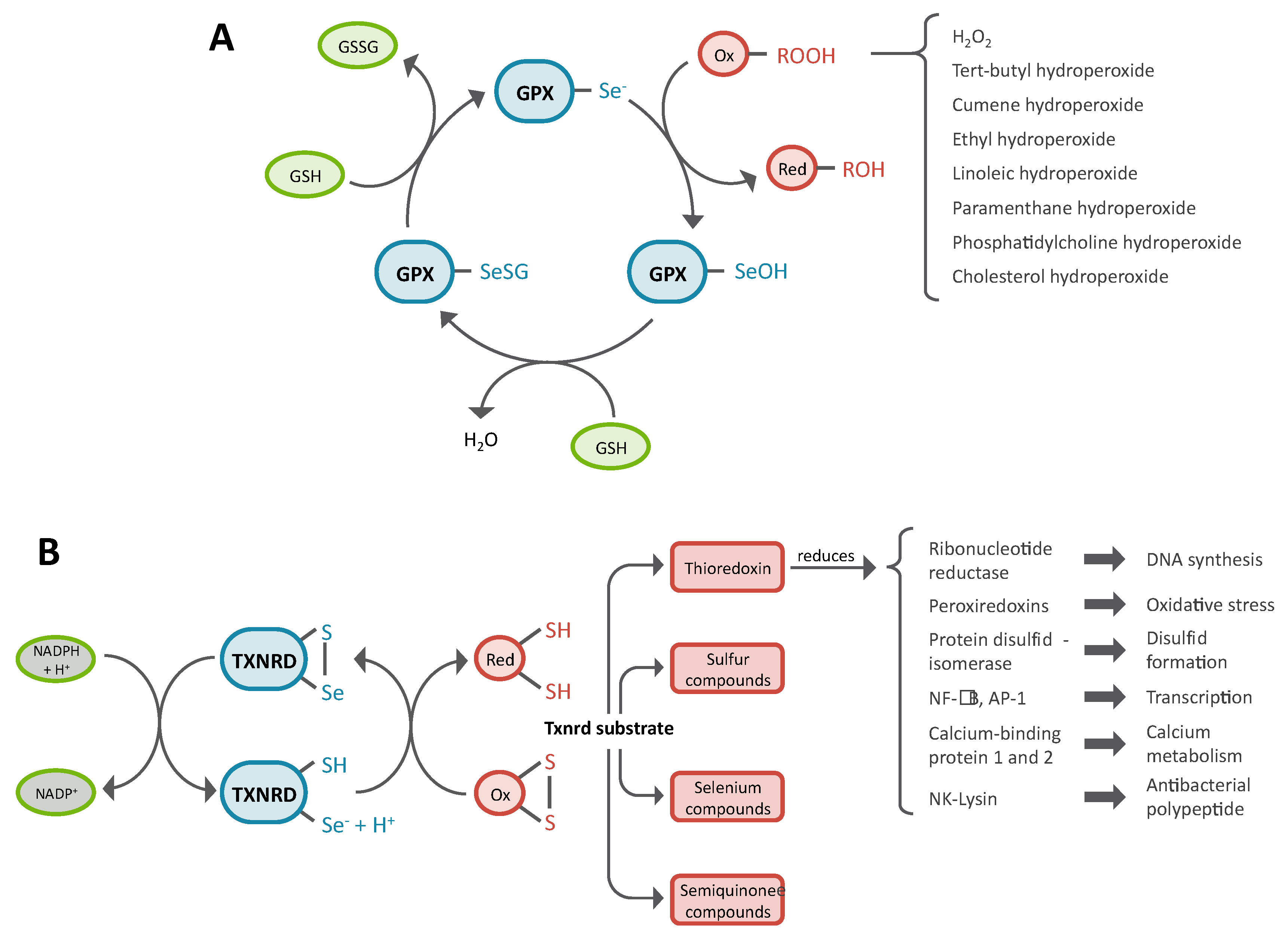
3.2. Role of Glutathione Peroxidases in Antioxidant Defense
3.3. Role of Thioredoxin Reductase in Antioxidant Defense, Redox Homeostasis and Redox Signaling
4. Selenium, Selenoproteins and Viral Replication
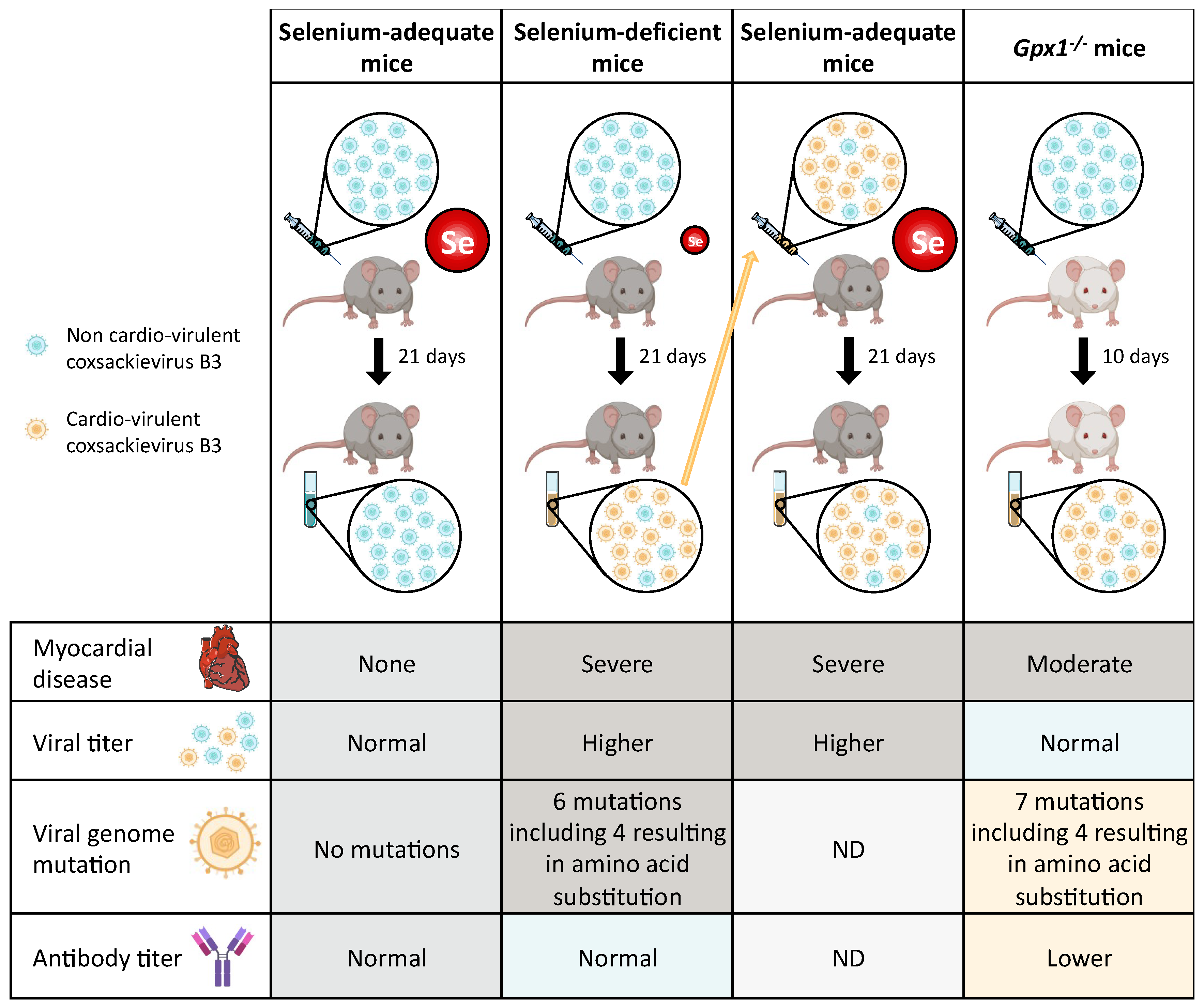
4.1. Coxsackie Virus
4.2. Influenza Virus (Orthomyxoviridae)
4.3. Human Immunodeficiency Virus (HIV)
4.4. Hepatitis C Virus (HCV)
4.5. Other Viruses
5. Selenoproteins in Viral Genomes
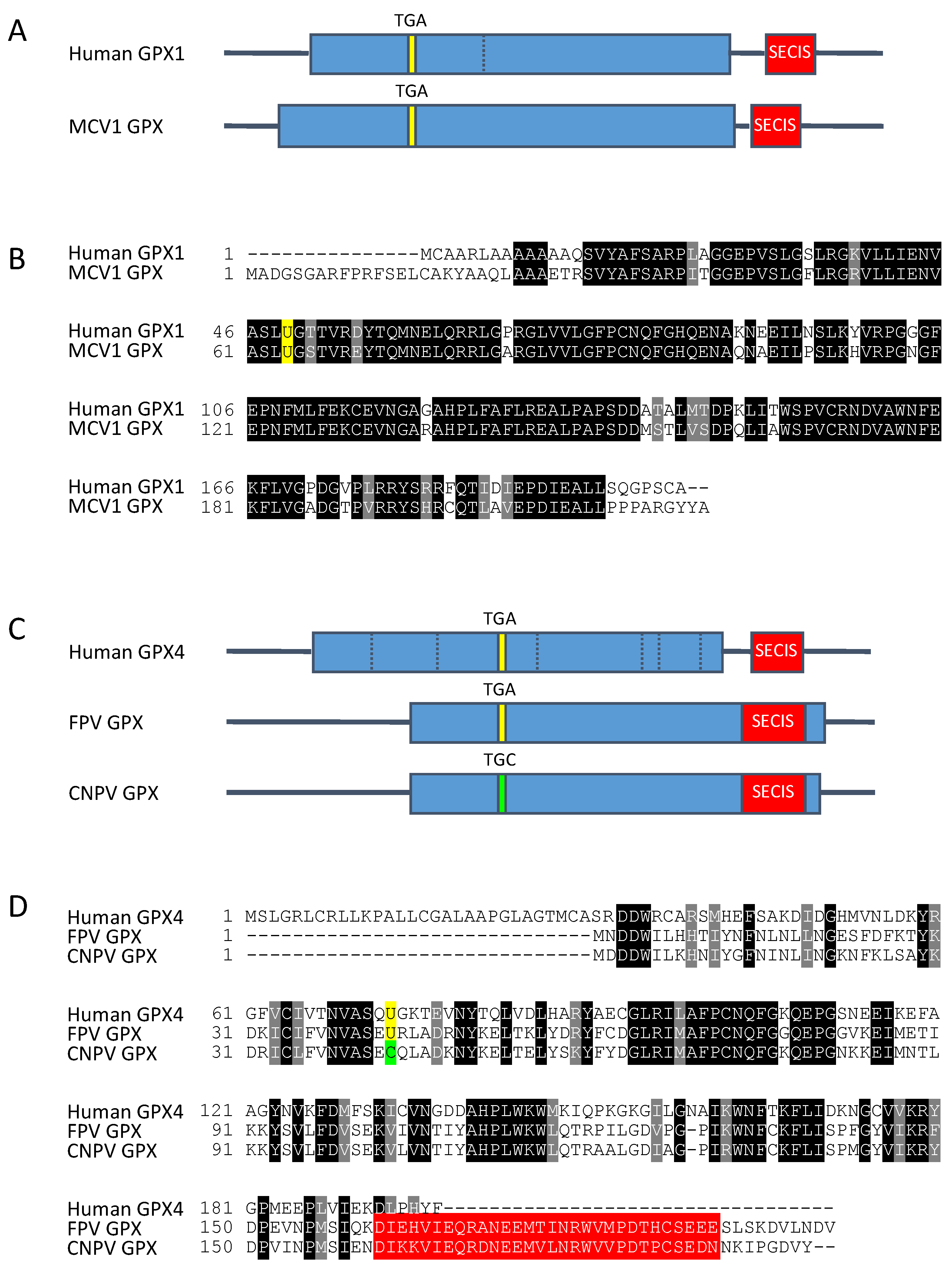
5.1. 1998: First Example of a Viral Selenoprotein Encoded in Molluscum Contagiosum Virus Genome
5.2. 2007: A Second Example of an Encoded Viral Selenoprotein in Fowlpox Virus Genome
5.3. Putative Selenoproteins in Other Viral Genomes
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef]
- Hatfield, D.L.; Tsuji, P.A.; Carlson, B.A.; Gladyshev, V.N. Selenium and selenocysteine: Roles in cancer, health, and development. Trends Biochem. Sci. 2014, 39, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, D.L.; Carlson, B.A.; Xu, X.M.; Mix, H.; Gladyshev, V.N. Selenocysteine incorporation machinery and the role of selenoproteins in development and health. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 97–142. [Google Scholar] [PubMed]
- Meplan, C.; Hesketh, J. Selenium and cancer: A story that should not be forgotten-insights from genomics. Cancer Treat. Res. 2014, 159, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Papp, L.V.; Holmgren, A.; Khanna, K.K. Selenium and selenoproteins in health and disease. Antioxid Redox Signal 2010, 12, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Whanger, P.D. Selenium and its relationship to cancer: An update. Br. J. Nutr. 2004, 91, 11–28. [Google Scholar] [CrossRef]
- Kurokawa, S.; Berry, M.J. Selenium. Role of the essential metalloid in health. Met. Ions Life Sci. 2013, 13, 499–534. [Google Scholar] [CrossRef]
- Vindry, C.; Ohlmann, T.; Chavatte, L. Selenium metabolism, regulation, and sex differences in mammals. In Selenium, Molecular and Integartive Toxicology; Michalke, B., Ed.; Springer International Publishing AG, Part of Springer Nature: Cham, Switzerland, 2018; pp. 89–107. [Google Scholar]
- Touat-Hamici, Z.; Legrain, Y.; Sonet, J.; Bulteau, A.-L.; Chavatte, L. Alteration of selenoprotein expression during stress and in aging. In Selenium: Its Molecular Biology and Role in Human Health, 4th ed.; Hatfield, D.L., Su, D., Tsuji, P.A., Gladyshev, V.N., Eds.; Springer Science+Business Media, LLC: New York, NY, USA, 2016; pp. 539–551. [Google Scholar]
- Sonet, J.; Bulteau, A.-L.; Chavatte, L. Selenium and Selenoproteins in Human Health and Diseases. In Metallomics: Analytical Techniques and Speciation Methods; Michalke, B., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; pp. 364–381. [Google Scholar] [CrossRef]
- Latrèche, L.; Chavatte, L. Selenium incorporation into selenoproteins, implications in human health. Met. Ions Biol. Med. X 2008, 10, 731–737. [Google Scholar]
- Avery, J.C.; Hoffmann, P.R. Selenium, Selenoproteins, and Immunity. Nutrients 2018, 10, 1203. [Google Scholar] [CrossRef]
- Jackson, M.I.; Combs, G.F. Selenium as a Cancer Preventive Agent. In Selenium: Its Molecular Biology and Role in Human Health, 3rd ed.; Hatfield, D.L., Berry, M.J., Gladyshev, V.N., Eds.; Springer: New York, NY, USA, 2012; pp. 313–323. [Google Scholar]
- Vindry, C.; Ohlmann, T.; Chavatte, L. Translation regulation of mammalian selenoproteins. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2480–2492. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Lee, B.J.; Tsuji, P.A.; Tobe, R.; Park, J.M.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L. Selenocysteine tRNA [Ser]Sec: From Nonsense Suppressor tRNA to the Quintessential Constituent in Selenoprotein Biosynthesis. In Selenium: Its Molecular Biology and Role in Human Health, 4th ed.; Hatfield, D.L., Tsuji, P.A., Gladyshev, V.N., Eds.; Springer Science+Business Media, LLC: New York, NY, USA, 2016; pp. 3–12. [Google Scholar]
- Bulteau, A.-L.; Chavatte, L. Update on selenoprotein biosynthesis. Antioxid. Redox Signal. 2015, 23, 775–794. [Google Scholar] [CrossRef] [PubMed]
- Donovan, J.; Copeland, P.R. Threading the needle: Getting selenocysteine into proteins. Antioxid. Redox Signal. 2010, 12, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Berry, M.J. Eukaryotic selenoprotein synthesis: Mechanistic insight incorporating new factors and new functions for old factors. IUBMB Life 2008, 60, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Papp, L.V.; Lu, J.; Holmgren, A.; Khanna, K.K. From selenium to selenoproteins: Synthesis, identity, and their role in human health. Antioxid. Redox Signal. 2007, 9, 775–806. [Google Scholar] [CrossRef] [PubMed]
- Allmang, C.; Krol, A. Selenoprotein synthesis: UGA does not end the story. Biochimie 2006, 88, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D.M.; Copeland, P.R. Mechanism and regulation of selenoprotein synthesis. Annu. Rev. Nutr. 2003, 23, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, D.L.; Gladyshev, V.N. How selenium has altered our understanding of the genetic code. Mol. Cell. Biol. 2002, 22, 3565–3576. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2012, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Dagnell, M.; Schmidt, E.E.; Arner, E.S.J. The A to Z of modulated cell patterning by mammalian thioredoxin reductases. Free Radic. Biol. Med. 2018, 115, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.J. Selective Evaluation of Thioredoxin Reductase Enzymatic Activities. Methods Mol. Biol. 2018, 1661, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S. Focus on mammalian thioredoxin reductases—Important selenoproteins with versatile functions. Biochim. Biophys. Acta 2009, 1790, 495–526. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Fomenko, D.E.; Novoselov, S.V.; Natarajan, S.K.; Lee, B.C.; Koc, A.; Carlson, B.A.; Lee, T.H.; Kim, H.Y.; Hatfield, D.L.; Gladyshev, V.N. MsrB1 (methionine-R-sulfoxide reductase 1) knock-out mice: Roles of MsrB1 in redox regulation and identification of a novel selenoprotein form. J. Biol. Chem. 2009, 284, 5986–5993. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; Pasqua, T.; Boukhzar, L.; Anouar, Y.; Angelone, T. Progress in the emerging role of selenoproteins in cardiovascular disease: Focus on endoplasmic reticulum-resident selenoproteins. Cell Mol. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pitts, M.W.; Hoffmann, P.R. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium 2018, 70, 76–86. [Google Scholar] [CrossRef]
- Addinsall, A.B.; Wright, C.R.; Andrikopoulos, S.; van der Poel, C.; Stupka, N. Emerging roles of endoplasmic reticulum-resident selenoproteins in the regulation of cellular stress responses and the implications for metabolic disease. Biochem. J. 2018, 475, 1037–1057. [Google Scholar] [CrossRef]
- Molteni, C.G.; Principi, N.; Esposito, S. Reactive oxygen and nitrogen species during viral infections. Free Radic Res. 2014, 48, 1163–1169. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10 (Suppl.), S18–S25. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Mishra, S.K.; Pant, H.C. Oxidative stress in neurodegeneration. Adv. Pharmacol. Sci. 2011, 2011, 572634. [Google Scholar] [CrossRef] [PubMed]
- Paravicini, T.M.; Touyz, R.M. Redox signaling in hypertension. Cardiovasc. Res. 2006, 71, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Yankner, B.A. The aging stress response. Mol. Cell 2010, 40, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Leto, T.L. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.K. What really happens in the neutrophil phagosome? Free Radic Biol. Med. 2012, 53, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Segal, A.W. The function of the NADPH oxidase of phagocytes and its relationship to other NOXs in plants, invertebrates, and mammals. Int. J. Biochem. Cell Biol. 2008, 40, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.H.; Devadas, S.; Kwon, J.; Pinto, L.A.; Williams, M.S. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat. Immunol. 2004, 5, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Shatynski, K.E.; Chen, H.; Morand, S.; de Deken, X.; Miot, F.; Leto, T.L.; Williams, M.S. The nonphagocytic NADPH oxidase Duox1 mediates a positive feedback loop during T cell receptor signaling. Sci. Signal. 2010, 3, ra59. [Google Scholar] [CrossRef] [PubMed]
- Thayer, T.C.; Delano, M.; Liu, C.; Chen, J.; Padgett, L.E.; Tse, H.M.; Annamali, M.; Piganelli, J.D.; Moldawer, L.L.; Mathews, C.E. Superoxide production by macrophages and T cells is critical for the induction of autoreactivity and type 1 diabetes. Diabetes 2011, 60, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.M.; Thayer, T.C.; Steele, C.; Cuda, C.M.; Morel, L.; Piganelli, J.D.; Mathews, C.E. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J. Immunol. 2010, 185, 5247–5258. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [PubMed]
- Reshi, M.L.; Su, Y.-C.; Hong, J.-R. RNA Viruses: ROS-Mediated Cell Death. Int. J. Cell Biol. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Peterhans, E. Reactive oxygen species and nitric oxide in viral diseases. Biol. Trace Elem. Res. 1997, 56, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.A.; Levander, O.A.; Handy, J. Selenium deficiency and viral infection. J. Nutr. 2003, 133, 1463S–1467S. [Google Scholar] [CrossRef] [PubMed]
- Baruchel, S.; Wainberg, M.A. The role of oxidative stress in disease progression in individuals infected by the human immunodeficiency virus. J. Leukoc. Biol. 1992, 52, 111–114. [Google Scholar] [CrossRef]
- Casola, A.; Burger, N.; Liu, T.; Jamaluddin, M.; Brasier, A.R.; Garofalo, R.P. Oxidant tone regulates RANTES gene expression in airway epithelial cells infected with respiratory syncytial virus. Role in viral-induced interferon regulatory factor activation. J. Biol. Chem. 2001, 276, 19715–19722. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.; Tian, B.; Boldogh, I.; Garofalo, R.P.; Brasier, A.R. Respiratory syncytial virus infection induces a reactive oxygen species-MSK1-phospho-Ser-276 RelA pathway required for cytokine expression. J. Virol. 2009, 83, 10605–10615. [Google Scholar] [CrossRef] [PubMed]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [PubMed]
- Seet, R.C.; Lee, C.Y.; Lim, E.C.; Quek, A.M.; Yeo, L.L.; Huang, S.H.; Halliwell, B. Oxidative damage in dengue fever. Free Radic. Biol. Med. 2009, 47, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Oberley, L.W.; Murhammer, D.W. Evidence of oxidative stress following the viral infection of two lepidopteran insect cell lines. Free Radic. Biol. Med. 2001, 31, 1448–1455. [Google Scholar] [CrossRef]
- Waris, G.; Siddiqui, A. Hepatitis C virus stimulates the expression of cyclooxygenase-2 via oxidative stress: Role of prostaglandin E2 in RNA replication. J. Virol. 2005, 79, 9725–9734. [Google Scholar] [CrossRef] [PubMed]
- Ciriolo, M.R.; Palamara, A.T.; Incerpi, S.; Lafavia, E.; Bue, M.C.; De Vito, P.; Garaci, E.; Rotilio, G. Loss of GSH, oxidative stress, and decrease of intracellular pH as sequential steps in viral infection. J. Biol. Chem. 1997, 272, 2700–2708. [Google Scholar] [CrossRef] [PubMed]
- Garaci, E.; Palamara, A.T.; Ciriolo, M.R.; D’Agostini, C.; Abdel-Latif, M.S.; Aquaro, S.; Lafavia, E.; Rotilio, G. Intracellular GSH content and HIV replication in human macrophages. J. Leukoc. Biol. 1997, 62, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Palamara, A.T.; Perno, C.F.; Ciriolo, M.R.; Dini, L.; Balestra, E.; D’Agostini, C.; Di Francesco, P.; Favalli, C.; Rotilio, G.; Garaci, E. Evidence for antiviral activity of glutathione: In vitro inhibition of herpes simplex virus type 1 replication. Antivir. Res. 1995, 27, 237–253. [Google Scholar] [CrossRef]
- Flores, S.C.; Marecki, J.C.; Harper, K.P.; Bose, S.K.; Nelson, S.K.; McCord, J.M. Tat protein of human immunodeficiency virus type 1 represses expression of manganese superoxide dismutase in HeLa cells. Proc. Natl. Acad. Sci. USA 1993, 90, 7632–7636. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.; Roederer, M.; Herzenberg, L.A.; Herzenberg, L.A. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1990, 87, 9943–9947. [Google Scholar] [CrossRef] [PubMed]
- Li, C. Selenium deficiency and endemic heart failure in China: A case study of biogeochemistry for human health. Ambio 2007, 36, 90–93. [Google Scholar] [CrossRef]
- Touat-Hamici, Z.; Bulteau, A.L.; Bianga, J.; Jean-Jacques, H.; Szpunar, J.; Lobinski, R.; Chavatte, L. Selenium-regulated hierarchy of human selenoproteome in cancerous and immortalized cells lines. Biochim. Biophys. Acta Gen. Subj. 2018. [Google Scholar] [CrossRef] [PubMed]
- Touat-Hamici, Z.; Legrain, Y.; Bulteau, A.-L.; Chavatte, L. Selective up-regulation of human selenoproteins in response to oxidative stress. J. Biol. Chem. 2014, 289, 14750–14761. [Google Scholar] [CrossRef] [PubMed]
- Legrain, Y.; Touat-Hamici, Z.; Chavatte, L. Interplay between selenium levels, selenoprotein expression, and replicative senescence in WI-38 human fibroblasts. J. Biol. Chem. 2014, 289, 6299–6310. [Google Scholar] [CrossRef] [PubMed]
- Latreche, L.; Duhieu, S.; Touat-Hamici, Z.; Jean-Jean, O.; Chavatte, L. The differential expression of glutathione peroxidase 1 and 4 depends on the nature of the SECIS element. RNA Biol. 2012, 9, 681–690. [Google Scholar] [CrossRef]
- Kuhbacher, M.; Bartel, J.; Hoppe, B.; Alber, D.; Bukalis, G.; Brauer, A.U.; Behne, D.; Kyriakopoulos, A. The brain selenoproteome: Priorities in the hierarchy and different levels of selenium homeostasis in the brain of selenium-deficient rats. J. Neurochem. 2009, 110, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Latreche, L.; Jean-Jean, O.; Driscoll, D.M.; Chavatte, L. Novel structural determinants in human SECIS elements modulate the translational recoding of UGA as selenocysteine. Nucleic Acids Res. 2009, 37, 5868–5880. [Google Scholar] [CrossRef]
- Lin, H.C.; Ho, S.C.; Chen, Y.Y.; Khoo, K.H.; Hsu, P.H.; Yen, H.C. SELENOPROTEINS. CRL2 aids elimination of truncated selenoproteins produced by failed UGA/Sec decoding. Science 2015, 349, 91–95. [Google Scholar] [CrossRef]
- Howard, M.T.; Carlson, B.A.; Anderson, C.B.; Hatfield, D.L. Translational redefinition of UGA codons is regulated by selenium availability. J. Biol. Chem. 2013, 288, 19401–19413. [Google Scholar] [CrossRef]
- Dalley, B.K.; Baird, L.; Howard, M.T. Studying Selenoprotein mRNA Translation Using RNA-Seq and Ribosome Profiling. Methods Mol. Biol. 2018, 1661, 103–123. [Google Scholar] [CrossRef]
- Zhao, W.; Bohleber, S.; Schmidt, H.; Seeher, S.; Howard, M.T.; Braun, D.; Arndt, S.; Reuter, U.; Wende, H.; Birchmeier, C.; et al. Ribosome profiling of selenoproteins in vivo reveals consequences of pathogenic Secisbp2 missense mutations. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Rebsch, C.M.; Kinzy, S.A.; Fletcher, J.E.; Copeland, P.R. Efficiency of mammalian selenocysteine incorporation. J. Biol. Chem. 2004, 279, 37852–37859. [Google Scholar] [CrossRef] [PubMed]
- Low, S.C.; Grundner-Culemann, E.; Harney, J.W.; Berry, M.J. SECIS-SBP2 interactions dictate selenocysteine incorporation efficiency and selenoprotein hierarchy. EMBO J. 2000, 19, 6882–6890. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, M.; Ridge, P.G.; Zhang, Y.; Lobanov, A.V.; Pringle, T.H.; Guigo, R.; Hatfield, D.L.; Gladyshev, V.N. Composition and evolution of the vertebrate and mammalian selenoproteomes. PLoS ONE 2012, 7, e33066. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, A.V.; Hatfield, D.L.; Gladyshev, V.N. Eukaryotic selenoproteins and selenoproteomes. Biochim. Biophys. Acta 2009, 1790, 1424–1428. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigo, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Takebe, G.; Yarimizu, J.; Saito, Y.; Hayashi, T.; Nakamura, H.; Yodoi, J.; Nagasawa, S.; Takahashi, K. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J. Biol. Chem. 2002, 277, 41254–41258. [Google Scholar] [CrossRef]
- Toppo, S.; Flohe, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim. Biophys. Acta 2009, 1790, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Cheng, W.H.; Ho, Y.S.; Ross, D.A.; Valentine, B.A.; Combs, G.F.; Lei, X.G. Cellular glutathione peroxidase knockout mice express normal levels of selenium-dependent plasma and phospholipid hydroperoxide glutathione peroxidases in various tissues. J. Nutr. 1997, 127, 1445–1450. [Google Scholar] [CrossRef]
- Esworthy, R.S.; Aranda, R.; Martin, M.G.; Doroshow, J.H.; Binder, S.W.; Chu, F.F. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G848–G855. [Google Scholar] [CrossRef]
- Ho, Y.S.; Magnenat, J.L.; Bronson, R.T.; Cao, J.; Gargano, M.; Sugawara, M.; Funk, C.D. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem. 1997, 272, 16644–16651. [Google Scholar] [CrossRef] [PubMed]
- De Haan, J.B.; Bladier, C.; Griffiths, P.; Kelner, M.; O’Shea, R.D.; Cheung, N.S.; Bronson, R.T.; Silvestro, M.J.; Wild, S.; Zheng, S.S.; et al. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J. Biol. Chem. 1998, 273, 22528–22536. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; Ho, Y.S.; Valentine, B.A.; Ross, D.A.; Combs, G.F., Jr.; Lei, X.G. Cellular glutathione peroxidase is the mediator of body selenium to protect against paraquat lethality in transgenic mice. J. Nutr. 1998, 128, 1070–1076. [Google Scholar] [CrossRef]
- Muller, M.F.; Florian, S.; Pommer, S.; Osterhoff, M.; Esworthy, R.S.; Chu, F.F.; Brigelius-Flohe, R.; Kipp, A.P. Deletion of glutathione peroxidase-2 inhibits azoxymethane-induced colon cancer development. PLoS ONE 2013, 8, e72055. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2013. [Google Scholar] [CrossRef]
- Beck, M.A.; Shi, Q.; Morris, V.C.; Levander, O.A. Benign coxsackievirus damages heart muscle in iron-loaded vitamin E-deficient mice. Free Radic. Biol. Med. 2005, 38, 112–116. [Google Scholar] [CrossRef]
- Beck, M.A.; Williams-Toone, D.; Levander, O.A. Coxsackievirus B3-resistant mice become susceptible in Se/vitamin E deficiency. Free Radic. Biol. Med. 2003, 34, 1263–1270. [Google Scholar] [CrossRef]
- Beck, M.A.; Nelson, H.K.; Shi, Q.; Van Dael, P.; Schiffrin, E.J.; Blum, S.; Barclay, D.; Levander, O.A. Selenium deficiency increases the pathology of an influenza virus infection. FASEB J. 2001, 15, 1481–1483. [Google Scholar] [CrossRef]
- Beck, M.A.; Matthews, C.C. Micronutrients and host resistance to viral infection. Proc. Nutr. Soc. 2000, 59, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.A. Nutritionally induced oxidative stress: Effect on viral disease. Am. J. Clin. Nutr. 2000, 71, 1676S–1681S. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.A.; Kolbeck, P.C.; Rohr, L.H.; Shi, Q.; Morris, V.C.; Levander, O.A. Benign human enterovirus becomes virulent in selenium-deficient mice. J. Med. Virol. 1994, 43, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.A.; Shi, Q.; Morris, V.C.; Levander, O.A. Rapid genomic evolution of a non-virulent coxsackievirus B3 in selenium-deficient mice results in selection of identical virulent isolates. Nat. Med. 1995, 1, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Jubelt, B.; Lipton, H.L. Enterovirus/picornavirus infections. Handb. Clin. Neurol. 2014, 123, 379–416. [Google Scholar] [CrossRef] [PubMed]
- Muehlenbachs, A.; Bhatnagar, J.; Zaki, S.R. Tissue tropism, pathology and pathogenesis of enterovirus infection. J. Pathol. 2015, 235, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.Q.; Chen, J.S.; Wen, Z.M.; Ge, K.Y.; Zhu, L.Z.; Chen, X.C.; Chen, X.S. The role of selenium in Keshan disease. Adv. Nutr. Res. 1984, 6, 203–231. [Google Scholar] [PubMed]
- Loscalzo, J. Keshan disease, selenium deficiency, and the selenoproteome. N. Engl. J. Med. 2014, 370, 1756–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, Y.; Chen, H. Detection of enteroviral RNA in paraffin-embedded myocardial tissue from patients with Keshan by nested PCR. Zhonghua Yi Xue Za Zhi 1995, 75, 344–345. [Google Scholar] [PubMed]
- Li, Y.; Peng, T.; Yang, Y.; Niu, C.; Archard, L.C.; Zhang, H. High prevalence of enteroviral genomic sequences in myocardium from cases of endemic cardiomyopathy (Keshan disease) in China. Heart 2000, 83, 696–701. [Google Scholar] [CrossRef]
- Liu, Y.; Chiba, M.; Inaba, Y.; Kondo, M. Keshan disease—A review from the aspect of history and etiology. Nihon Eiseigaku Zasshi 2002, 56, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.A. Selenium and host defence towards viruses. Proc. Nutr. Soc. 1999, 58, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.A.; Esworthy, R.S.; Ho, Y.; Chu, F. Glutathione peroxidase protects mice from viral-induced myocarditis. FASEB J. 1998, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.A. Rapid genomic evolution of a non-virulent coxsackievirus B3 in selenium-deficient mice. Biomed. Environ. Sci. 1997, 10, 307–315. [Google Scholar] [PubMed]
- Beck, M.A. Increased virulence of coxsackievirus B3 in mice due to vitamin E or selenium deficiency. J. Nutr. 1997, 127, 966S–970S. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.A.; Kolbeck, P.C.; Shi, Q.; Rohr, L.H.; Morris, V.C.; Levander, O.A. Increased virulence of a human enterovirus (coxsackievirus B3) in selenium-deficient mice. J. Infect. Dis. 1994, 170, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.T.; Rader, V.; Ralston, N.V. The roles of selenium and mercury in the pathogenesis of viral cardiomyopathy. Congest Heart Fail 2007, 13, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Pleschka, S. Overview of influenza viruses. Curr. Top. Microbiol. Immunol. 2013, 370, 1–20. [Google Scholar] [CrossRef]
- Lim, J.Y.; Oh, E.; Kim, Y.; Jung, W.W.; Kim, H.S.; Lee, J.; Sul, D. Enhanced oxidative damage to DNA, lipids, and proteins and levels of some antioxidant enzymes, cytokines, and heat shock proteins in patients infected with influenza H1N1 virus. Acta Virol. 2014, 58, 253–260. [Google Scholar] [CrossRef]
- Ng, M.P.; Lee, J.C.; Loke, W.M.; Yeo, L.L.; Quek, A.M.; Lim, E.C.; Halliwell, B.; Seet, R.C. Does influenza A infection increase oxidative damage? Antioxid. Redox Signal. 2014, 21, 1025–1031. [Google Scholar] [CrossRef]
- Erkekoglu, P.; Asci, A.; Ceyhan, M.; Kizilgun, M.; Schweizer, U.; Atas, C.; Kara, A.; Kocer Giray, B. Selenium levels, selenoenzyme activities and oxidant/antioxidant parameters in H1N1-infected children. Turk. J. Pediatr. 2013, 55, 271–282. [Google Scholar] [PubMed]
- Buffinton, G.D.; Christen, S.; Peterhans, E.; Stocker, R. Oxidative stress in lungs of mice infected with influenza A virus. Free Radic. Res. Commun. 1992, 16, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Hennet, T.; Peterhans, E.; Stocker, R. Alterations in antioxidant defences in lung and liver of mice infected with influenza A virus. J. Gen. Virol. 1992, 73 Pt 1, 39–46. [Google Scholar] [CrossRef]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell Microbiol. 2015, 17, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Lowther, S.; Stambas, J. Inhibition of reactive oxygen species production ameliorates inflammation induced by influenza A viruses via upregulation of SOCS1 and SOCS3. J. Virol. 2015, 89, 2672–2683. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.K.; Shi, Q.; Van Dael, P.; Schiffrin, E.J.; Blum, S.; Barclay, D.; Levander, O.A.; Beck, M.A. Host nutritional selenium status as a driving force for influenza virus mutations. FASEB J. 2001, 15, 1846–1848. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, I.; Zhang, W.; Brighton, L.E.; Carson, J.L.; Styblo, M.; Beck, M.A. Selenium deficiency alters epithelial cell morphology and responses to influenza. Free Radic. Biol. Med. 2007, 42, 1826–1837. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, P.A.; Zhong, N.; Carlson, B.A.; Perella, C.M.; Hatfield, D.L.; Beck, M.A. Decreased selenoprotein expression alters the immune response during influenza virus infection in mice. J. Nutr. 2007, 137, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Stýblo, M.; Walton, F.S.; Harmon, A.W.; Sheridan, P.A.; Beck, M.A. Activation of superoxide dismutase in selenium-deficient mice infected with influenza virus. J. Trace Elem. Med. Biol. 2007, 21, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Beck, M.A. Selenium deficiency induced an altered immune response and increased survival following influenza A/Puerto Rico/8/34 infection. Exp. Biol. Med. 2007, 232, 412–419. [Google Scholar]
- Ferguson, M.R.; Rojo, D.R.; von Lindern, J.J.; O’Brien, W.A. HIV-1 replication cycle. Clin. Lab. Med. 2002, 22, 611–635. [Google Scholar] [CrossRef]
- Nyamweya, S.; Hegedus, A.; Jaye, A.; Rowland-Jones, S.; Flanagan, K.L.; Macallan, D.C. Comparing HIV-1 and HIV-2 infection: Lessons for viral immunopathogenesis. Rev. Med. Virol. 2013, 23, 221–240. [Google Scholar] [CrossRef] [PubMed]
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Samji, H.; Cescon, A.; Hogg, R.S.; Modur, S.P.; Althoff, K.N.; Buchacz, K.; Burchell, A.N.; Cohen, M.; Gebo, K.A.; Gill, M.J.; et al. Closing the gap: Increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS ONE 2013, 8, e81355. [Google Scholar] [CrossRef] [PubMed]
- Pace, G.W.; Leaf, C.D. The role of oxidative stress in HIV disease. Free Radic. Biol. Med. 1995, 19, 523–528. [Google Scholar] [CrossRef]
- Hoffmann, F.W.; Hashimoto, A.C.; Shafer, L.A.; Dow, S.; Berry, M.J.; Hoffmann, P.R. Dietary selenium modulates activation and differentiation of CD4+ T cells in mice through a mechanism involving cellular free thiols. J. Nutr. 2010, 140, 1155–1161. [Google Scholar] [CrossRef]
- Huang, Z.; Rose, A.H.; Hoffmann, P.R. The role of selenium in inflammation and immunity: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2012, 16, 705–743. [Google Scholar] [CrossRef]
- Bogden, J.D.; Oleske, J.M. The essential trace minerals, immunity, and progression of HIV-1 infection. Nutr. Res. 2007, 27, 69–77. [Google Scholar] [CrossRef]
- Stone, C.A.; Kawai, K.; Kupka, R.; Fawzi, W.W. Role of selenium in HIV infection. Nutr. Rev. 2010, 68, 671–681. [Google Scholar] [CrossRef]
- Chandrasekhar, A.; Gupta, A. Nutrition and disease progression pre-highly active antiretroviral therapy (HAART) and post-HAART: Can good nutrition delay time to HAART and affect response to HAART? Am. J. Clin. Nutr. 2011, 94, 1703S–1715S. [Google Scholar] [CrossRef]
- Pitney, C.L.; Royal, M.; Klebert, M. Selenium supplementation in HIV-infected patients: Is there any potential clinical benefit? J. Assoc. Nurses AIDS Care 2009, 20, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.E.; Klaus, J.R.; Llabre, M.M.; Gonzalez, A.; Lawrence, P.J.; Maher, K.J.; Greeson, J.M.; Baum, M.K.; Shor-Posner, G.; Skyler, J.S.; et al. Suppression of human immunodeficiency virus type 1 viral load with selenium supplementation: A randomized controlled trial. Arch. Intern. Med. 2007, 167, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Baum, M.K.; Campa, A.; Lai, S.; Sales Martinez, S.; Tsalaile, L.; Burns, P.; Farahani, M.; Li, Y.; van Widenfelt, E.; Page, J.B.; et al. Effect of micronutrient supplementation on disease progression in asymptomatic, antiretroviral-naive, HIV-infected adults in Botswana: A randomized clinical trial. JAMA 2013, 310, 2154–2163. [Google Scholar] [CrossRef] [PubMed]
- Kamwesiga, J.; Mutabazi, V.; Kayumba, J.; Tayari, J.C.; Uwimbabazi, J.C.; Batanage, G.; Uwera, G.; Baziruwiha, M.; Ntizimira, C.; Murebwayire, A.; et al. Effect of selenium supplementation on CD4+ T-cell recovery, viral suppression and morbidity of HIV-infected patients in Rwanda: A randomized controlled trial. AIDS 2015, 29, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.A.; Sitole, L.J.; Meyer, D. HIV/HAART-associated oxidative stress is detectable by metabonomics. Mol. Biosyst. 2017, 13, 2202–2217. [Google Scholar] [CrossRef] [PubMed]
- De Menezes Barbosa, E.G.; Junior, F.B.; Machado, A.A.; Navarro, A.M. A longer time of exposure to antiretroviral therapy improves selenium levels. Clin. Nutr. 2015, 34, 248–251. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Stadtman, T.C.; Hatfield, D.L.; Jeang, K.T. Levels of major selenoproteins in T cells decrease during HIV infection and low molecular mass selenium compounds increase. Proc. Natl. Acad. Sci. USA 1999, 96, 835–839. [Google Scholar] [CrossRef]
- Davis, G.L. Treatment of chronic hepatitis C. BMJ 2001, 323, 1141–1142. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and oxidative stress in the liver. Viruses 2013, 5, 439–469. [Google Scholar] [CrossRef]
- Boya, P.; de la Pena, A.; Beloqui, O.; Larrea, E.; Conchillo, M.; Castelruiz, Y.; Civeira, M.P.; Prieto, J. Antioxidant status and glutathione metabolism in peripheral blood mononuclear cells from patients with chronic hepatitis C. J. Hepatol. 1999, 31, 808–814. [Google Scholar] [CrossRef]
- De Maria, N.; Colantoni, A.; Fagiuoli, S.; Liu, G.J.; Rogers, B.K.; Farinati, F.; Van Thiel, D.H.; Floyd, R.A. Association between reactive oxygen species and disease activity in chronic hepatitis C. Free Radic. Biol. Med. 1996, 21, 291–295. [Google Scholar] [CrossRef]
- Bianchi, G.; Marchesini, G.; Brizi, M.; Rossi, B.; Forlani, G.; Boni, P.; Melchionda, N.; Thomaseth, K.; Pacini, G. Nutritional effects of oral zinc supplementation in cirrhosis. Nutr. Res. 2000, 20, 1079–1089. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS ONE 2011, 6, e24957. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Polyak, S.J.; Bano, N.; Qiu, W.C.; Carithers, R.L.; Shuhart, M.; Gretch, D.R.; Das, A. Hepatitis C virus induces oxidative stress, DNA damage and modulates the DNA repair enzyme NEIL1. J. Gastroenterol. Hepatol. 2010, 25, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Bureau, C.; Bernad, J.; Chaouche, N.; Orfila, C.; Beraud, M.; Gonindard, C.; Alric, L.; Vinel, J.P.; Pipy, B. Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J. Biol. Chem. 2001, 276, 23077–23083. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mediavilla, M.V.; Sanchez-Campos, S.; Gonzalez-Perez, P.; Gomez-Gonzalo, M.; Majano, P.L.; Lopez-Cabrera, M.; Clemente, G.; Garcia-Monzon, C.; Gonzalez-Gallego, J. Differential contribution of hepatitis C virus NS5A and core proteins to the induction of oxidative and nitrosative stress in human hepatocyte-derived cells. J. Hepatol. 2005, 43, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Ko, W.S.; Guo, C.H.; Yeh, M.S.; Lin, L.Y.; Hsu, G.S.; Chen, P.C.; Luo, M.C.; Lin, C.Y. Blood micronutrient, oxidative stress, and viral load in patients with chronic hepatitis C. World J. Gastroenterol. 2005, 11, 4697–4702. [Google Scholar] [CrossRef] [PubMed]
- Look, M.P.; Rockstroh, J.K.; Rao, G.S.; Kreuzer, K.A.; Barton, S.; Lemoch, H.; Sudhop, T.; Hoch, J.; Stockinger, K.; Spengler, U.; et al. Serum selenium, plasma glutathione (GSH) and erythrocyte glutathione peroxidase (GSH-Px)-levels in asymptomatic versus symptomatic human immunodeficiency virus-1 (HIV-1)-infection. Eur. J. Clin. Nutr. 1997, 51, 266–272. [Google Scholar] [CrossRef]
- Chusri, P.; Kumthip, K.; Hong, J.; Zhu, C.; Duan, X.; Jilg, N.; Fusco, D.N.; Brisac, C.; Schaefer, E.A.; Cai, D.; et al. HCV induces transforming growth factor beta1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci. Rep. 2016, 6, 22487. [Google Scholar] [CrossRef]
- Guerriero, E.; Accardo, M.; Capone, F.; Colonna, G.; Castello, G.; Costantini, S. Assessment of the Selenoprotein M (SELM) over-expression on human hepatocellular carcinoma tissues by immunohistochemistry. Eur. J. Histochem. 2014, 58, 2433. [Google Scholar] [CrossRef]
- Tsai, K.N.; Kuo, C.F.; Ou, J.J. Mechanisms of Hepatitis B Virus Persistence. Trends Microbiol. 2018, 26, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Karayiannis, P. Hepatitis B virus: Virology, molecular biology, life cycle and intrahepatic spread. Hepatol. Int. 2017, 11, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Balamtekin, N.; Kurekci, A.E.; Atay, A.; Kalman, S.; Okutan, V.; Gokcay, E.; Aydin, A.; Sener, K.; Safali, M.; Ozcan, O. Plasma levels of trace elements have an implication on interferon treatment of children with chronic hepatitis B infection. Biol. Trace Elem. Res. 2010, 135, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Abediankenari, S.; Ghasemi, M.; Nasehi, M.M.; Abedi, S.; Hosseini, V. Determination of trace elements in patients with chronic hepatitis B. Acta Med. Iran. 2011, 49, 667–669. [Google Scholar] [PubMed]
- Khan, M.S.; Dilawar, S.; Ali, I.; Rauf, N. The possible role of selenium concentration in hepatitis B and C patients. Saudi J. Gastroenterol. Off. J. Saudi Gastroenterol. Assoc. 2012, 18, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zhi, X.; Sun, G.; Guo, W.; Huang, Y.; Sun, W.; Tian, X.; Zhao, F.; Hu, K. Sodium selenite suppresses hepatitis B virus transcription and replication in human hepatoma cell lines. J. Med. Virol. 2016, 88, 653–663. [Google Scholar] [CrossRef]
- Yu, S.Y.; Zhu, Y.J.; Li, W.G. Protective role of selenium against hepatitis B virus and primary liver cancer in Qidong. Biol. Trace Elem. Res. 1997, 56, 117–124. [Google Scholar] [CrossRef]
- Finsterbusch, T.; Mankertz, A. Porcine circoviruses—Small but powerful. Virus Res. 2009, 143, 177–183. [Google Scholar] [CrossRef]
- Pan, Q.; Huang, K.; He, K.; Lu, F. Effect of different selenium sources and levels on porcine circovirus type 2 replication in vitro. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2008, 22, 143–148. [Google Scholar] [CrossRef]
- Chen, X.; Ren, F.; Hesketh, J.; Shi, X.; Li, J.; Gan, F.; Huang, K. Selenium blocks porcine circovirus type 2 replication promotion induced by oxidative stress by improving GPx1 expression. Free Radic. Biol. Med. 2012, 53, 395–405. [Google Scholar] [CrossRef]
- Gan, F.; Hu, Z.; Huang, Y.; Xue, H.; Huang, D.; Qian, G.; Hu, J.; Chen, X.; Wang, T.; Huang, K. Overexpression of pig selenoprotein S blocks OTA-induced promotion of PCV2 replication by inhibiting oxidative stress and p38 phosphorylation in PK15 cells. Oncotarget 2016, 7, 20469–20485. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xu, J.; Qian, G.; Hamid, M.; Gan, F.; Chen, X.; Huang, K. Selenizing astragalus polysaccharide attenuates PCV2 replication promotion caused by oxidative stress through autophagy inhibition via PI3K/AKT activation. Int. J. Biol. Macromol. 2018, 108, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Qian, G.; Liu, D.; Hu, J.; Gan, F.; Hou, L.; Zhai, N.; Chen, X.; Huang, K. SeMet attenuates OTA-induced PCV2 replication promotion by inhibiting autophagy by activating the AKT/mTOR signaling pathway. Vet. Res. 2018, 49, 15. [Google Scholar] [CrossRef]
- Sumba, P.O.; Kabiru, E.W.; Namuyenga, E.; Fiore, N.; Otieno, R.O.; Moormann, A.M.; Orago, A.S.; Rosenbaum, P.F.; Rochford, R. Microgeographic variations in Burkitt’s lymphoma incidence correlate with differences in malnutrition, malaria and Epstein-Barr virus. Br. J. Cancer 2010, 103, 1736–1741. [Google Scholar] [CrossRef]
- Jian, S.-W.; Mei, C.-E.; Liang, Y.-N.; Li, D.; Chen, Q.-L.; Luo, H.-L.; Li, Y.-Q.; Cai, T.-Y. Influence of selenium-rich rice on transformation of umbilical blood B lymphocytes by Epstein-Barr virus and Epstein-Barr virus early antigen expression. Ai Zheng = Aizheng = Chin. J. Cancer 2003, 22, 26–29. [Google Scholar]
- Taylor, E.W.; Nadimpalli, R.G.; Ramanathan, C.S. Genomic structures of viral agents in relation to the biosynthesis of selenoproteins. Biol. Elem. Res. 1997, 56, 63–91. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Kharaeva, Z.; Raskovic, D.; Pastore, P.; Luci, A.; Korkina, L. Coenzyme Q(10), vitamin E, selenium, and methionine in the treatment of chronic recurrent viral mucocutaneous infections. Nutrition (Burbank) 2012, 28, 509–514. [Google Scholar] [CrossRef]
- Sartori, G.; Jardim, N.S.; Marcondes Sari, M.H.; Dobrachinski, F.; Pesarico, A.P.; Rodrigues, L.C.; Cargnelutti, J.; Flores, E.F.; Prigol, M.; Nogueira, C.W. Antiviral Action of Diphenyl Diselenide on Herpes Simplex Virus 2 Infection in Female BALB/c Mice. J. Cell. Biochem. 2016, 117, 1638–1648. [Google Scholar] [CrossRef]
- Reffett, J.K.; Spears, J.W.; Brown, T.T. Effect of dietary selenium on the primary and secondary immune response in calves challenged with infectious bovine rhinotracheitis virus. J. Nutr. 1988, 118, 229–235. [Google Scholar] [CrossRef]
- Shisler, J.L.; Senkevich, T.G.; Berry, M.J.; Moss, B. Ultraviolet-induced cell death blocked by a selenoprotein from a human dermatotropic poxvirus. Science 1998, 279, 102–105. [Google Scholar] [CrossRef]
- Mix, H.; Lobanov, A.V.; Gladyshev, V.N. SECIS elements in the coding regions of selenoprotein transcripts are functional in higher eukaryotes. Nucleic Acids Res. 2007, 35, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Polansky, H.; Itzkovitz, E.; Javaherian, A. Human papillomavirus (HPV): Systemic treatment with Gene-Eden-VIR/Novirin safely and effectively clears virus. Drug Des. Dev. Ther. 2017, 11, 575–583. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, G.; Yang, G.; Guan, G.; Zhang, Y.; Ren, W.; Yin, J.; Aguilar, Y.M.; Luo, W.; Fang, J.; Yu, X.; et al. Effect of Dietary Selenium Yeast Supplementation on Porcine Circovirus Type 2 (PCV2) Infections in Mice. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Gómez, R.M.; Berría, M.I.; Levander, O.A. Host selenium status selectively influences susceptibility to experimental viral myocarditis. Biol. Trace Elem. Res. 2001, 80, 23–31. [Google Scholar] [CrossRef]
- Sepúlveda, R.T.; Zhang, J.; Watson, R.R. Selenium supplementation decreases coxsackievirus heart disease during murine AIDS. Cardiovasc. Toxicol. 2002, 2, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Molin, Y.; Frisk, P.; Ilbäck, N.-G. Viral RNA kinetics is associated with changes in trace elements in target organs of Coxsackie virus B3 infection. Microbes Infect. 2009, 11, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Jun, E.J.; Ye, J.S.; Hwang, I.S.; Kim, Y.K.; Lee, H. Selenium deficiency contributes to the chronic myocarditis in coxsackievirus-infected mice. Acta Virol. 2011, 55, 23–29. [Google Scholar] [CrossRef]
- Zhang, W.; Ramanathan, C.S.; Nadimpalli, R.G.; Bhat, A.A.; Cox, A.G.; Taylor, E.W. Selenium-dependent glutathione peroxidase modules encoded by RNA viruses. Biol. Trace Elem. Res. 1999, 70, 97–116. [Google Scholar] [CrossRef]
- Cermelli, C.; Vinceti, M.; Scaltriti, E.; Bazzani, E.; Beretti, F.; Vivoli, G.; Portolani, M. Selenite inhibition of Coxsackie virus B5 replication: Implications on the etiology of Keshan disease. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2002, 16, 41–46. [Google Scholar] [CrossRef]
- Broome, C.S.; McArdle, F.; Kyle, J.A.M.; Andrews, F.; Lowe, N.M.; Hart, C.A.; Arthur, J.R.; Jackson, M.J. An increase in selenium intake improves immune function and poliovirus handling in adults with marginal selenium status. Am. J. Clin. Nutr. 2004, 80, 154–162. [Google Scholar] [CrossRef]
- Abou-Zeina, H.A.A.; Nasr, S.M.; Nassar, S.A.; Farag, T.K.; El-Bayoumy, M.K.; Ata, E.B.; Hassan, N.M.F.; Abdel-Aziem, S.H. Beneficial effects of antioxidants in improving health conditions of sheep infected with foot-and-mouth disease. Trop. Anim. Health Prod. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, H.B.; Benetucci, J.; Muzzio, E.; Redini, L.; Naveira, J.; Segura, M.; Weissenbacher, M.; Tang, A.M. High rates of serum selenium deficiency among HIV- and HCV-infected and uninfected drug users in Buenos Aires, Argentina. Public Health Nutr. 2012, 15, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Groenbaek, K.; Friis, H.; Hansen, M.; Ring-Larsen, H.; Krarup, H.B. The effect of antioxidant supplementation on hepatitis C viral load, transaminases and oxidative status: A randomized trial among chronic hepatitis C virus-infected patients. Eur. J. Gastroenterol. Hepatol. 2006, 18, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cox, A.G.; Taylor, E.W. Hepatitis C virus encodes a selenium-dependent glutathione peroxidase gene. Implications for oxidative stress as a risk factor in progression to hepatocellular carcinoma. Medizinische Klinik (Munich) 1999, 94 (Suppl. 3), 2–6. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Molina, Y.; Lo, Y.Y.; Cropp, B.; Nakano, C.; Yanagihara, R.; Nerurkar, V.R. In vitro effects of selenium deficiency on West Nile virus replication and cytopathogenicity. Virol. J. 2008, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Taylor, E.W. Structure and dynamics of a predicted ferredoxin-like selenoprotein in Japanese encephalitis virus. J. Mol. Graph. Model. 2004, 23, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.-Q.; Goeijenbier, M.; Zuo, S.-Q.; Wang, L.-P.; Liang, S.; Klein, S.L.; Li, X.-L.; Liu, K.; Liang, L.; Gong, P.; et al. The association between hantavirus infection and selenium deficiency in mainland China. Viruses 2015, 7, 333–351. [Google Scholar] [CrossRef]
- Al-Sonboli, N.; Al-Aghbari, N.; Al-Aryani, A.; Atef, Z.; Brabin, B.; Shenkin, A.; Roberts, E.; Harper, G.; Hart, C.A.; Cuevas, L.E. Micronutrient concentrations in respiratory syncytial virus and human metapneumovirus in Yemeni children. Ann. Trop. Paediatr. 2009, 29, 35–40. [Google Scholar] [CrossRef]
- Liu, X.; Yin, S.; Li, G. Effects of selenium supplement on acute lower respiratory tract infection caused by respiratory syncytial virus. Zhonghua Yu Fang Yi Xue Za Zhi [Chin. J. Prev. Med.] 1997, 31, 358–361. [Google Scholar]
- Taylor, E.W.; Ruzicka, J.A.; Premadasa, L.; Zhao, L. Cellular Selenoprotein mRNA Tethering via Antisense Interactions with Ebola and HIV-1 mRNAs May Impact Host Selenium Biochemistry. Curr. Top Med. Chem. 2016, 16, 1530–1535. [Google Scholar] [CrossRef]
- Moya, M.; Bautista, E.G.; Velázquez-González, A.; Vázquez-Gutiérrez, F.; Tzintzun, G.; García-Arreola, M.E.; Castillejos, M.; Hernández, A. Potentially-toxic and essential elements profile of AH1N1 patients in Mexico City. Sci. Rep. 2013, 3, 1284. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Sun, L.; Nan, Y.; Zhu, L.-Y. Protection from H1N1 influenza virus infections in mice by supplementation with selenium: A comparison with selenium-deficient mice. Biol. Trace Elem. Res. 2011, 141, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Shojadoost, B.; Kulkarni, R.R.; Yitbarek, A.; Laursen, A.; Taha-Abdelaziz, K.; Negash Alkie, T.; Barjesteh, N.; Quinteiro-Filho, W.M.; Smith, T.K.; Sharif, S. Dietary selenium supplementation enhances antiviral immunity in chickens challenged with low pathogenic avian influenza virus subtype H9N2. Vet. Immunol. Immunopathol. 2019, 207, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Deryabin, P.G.; Lvov, D.K.; Botikov, A.G.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Effects of a nutrient mixture on infectious properties of the highly pathogenic strain of avian influenza virus A/H5N1. BioFactors (Oxford) 2008, 33, 85–97. [Google Scholar] [CrossRef]
- Reffett, J.K.; Spears, J.W.; Brown, T.T. Effect of dietary selenium and vitamin E on the primary and secondary immune response in lambs challenged with parainfluenza3 virus. J. Anim. Sci. 1988, 66, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Favier, A.; Sappey, C.; Leclerc, P.; Faure, P.; Micoud, M. Antioxidant status and lipid peroxidation in patients infected with HIV. Chem. Biol. Interact. 1994, 91, 165–180. [Google Scholar] [CrossRef]
- Allavena, C.; Dousset, B.; May, T.; Dubois, F.; Canton, P.; Belleville, F. Relationship of trace element, immunological markers, and HIV1 infection progression. Biol. Trace Elem. Res. 1995, 47, 133–138. [Google Scholar] [CrossRef]
- Baum, M.K.; Shor-Posner, G.; Lai, S.; Zhang, G.; Lai, H.; Fletcher, M.A.; Sauberlich, H.; Page, J.B. High risk of HIV-related mortality is associated with selenium deficiency. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997, 15, 370–374. [Google Scholar] [CrossRef]
- Baum, M.K.; Shor-Posner, G.; Zhang, G.; Lai, H.; Quesada, J.A.; Campa, A.; Jose-Burbano, M.; Fletcher, M.A.; Sauberlich, H.; Page, J.B. HIV-1 infection in women is associated with severe nutritional deficiencies. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997, 16, 272–278. [Google Scholar] [CrossRef]
- Sabe, R.; Rubio, R.; Garcia-Beltran, L. Reference values of selenium in plasma in population from Barcelona. Comparison with several pathologies. J. Trace Elem. Med. Biol. 2002, 16, 231–237. [Google Scholar] [CrossRef]
- Kupka, R.; Msamanga, G.I.; Spiegelman, D.; Morris, S.; Mugusi, F.; Hunter, D.J.; Fawzi, W.W. Selenium status is associated with accelerated HIV disease progression among HIV-1-infected pregnant women in Tanzania. J. Nutr. 2004, 134, 2556–2560. [Google Scholar] [CrossRef] [PubMed]
- Ogunro, P.S.; Ogungbamigbe, T.O.; Elemie, P.O.; Egbewale, B.E.; Adewole, T.A. Plasma selenium concentration and glutathione peroxidase activity in HIV-1/AIDS infected patients: A correlation with the disease progression. Niger Postgrad. Med. J. 2006, 13, 1–5. [Google Scholar] [PubMed]
- Khalili, H.; Soudbakhsh, A.; Hajiabdolbaghi, M.; Dashti-Khavidaki, S.; Poorzare, A.; Saeedi, A.A.; Sharififar, R. Nutritional status and serum zinc and selenium levels in Iranian HIV infected individuals. BMC Infect. Dis. 2008, 8, 165. [Google Scholar] [CrossRef] [PubMed]
- Djinhi, J.; Tiahou, G.; Zirihi, G.; Lohoues, E.; Monde, A.; Camara, C.; Sess, E. Selenium deficiency and oxidative stress in asymptomatic HIV1-infected patients in Côte d’Ivoire. Bull. Soc. Pathol. Exotique (1990) 2009, 102, 11–13. [Google Scholar] [CrossRef]
- Okwara, E.C.; Meludu, S.C.; Okwara, J.E.; Enwere, O.O.; Diwe, K.C.; Amah, U.K.; Ubajaka, C.F.; Chukwulebe, A.E.; Ezeugwunne, I.P. Selenium, zinc and magnesium status of HIV positive adults presenting at a university teaching hospital in Orlu-Eastern Nigeria. Niger. J. Med. J. Natl. Assoc. Res. Doctors Niger. 2012, 21, 165–168. [Google Scholar]
- Akinboro, A.O.; Mejiuni, D.A.; Onayemi, O.; Ayodele, O.E.; Atiba, A.S.; Bamimore, G.M. Serum selenium and skin diseases among Nigerians with human immunodeficiency virus/acquired immune deficiency syndrome. HIV/AIDS 2013, 5, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Anyabolu, H.C.; Adejuyigbe, E.A.; Adeodu, O.O. Serum Micronutrient Status of Haart-Naïve, HIV Infected Children in South Western Nigeria: A Case Controlled Study. AIDS Res. Treat. 2014, 2014, 351043. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.A.; Talusan, K.; Hutton, N.; Yolken, R.H.; Caballero, B. Serum and plasma markers of nutritional status in children infected with the human immunodeficiency virus. J. Am. Diet. Assoc. 1997, 97, 1377–1381. [Google Scholar] [CrossRef]
- Bunupuradah, T.; Ubolyam, S.; Hansudewechakul, R.; Kosalaraksa, P.; Ngampiyaskul, C.; Kanjanavanit, S.; Wongsawat, J.; Luesomboon, W.; Pinyakorn, S.; Kerr, S.; et al. Correlation of selenium and zinc levels to antiretroviral treatment outcomes in Thai HIV-infected children without severe HIV symptoms. Eur. J. Clin. Nutr. 2012, 66, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Hileman, C.O.; Dirajlal-Fargo, S.; Lam, S.K.; Kumar, J.; Lacher, C.; Combs, G.F.; McComsey, G.A. Plasma Selenium Concentrations Are Sufficient and Associated with Protease Inhibitor Use in Treated HIV-Infected Adults. J. Nutr. 2015, 145, 2293–2299. [Google Scholar] [CrossRef] [PubMed]
- Baeten, J.M.; Mostad, S.B.; Hughes, M.P.; Overbaugh, J.; Bankson, D.D.; Mandaliya, K.; Ndinya-Achola, J.O.; Bwayo, J.J.; Kreiss, J.K. Selenium deficiency is associated with shedding of HIV-1—Infected cells in the female genital tract. J. Acquir. Immune Defic. Syndr. 2001, 26, 360–364. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kupka, R.; Msamanga, G.I.; Xu, C.; Anderson, D.; Hunter, D.; Fawzi, W.W. Relationship between plasma selenium concentrations and lower genital tract levels of HIV-1 RNA and interleukin type 1beta. Eur. J. Clin. Nutr. 2007, 61, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.Y.; Tang, A.M.; Forrester, J.E.; Huang, J.; Hendricks, K.M.; Knox, T.A.; Spiegelman, D.; Semba, R.D.; Woods, M.N. Micronutrient levels and HIV disease status in HIV-infected patients on highly active antiretroviral therapy in the Nutrition for Healthy Living cohort. J. Acquir. Immune Defic. Syndr. 2006, 43, 475–482. [Google Scholar] [CrossRef]
- Kupka, R.; Mugusi, F.; Aboud, S.; Msamanga, G.I.; Finkelstein, J.L.; Spiegelman, D.; Fawzi, W.W. Randomized, double-blind, placebo-controlled trial of selenium supplements among HIV-infected pregnant women in Tanzania: Effects on maternal and child outcomes. Am. J. Clin. Nutr. 2008, 87, 1802–1808. [Google Scholar] [CrossRef] [PubMed]
- Look, M.P.; Rockstroh, J.K.; Rao, G.S.; Barton, S.; Lemoch, H.; Kaiser, R.; Kupfer, B.; Sudhop, T.; Spengler, U.; Sauerbruch, T. Sodium selenite and N-acetylcysteine in antiretroviral-naive HIV-1-infected patients: A randomized, controlled pilot study. Eur. J. Clin. Invest. 1998, 28, 389–397. [Google Scholar] [CrossRef] [PubMed]
- McClelland, R.S.; Baeten, J.M.; Overbaugh, J.; Richardson, B.A.; Mandaliya, K.; Emery, S.; Lavreys, L.; Ndinya-Achola, J.O.; Bankson, D.D.; Bwayo, J.J.; et al. Micronutrient supplementation increases genital tract shedding of HIV-1 in women: Results of a randomized trial. J. Acquir. Immune Defic. Syndr. 2004, 37, 1657–1663. [Google Scholar] [CrossRef]
- Constans, J.; Delmas-Beauvieux, M.C.; Sergeant, C.; Peuchant, E.; Pellegrin, J.L.; Pellegrin, I.; Clerc, M.; Fleury, H.; Simonoff, M.; Leng, B.; et al. One-year antioxidant supplementation with beta-carotene or selenium for patients infected with human immunodeficiency virus: A pilot study. Clin. Infect. Dis. 1996, 23, 654–656. [Google Scholar] [CrossRef]
- Durosinmi, M.A.; Armistead, H.; Akinola, N.O.; Onayemi, O.; Adediran, I.A.; Olasode, O.A.; Elujoba, A.A.; Irinoye, O.; Ogun, S.A.; Odusoga, O.L.; et al. Selenium and aspirin in people living with HIV and AIDS in Nigeria. Niger. Postgrad. Med. J. 2008, 15, 215–218. [Google Scholar]
- Sudfeld, C.R.; Aboud, S.; Kupka, R.; Mugusi, F.M.; Fawzi, W.W. Effect of selenium supplementation on HIV-1 RNA detection in breast milk of Tanzanian women. Nutrition (Burbank) 2014, 30, 1081–1084. [Google Scholar] [CrossRef][Green Version]
- Kupka, R.; Mugusi, F.; Aboud, S.; Hertzmark, E.; Spiegelman, D.; Fawzi, W.W. Effect of selenium supplements on hemoglobin concentration and morbidity among HIV-1-infected Tanzanian women. Clin. Infect. Dis. Off. Pub. Infect. Dis. Soc. Am. 2009, 48, 1475–1478. [Google Scholar] [CrossRef]
- Richard, M.J.; Guiraud, P.; Didier, C.; Seve, M.; Flores, S.C.; Favier, A. Human immunodeficiency virus type 1 Tat protein impairs selenoglutathione peroxidase expression and activity by a mechanism independent of cellular selenium uptake: Consequences on cellular resistance to UV-A radiation. Arch. Biochem. Biophys. 2001, 386, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Sappey, C.; Legrand-Poels, S.; Best-Belpomme, M.; Favier, A.; Rentier, B.; Piette, J. Stimulation of glutathione peroxidase activity decreases HIV type 1 activation after oxidative stress. AIDS Res. Hum. Retrovir. 1994, 10, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Makropoulos, V.; Bruning, T.; Schulze-Osthoff, K. Selenium-mediated inhibition of transcription factor NF-kappa B and HIV-1 LTR promoter activity. Arch. Toxicol. 1996, 70, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Hatfield, D.; Maldarelli, F.; Lee, B.J.; Clouse, K.A. Selenium supplementation suppresses tumor necrosis factor alpha-induced human immunodeficiency virus type 1 replication in vitro. AIDS Res. Hum. Retrovir. 1997, 13, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, P.A.; Murray, J.; Folks, T.M.; Diamond, A.M. Antioxidant defenses influence HIV-1 replication and associated cytopathic effects. Free Radic. Biol. Med. 1998, 24, 1485–1491. [Google Scholar] [CrossRef]
- Kalantari, P.; Narayan, V.; Natarajan, S.K.; Muralidhar, K.; Gandhi, U.H.; Vunta, H.; Henderson, A.J.; Prabhu, K.S. Thioredoxin reductase-1 negatively regulates HIV-1 transactivating protein Tat-dependent transcription in human macrophages. J. Biol. Chem. 2008. [Google Scholar] [CrossRef] [PubMed]
- Benelli, J.L.; de Medeiros, R.M.; Matte, M.C.C.; de Melo, M.G.; de Matos Almeida, S.E.; Fiegenbaum, M. Role of SEP15 Gene Polymorphisms in the Time of Progression to AIDS. Gen. Test. Mol. Biomark. 2016, 20, 383–387. [Google Scholar] [CrossRef]
- Zhao, L.; Cox, A.G.; Ruzicka, J.A.; Bhat, A.A.; Zhang, W.; Taylor, E.W. Molecular modeling and in vitro activity of an HIV-1-encoded glutathione peroxidase. Proc. Natl. Acad. Sci. USA 2000, 97, 6356–6361. [Google Scholar] [CrossRef]
- Cohen, I.; Boya, P.; Zhao, L.; Metivier, D.; Andreau, K.; Perfettini, J.L.; Weaver, J.G.; Badley, A.; Taylor, E.W.; Kroemer, G. Anti-apoptotic activity of the glutathione peroxidase homologue encoded by HIV-1. Apoptosis 2004, 9, 181–192. [Google Scholar] [CrossRef]
- Xu, X.-M.; Carlson, B.A.; Grimm, T.A.; Kutza, J.; Berry, M.J.; Arreola, R.; Fields, K.H.; Shanmugam, I.; Jeang, K.-T.; Oroszlan, S.; et al. Rhesus monkey simian immunodeficiency virus infection as a model for assessing the role of selenium in AIDS. J. Acquir. Immune Defic. Syndr. (1999) 2002, 31, 453–463. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, J.; Xu, H.; Jiang, Y.; Zhu, G. Effect of selenium supplementation on mice infected with LP-BM5 MuLV, a murine AIDS model. Biol. Trace Elem. Res. 1997, 59, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Anstey, A.V.; Bugert, J.J. Molluscum contagiosum virus infection. Lancet Infect. Dis. 2013, 13, 877–888. [Google Scholar] [CrossRef]
- Schaffer, J.V.; Berger, E.M. Molluscum Contagiosum. JAMA Dermatol. 2016, 152, 1072. [Google Scholar] [CrossRef] [PubMed]
- McFadden, G. Even viruses can learn to cope with stress. Science 1998, 279, 40–41. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Rios, J.D.; Yang, Z.; Erlandson, K.J.; Cohen, J.I.; Martens, C.A.; Bruno, D.P.; Porcella, S.F.; Moss, B. Molluscum Contagiosum Virus Transcriptome in Abortively Infected Cultured Cells and a Human Skin Lesion. J. Virol. 2016, 90, 4469–4480. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.W.; Bhat, A.; Nadimpalli, R.G.; Zhang, W.; Kececioglu, J. HIV-1 encodes a sequence overlapping env gp41 with highly significant similarity to selenium-dependent glutathione peroxidases. J. Acquir. Immune Def. Syndr. Hum. Retrovirol. Off. Pub. Int. Retrovirol. Assoc. 1997, 15, 393–394. [Google Scholar] [CrossRef][Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Genome Structure | Virus Family | Virus | Epidemiological Study | Epidemiological Intervention | In Vitro Study | In Vivo Study | Viral Selenoprot |
|---|---|---|---|---|---|---|---|---|
| I | Double-stranded DNA | Herpesviridae | Epstein-Barr virus (EBV) | ↓ GPX activity associated with ↑ viral load [169] | CT = ombilical blood mononuclear cells SS = Se-rich rice extract Inhibits EBV mediated cell transformation [170] | [171] | ||
| Herpes Simplex Virus 2 (HSV-2) | SS = Selenium aspartate + multisupplementation Faster healing, ↓ in viral load and ↑ in antiviral cytokines [172] | CT = Vero cells SS = Diphenyl diselenide ↓ infectivity [173] | AM = BALB/c Mice SS = Diphenyl diselenide ↓ histological damages and viral load ↑ levels of TNFalpha and IFNgamma [173] | |||||
| Human Herpesvirus 3 (HHV3) | SS = Selenium aspartate + multisupplementation Faster healing, ↓ in viral load and ↑ in antiviral cytokines [172] | |||||||
| Cytomegalovirus (CMV) | [171] | |||||||
| Infectious bovine rhinotracheitis (IBRV) | SS = Sodium selenite ↑ GPX activity after infection in Se group ↑ IgM after infection in Se group ↑ antibody titer after infection in Se group [174] | |||||||
| Poxviridae | Molluscum contagiosum virus (MCV) | [171,175] | ||||||
| Fowlpox virus (FWPV) | [176] | |||||||
| Papovaviridae | Human Papillomavirus (HPV) | SS = Selenium aspartate + multisupplementation Faster healing, ↓ in viral load and ↑ in antiviral cytokines [172,177] | ||||||
| II | Single-stranded DNA | Circoviridae | Porcine Circovirus type 2 (PCV2) | CT = PK15 cells SS = selenite, selenocarrageenan and selenomethionine Selenomethionine inhibits replication of PCV2 via Gpx1 and oxidative stress [164,165] CT = PK15 cells SELENOS overexpression may ↓ viral replication via oxidative stress [166] CT = PK15 cells SS = selenizing astragalus polysaccharide and selenomethionine ↓ PCV2 replication through autophagy ↓ [167,168] | AM = KunMing Mice SS = Selenized yeast ↓ TNFalpha, viral load and histological damages [178] | |||
| IV | Positive-sense single-stranded RNA | Picornaviridae | Coxsackievirus B3 (CVB3) | AM = C3H/HeJ Mice SS = Selenite Apparition of a more virulent strain after infection of a selenium or vitamin E deficient host due to viral mutations [98,99,110,111,179] AM = Gpx1-/- Mice Apparition of a more virulent strain after infection of GPX1 deficient mice [108] AM = C57Bl/6 Mice SS = not specified Co-infection with a retrovirus lead to a more virulent pathology Selenium supplementation reverse the effect [180] AM = Balb/c Mice Viral load is associated with selenium status in tissues [181] AM = Mice SS = commercial Se repleted ‘feedstuff’ Se deficient mice exhibit a higher mortality, histopathological pathogenicity and viral load [182] | [171,183] | |||
| Coxsackievirus B4 (CVB5) | [183] | |||||||
| Coxsackievirus B5 (CVB5) | CT = Vero Cells SS = selenite, selenate and selenomethionine Selenite ↓ viral replication via thiol interaction [184] | |||||||
| Live attenuated poliomyelitis vaccine | SS = Sodium selenite ↑ GPX activity after infection in Se group ↑ antiviral cytokines ↑ TH4 response Faster clearance Mutations in the viral particles [185] | |||||||
| Foot-and-mouth disease virus (FMDV) | SS = selenium enriched yeast ↑ GPX activity after infection in Se group ↓ DNA damage [186] | |||||||
| V | Negative-sense single-stranded RNA | Flaviviridae | Hepatite C virus (HCV) | ↓ Se in infected people [152,160,187] ↓ GPX activity in infected people [152] No variation in GPX activity with infection [145] | SS = Selenomethionnine ↑ GPX activity after infection in Se group No effect on viral load [188] | [183,189] | ||
| West nile virus (WNV) | CT = Vero cells Selenium deficiency induces higher cell death and cytopathic effects but has no impacts on viral production [190] | |||||||
| Japanese encephalitis virus (JEV) | [191] | |||||||
| Bunyaviridae | Hantaan virus (HTNV) or Seoul virus (SEOV) | ↑ incidence of the infection with ↓ Se [192] | CT = HUVEC SS = sodium selenite ↓ viral replication with a low MOI [192] | |||||
| Respiratory syncytial virus (RSV) | ↓ Se in infected people [193] | SS = Sodium selenite Faster healing in Se group [194] | ||||||
| Filoviridae | Ebola virus (EBOV) | [195] | ||||||
| Orthomyxoviridae | Influenza A/Bangkok/1/79 (H3N2) | CT = Differenciated human bronchial epithelial cells Se deficiency ↑ mucus production, influenza-induced apoptosis and modifies cytokine expression [122] | AM = C57Bl/6J SS = Selenite Deficiency leads to an ↑ in inflammation and pathology and an altered cytokine expression No changes in viral load [95] Apparition of a more virulent strain after infection of a selenium deficient host due to mutations [121] ↑ SOD activity in selenium deficient group [124] AM = transgenic mice carrying a mutant Sec-tRNA[Ser]Sec No impact on lung pathology [123] | |||||
| Influenza A/Puerto Rico/8/34 (H3N2) | AM = C57Bl/6J SS = Selenite Deficiency leads to altered immune response responsible of less death No change in viral load [125] | |||||||
| Influenza A (H1N1) | ↓ Se in infected people [116,196] ↓ GPX activity in infected people [116] | AM = KunMing Mice SS = Selenite Reduces mortality, ↑ levels of TNFalpha and IFNgamma No change in viral load [197] | ||||||
| Avian influenza (H9N2) | SS sodium enriched yeast or sodium selenite ↓ viral shedding ↑ ISG expression and IFN [198] | |||||||
| Avian Influenza A/duck/Novosibirst56/05 (H5N1) | CT = RK, BHK21 and Vero E6 cells SS = nutrient mixture containing selenium ↓ viral replication in late stages [199] | |||||||
| Paramyxoviridae | Parainfluenza-3 (PI3) | SS = sodium selenite ↑ GPX activity after infection in Se group ↑ IgM after infection in Se group antibody titer after infection in Se group [200] | ||||||
| Human metapneumovirus (HMPV) | ↓ Se in infected people [193] | |||||||
| Measles virus (MV) | [183] | |||||||
| VI | Single-stranded RNA with a DNA intermediate | Retroviridae | Human immuno-deficiency virus 1 (HIV-1) | ↓ Se in infected people [153,187,201,202,203,204,205,206,207,208,209,210,211,212] No significant variation of Se level in infected people [213,214,215] Low selenium level associated with low CD4 count [153,203,206,207,211] Low selenium level associated with a higher progression to AIDS [201,202,203,204,206] Low selenium level associated with vaginal shedding of HIV [216] High selenium level associated with vaginal shedding of HIV [217] No significant variation of Se level in infected people treated with HAART [218] More skin desease in Se deficient HIV infected people [211] | No change in viral load SS = selenized yeast [137,139] SS = selenomethionine [219] SS = Sodium selenite [220] SS = not indicated [138,221] ↑ in CD4 count in Se group SS = selenized yeast yeast [137,139] SS = Sodium selenite [220] SS = not indicated [138,221] No change in CD4 count SS = selenomethionine [219,222] SS = not indicated [223] ↑ of viral shedding in Se group SS = selenomethionine [224] SS = not indicated [221] Se supplementation improves child survival if the mother is infected SS = selenomethionine [219] Se supplementation decreases diarrheal morbidity SS = selenomethionine [225] | CT = Jurkat and HeLa cells Infection or TAT expression ↓ some selenoproteins but ↑ low molecular mass selenocompounds [142,226] CT = ACH2, Jurkat, ESb-L, KK1, U1 cells and monocytes SS = selenite Prevents HIV transcription by TNF alpha mediated NFkappaB activiation in chronically infected cells [227,228,229] CT = SupT1 GPX1 overexpression ↑ viral replication and cytopathic effects and inversely [230] CT = U937, monocytes derived macrophages TXNRD1 negatively regulates TAT activity by targeting disulfides bonds [231] | In patients, a polymorphism a SELENOF is associated with a shorter time of progression to AIDS [232] | [195,233,234] |
| Human immuno-deficiency virus 2 (HIV-2) | [183] | |||||||
| Simian immuno-deficiency virus (SIV) | ↓ Se in infected monkeys [235] | CT = CEM and Jurkat cells Infection leads to a ↓ in selenoprotein expression and an ↑ in low molecular mass selenocompounds TAT transfection leads to a ↓ in GPX and SELENOF but an ↑ in TXNRD1 expression [235] | ||||||
| Murine Leukemia virus (MuLV) | SS = Sodium selenite ↑ GPX activity after infection in Se group [236] | [171] | ||||||
| VII | Double-stranded DNA with a single stranded RNA intermediate | Hepadnaviridae | Hepatitis B (HBV) | ↓ Se in infected people [158,160] {↑ Se associated with less hepatic damages Abediankenari, 2011 #4334} | SS = selenized table salt or selenized yeast Lower cancer induced by HBV incidence [162] | CT = HepG2 and HuH7 SS = sodium selenite Suppresses HBV replication, transcription and protein expression [161] | [171] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guillin, O.M.; Vindry, C.; Ohlmann, T.; Chavatte, L. Selenium, Selenoproteins and Viral Infection. Nutrients 2019, 11, 2101. https://doi.org/10.3390/nu11092101
Guillin OM, Vindry C, Ohlmann T, Chavatte L. Selenium, Selenoproteins and Viral Infection. Nutrients. 2019; 11(9):2101. https://doi.org/10.3390/nu11092101
Chicago/Turabian StyleGuillin, Olivia M., Caroline Vindry, Théophile Ohlmann, and Laurent Chavatte. 2019. "Selenium, Selenoproteins and Viral Infection" Nutrients 11, no. 9: 2101. https://doi.org/10.3390/nu11092101
APA StyleGuillin, O. M., Vindry, C., Ohlmann, T., & Chavatte, L. (2019). Selenium, Selenoproteins and Viral Infection. Nutrients, 11(9), 2101. https://doi.org/10.3390/nu11092101