1. Introduction
Bone diseases are caused by disharmony in the bone remodeling process, which is mediated by bone-formative osteoblasts and bone-resorbing osteoclasts [
1]. Excessive activation of osteoclasts, which leads to excessive bone destruction, is associated with low concentration levels of estrogen in post-menopausal women or excessive inflammatory responses in pathological circumstances [
2,
3].
Osteoclasts are derived from bone-marrow macrophages and are responsible for the removal of old bone tissue by secreting acid and collagenase [
4,
5]. Osteoclastogenesis needs two factors, macrophage colony-stimulating factor (M-CSF) for proliferation and survival, and receptor activator of nuclear factor kappa-Β ligand (RANKL) for differentiation and function [
5]. M-CSF has a crucial role in sustaining the survival and proliferation of osteoclast precursor cells [
6]. The binding of M-CSF to c-Fms on the osteoclast cell surface leads to the recruitment of c-Cbl and phosphatidylinositol 3-kinase (PI3K) complex. This complex activates the Akt pathway and causes c-Fms ubiquitination [
7], which results in an increase in tyrosine phosphorylation and activation. Besides, M-CSF augments Grb2, which mediates the phosphorylation of ERK [
8]. Thus, the M-CSF-induced activation of c-Fms plays a crucial role in the proliferation and survival of osteoclast precursor cells through the PI3K/Akt and ERK pathways [
7,
8,
9]. The binding of RANKL to its cognate receptor RANK results in the activation of signaling pathways, including components of the mitogen-activated protein kinase (MAPK) pathway, such as c-Jun N-terminal protein kinase (JNK), p38, and ERK through tumor necrosis factor receptor-associated factor 6 (TRAF6) [
9]. It has also been reported that the JNK signaling pathway plays an important role in the regulation of apoptosis [
10]. Nuclear factor-κB (NF-κB) is one of the key transcription factors for osteoclast differentiation, which is activated by RANKL. RANKL phosphorylates NF-κB inhibitor (IκB), then activates ubiquitin-dependent proteasomal degradation of IκB, resulting in activation of transcription after NF-κB is translocated from the cytoplasm to the nucleus [
11,
12]. Activation of the TRAF6 family stimulates the auto-amplification of nuclear factor of activated T cells (NFAT)c1, the key factor for osteoclastogenesis [
13], which induces the expression of osteoclast-specific marker genes, such as osteoclast-associated receptor (OSCAR), cathepsin K (CtsK), and tartrate-resistant acid phosphatase (TRAP).
Grape seed proanthocyanidin extract (GSPE), which is extracted from European red grape seed
Vitis vinifera, is a commercially available medicine for the treatment of venous and lymphatic dysfunction [
14,
15]. Grapes have healthful properties with ingredients like phenolic bioflavonoids that are powerful antioxidants [
15,
16]. Related studies on the effects of GSPE in animals have demonstrated the potent anti-inflammatory properties of proanthocyanidins and their impact on experimental inflammation [
17]. Recent studies on GSPE have also reported its protective effects on monosodium iodoacetate-induced arthritis and collagen-induced arthritis [
18,
19,
20]. However, the effects of GSPE on osteoclast proliferation, differentiation, and apoptosis in bone metabolism have not yet been researched.
We investigated whether GSPE protects against lipopolysaccharide (LPS)-induced bone loss in mouse to identify whether it could be used as a candidate medicine for the possible treatment of bone metabolic disease. We also studied its effects on the proliferation, differentiation, and apoptosis of osteoclasts and the related mechanism of action in vitro.
2. Materials and Methods
2.1. Mice, Reagents and Antibodies
Five-week old male ICR mice were purchased from Samtako Bio Korea (Osan, Korea). The mice were kept in a temperature-controlled (22–24 °C) and humidity-controlled (55–60%) environment with a 12 h light/dark cycle. All experiments were performed in accordance with the guidelines of the Institute Committee of Wonkwang University for animal experimentation (Approval Number: WKU20-39). GSPE was obtained from the Hanlim pharmaceutical company (Seoul, Korea), and was prepared as a stock solution of 25 mg/mL in distilled water (DW) and stored at −20 °C. Anti-β-actin monoclonal antibody and LPS were obtained from Sigma (St. Louis, MO, USA). LPS was dissolved in PBS at 5 mg/mL and stored it at −20 °C. Soluble, recombinant human M-CSF and human RANKL were obtained from PeproTech EC Ltd. (London, UK). Anti-p38, anti-phospho-p38, anti-JNK, anti-phospho-JNK, anti-Akt, anti-phospho-Akt, anti-phospho-ERK, anti-ERK, anti-PLCγ2, anti-phospho-PLCγ2, anti-Btk, anti-IκB, and anti-phospho-IκB antibodies were purchased from Cell Signaling Technology Inc. (Beverly, MA, USA). Anti-phospho-Btk was purchased from GeneTex (Irvine, CA, USA). Anti-c-Fos and anti-NFATc1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Secondary antibodies, including horseradish peroxidase-conjugated anti-mouse IgG and anti-rabbit IgG were obtained from Enzo Life Sciences (Farmingdale, NY, USA). Fetal bovine serum (FBS), α-minimum essential medium (α-MEM), and penicillin/streptomycin antibiotics were purchased from Gibco BRL (Grand Island, NY, USA). All other chemicals were of analytical grade or complied with the standards required for cell culture.
2.2. In Vitro Osteoclastogenesis Assay
Bone marrow cells (BMCs) were obtained from 5-week old male ICR mice by flushing the femurs and tibias with α-MEM supplemented with 10% FBS and 1% antibiotics. To obtain bone marrow macrophages (BMMs), BMCs were seeded on culture dishes in α-MEM supplemented with 10% FBS and 10 ng/mL M-CSF and cultured for 1 day. Non-adherent cells were transferred to 10 cm petri dishes and further cultured in the presence of 30 ng/mL M-CSF for 3 days. After removal of the non-adherent cells, the adherent cells were used as the osteoclast precursors. To generate osteoclasts from these osteoclast precursors, the cells were seeded in a 48-well plate (3.5 × 104 cells/well) in complete medium, containing 30 ng/mL M-CSF and 100 ng/mL RANKL, and cultured for 4 days with or without GSPE. The cells were fixed in 3.7% formalin for 10 min, permeabilized with 0.1% Triton X-100 (Sigma, St. Louis, MO, USA), and then stained with TRAP (Sigma). TRAP-positive multinucleated cells with more than 3 nuclei were counted as osteoclasts.
2.3. Fluorescence Microscopy Analysis
BMMs were seeded in a 24-well plate (3.5 × 104 cells/well) in complete medium containing 30 ng/mL M-CSF and cultured for 48 h with or without GSPE. The cells were fixed in 3.7% formalin for 10 min, permeabilized with 0.1% Triton X-100, and then stained with Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) using the ApopTag® Fluorescein In Situ Apoptosis Detection kit (Millipore, Darmstadt, Germany).
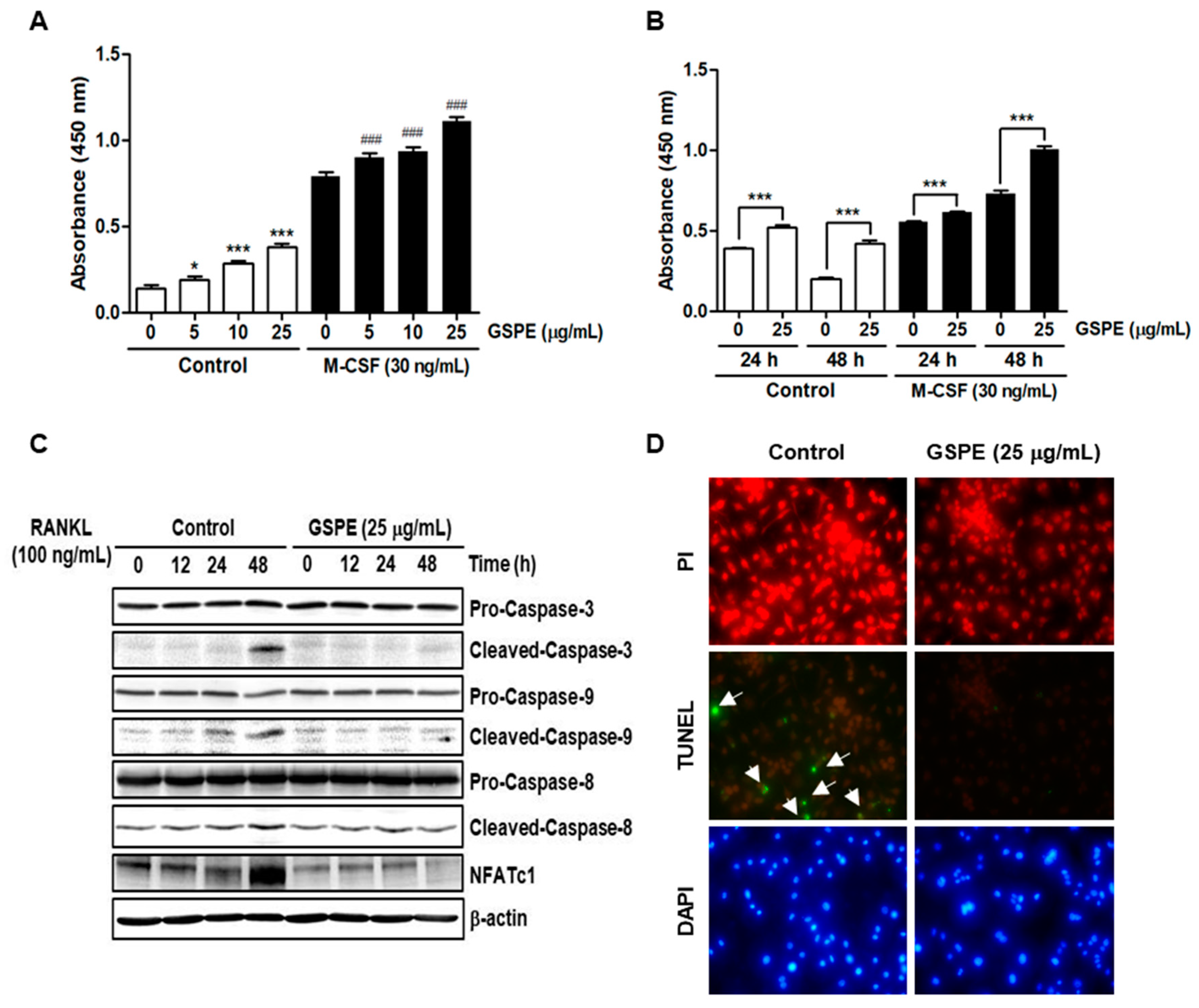
2.4. Proliferation Assay
XTT assay was performed to examine the effects of GSPE on the proliferation of osteoclast precursors. BMMs were seeded into 96-well plates at a density of 1 × 104 cells/well with various concentrations of GSPE and incubated for 3 days in the presence of 30 ng/mL M-CSF. Following that, 50 μL XTT solution was added to each well and incubated for 4 h. The plate was read at 450 nm using an ELISA reader (Molecular Devices, Sunnyvale, CA, USA).
2.5. Pit Formation Assay
For the assay, 1 × 107 BMCs cells and 1 × 106 primary osteoblast cells were seeded on collagen gel-coated culture dishes and cultured for 9 days in the presence of 10 nM 1,25-dihydroxyvitamin D3 (Sigma) and 1 μM prostaglandin E2 (PGE2) (Sigma). The co-cultured cells were detached by treatment with 0.1% collagenase at 37 °C for 10 min and then re-plated onto hydroxyapatite (HA)-coated plates (Corning Inc., Corning, NY, USA) and dentin slices. The cells were incubated on the plates with or without GSPE. After 24 and 48 h, the cells were removed using 10% sodium hypochlorite and the total resorption pits were photographed and analyzed using Image-Pro Plus version 4.0 (Media Cybernetics, Silver Spring, MD, USA).
2.6. Gene Expression Analysis Using Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA), and the cDNA was synthesized at 42 °C for 1 h using 1 μg RNA, oligo (dT), and an M-MLV RT kit (Invitrogen). Real-time RT-PCR was carried out on an Exicycler 96 Real-Time Quantitative Thermal Block (Bioneer Co., Daejeon, Korea) in a 20 μL reaction mixture containing 10 μL of SYBR
® Green Premix (Bioneer Co.), 1 μL of each primer (10 pmol), and 1 μL of cDNA. Primer sets used are shown in
Table 1. The specificity of the SYBR green assays was confirmed using melting-point analysis. The mRNA levels of genes were calculated from the cycle threshold (Ct) value using the 2(-Delta pool Ct) method [
21]. All reactions were run in triplicate and the data were normalized to
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels.
2.7. Western Blot Analyses
Whole-cell lysates were prepared using lysis buffer containing 50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1 mM sodium fluoride, 1 mM sodium vanadate, 1% deoxycholate, and protease inhibitors. Whole-cell lysates was centrifuged at 13,500× g for 15 min, and then the supernatant was collected. The protein concentration was measured using a DC Protein Assay kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). Equal amounts of protein (20–30 µg) were run on 10% SDS-PAGE gels and transferred by electroblotting onto polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h, followed by blotting with the primary antibodies for 2 h at room temperature. After washing the membranes with TBST, they were incubated for 1 h with horseradish peroxidase-conjugated sheep anti-mouse or donkey anti-rabbit immunoglobulin antibodies. The specific signals were detected using the Western Blotting Detection Reagent kit (Millipore).
2.8. Luciferase Assay
293R cells were plated in 24-well plates as triplicates for each condition and then transfected with differing amounts of the following cDNAs: NF-kB luciferase and β-galactosidase. After 48 h, the transfected cells were lysed using Cell Culture Lysis Reagent (Promega, Madison, WI, USA), and luciferase activity was measured using the Luciferase Assay System from Promega. Luciferase activity was normalized to the β-galactosidase activity in each sample.
2.9. Retrovirus Preparation and Infection
Packaging of the retroviral vectors pMX-IRES-EGFP and pMX-constitutively active (CA)-Iκκβ-IRES-EGFP were transfected by transient transfection of these pMX vectors into Plat-E retroviral packaging cells using X-tremeGENE 9 (Roche, Nutley, NJ, USA), according to the manufacturer’s protocol. After incubation in fresh medium for 2 days, the culture supernatants of the retrovirus-producing cells were collected. For retroviral infection, non-adherent BMCs were cultured in 30 ng/mL M-CSF. After 2 days incubation, the BMMs were infected with the viral supernatants of the pMX-IRES-EGFP and pMX-CA-Iκκβ-IRES-EGFP virus-producing Plat-E cells together with 10 mg/mL polybrene and 30 ng/mL M-CSF for 6 h. The infection efficiency of the retroviruses was determined by green fluorescent protein (GFP) expression and was always found to be greater than 80%. After infection, the BMMs were induced to differentiate in the presence of 30 ng/mL M-CSF and 100 ng/mL RANKL for 4 days. Osteoclast formation was detected using TRAP staining.
2.10. Model of LPS-Induced Bone Loss and Treatment
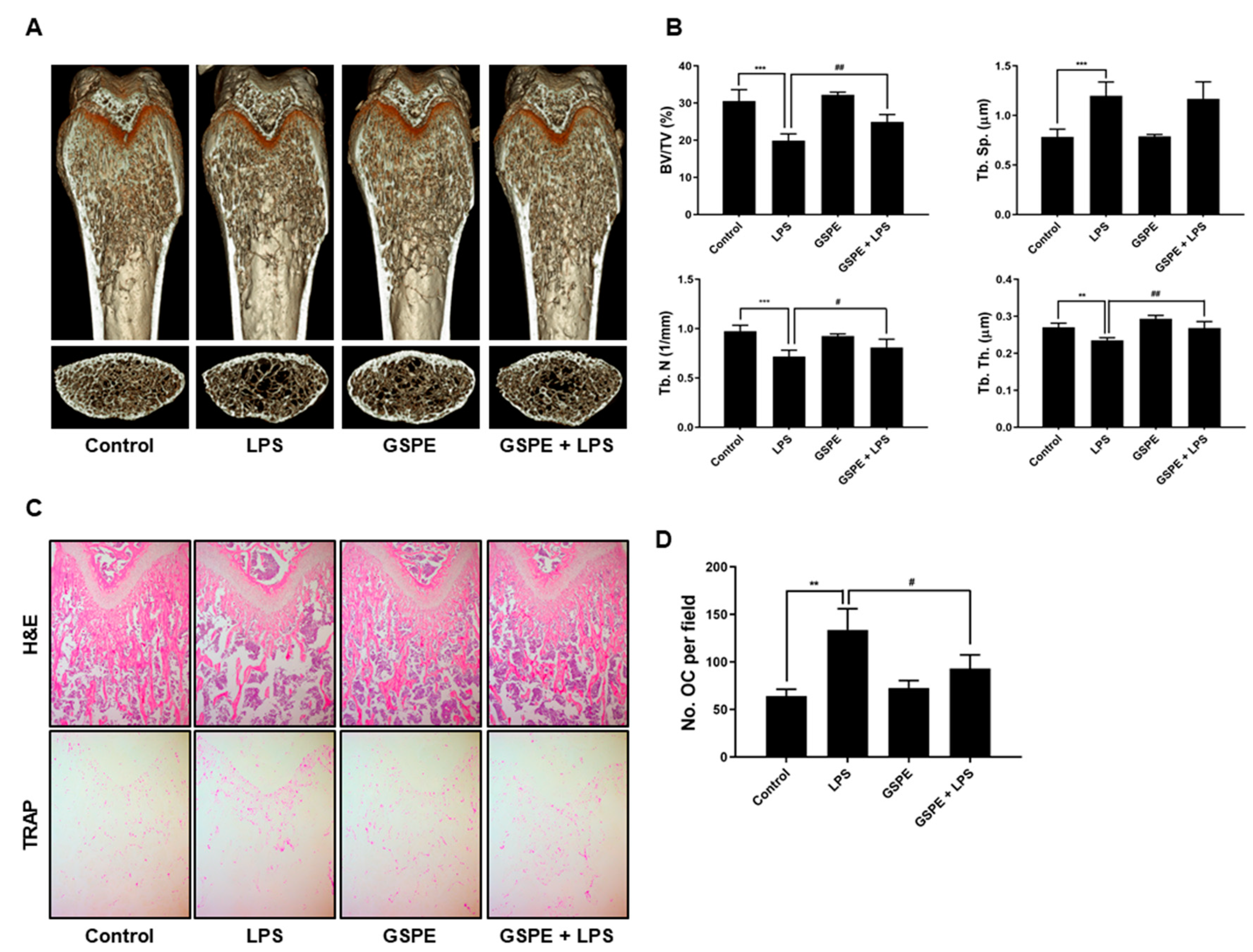
To study the effect of GSPE on LPS-induced osteoclast formation in vivo, ICR mice were divided into four experimental groups composed of 5 mice each: PBS-treated (control), only LPS-treated, only GSPE-treated, and LPS+GSPE-treated groups. Further, 200 mg/kg GSPE or PBS was injected intraperitoneally 1 day before the LPS (5 mg/kg) injection. GSPE or PBS was administered orally every other day for 8 days. LPS was injected intraperitoneally on days 1 and 4. The mice were euthanized after 8 days and the left and right femurs were analyzed using high-resolution micro-computed tomography (micro-CT). The femur metaphysic regions were scanned using high-resolution micro-CT (NFR-Polaris-S160; Nanofocus Ray, Iksan, Korea) with a source voltage of 55 kVp, current of 60 µA, and 6 µm isotropic resolution. Femur scans were performed from the growth plate until 2 mm proximally, with 720 sections per scan. Bone histomorphometric analyses were performed with the micro-CT data using INFINITT-Xelis software (INFINITT Healthcare, Seoul, Korea). The structural parameters analyzed included trabecular bone volume/total volume (BV/TV, %), trabecular thickness (Tb.Th, μm), trabecular separation (Tb.Sp, μm), and trabecular number (Tb.N, 1/mm). The femurs were fixed in 4% paraformaldehyde (Sigma) for 1 day, decalcified for 2 weeks in 12% EDTA, and then embedded in paraffin. Sections (5 µm thick) were prepared using Leica microtome RM2145 (Leica Microsystems, Bannockburn, IL, USA) and stained with hematoxylin and eosin (H&E) and TRAP. Representative images were captured using a light microscope and the number of osteoclasts per field was counted using Image Pro-Plus software version 4.0 (Media Cybernetics, Silver Spring, MD, USA).
2.11. Statistical Analyses
Experiments were conducted at least three times and the data are expressed as mean ± standard deviation or mean ± standard error. All statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). Student’s t-test was used to compare the parameters between 2 groups, while the analysis of variance (ANOVA) test followed by the Tukey post-hoc test was used to compare the parameters among 3 groups. Data with p values < 0.05 were considered statistically significant.
4. Discussion
GSPE has various pharmacological activities including anti-inflammatory, anti-oxidative, anti-bacterial, anti-viral, vasodilatory, anti-allergic, cardioprotective, and anti-carcinogenic activities [
14,
15,
16,
17]. In particular, GSPE has been reported to have a protective and therapeutic effect on osteoporosis [
22], bone necrosis [
23], and inflammatory autoimmune arthritis [
18,
19,
20]; it has also been shown to increase bone density and strength [
24,
25]. Based on this evidence, we tried to elucidate the role and mechanism of osteoclasts in the protective effect of GSPE in bone loss. In this study, we determined the protective effect of GSPE on LPS-induced inflammatory bone loss in vivo. In addition, we investigated the effects of GSPE on osteoclast proliferation, differentiation, apoptosis, and bone resorbing function, and its underlying signaling mechanisms.
Since osteoclasts have a short lifespan, any regulation that prolongs their viability can increase osteoclast activity [
26]. The number and activity of osteoclast is determined by the ability to proliferate, survive, and differentiate osteoclast precursors [
27]. M-CSF acts primarily to promote the proliferation and survival of osteoclast precursors during the osteoclastogenesis process [
28]. RANKL acts as a major factor in the differentiation of osteoclast precursors into mature osteoclasts [
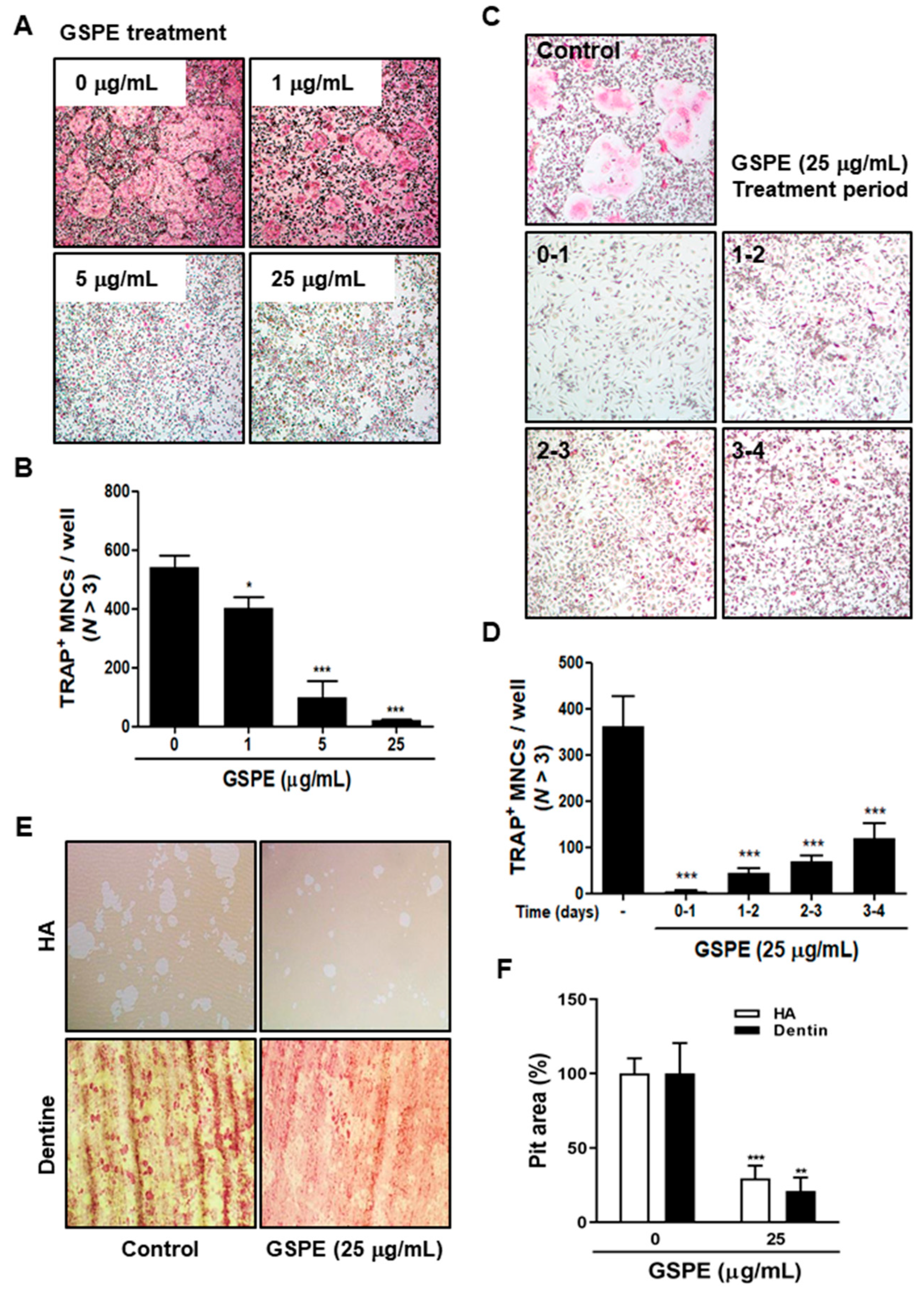
28]. Our data suggest that GSPE strongly inhibited RANKL-induced osteoclast differentiation, especially when added in the early stage of culture (
Figure 2A–D). In addition, GSPE attenuated mature osteoclastic bone resorption (
Figure 2E,F). Osteoclastogenesis is a multi-step process involving commitment, fusion, and activation [
29,
30]. Osteoclast differentiation is mainly triggered by RANKL and RANK on the osteoclast precursor and amplified by downstream transduction signaling. In particular, NFATc1 is a master transcription factor of osteoclast differentiation that promotes the expression of osteoclast-specific genes. NFATc1 play an important role in the expression of osteoclast-specific genes, such as
Integrin-β3,
OSCAR,
MMP9,
Ctr,
Atp6v0d2, and
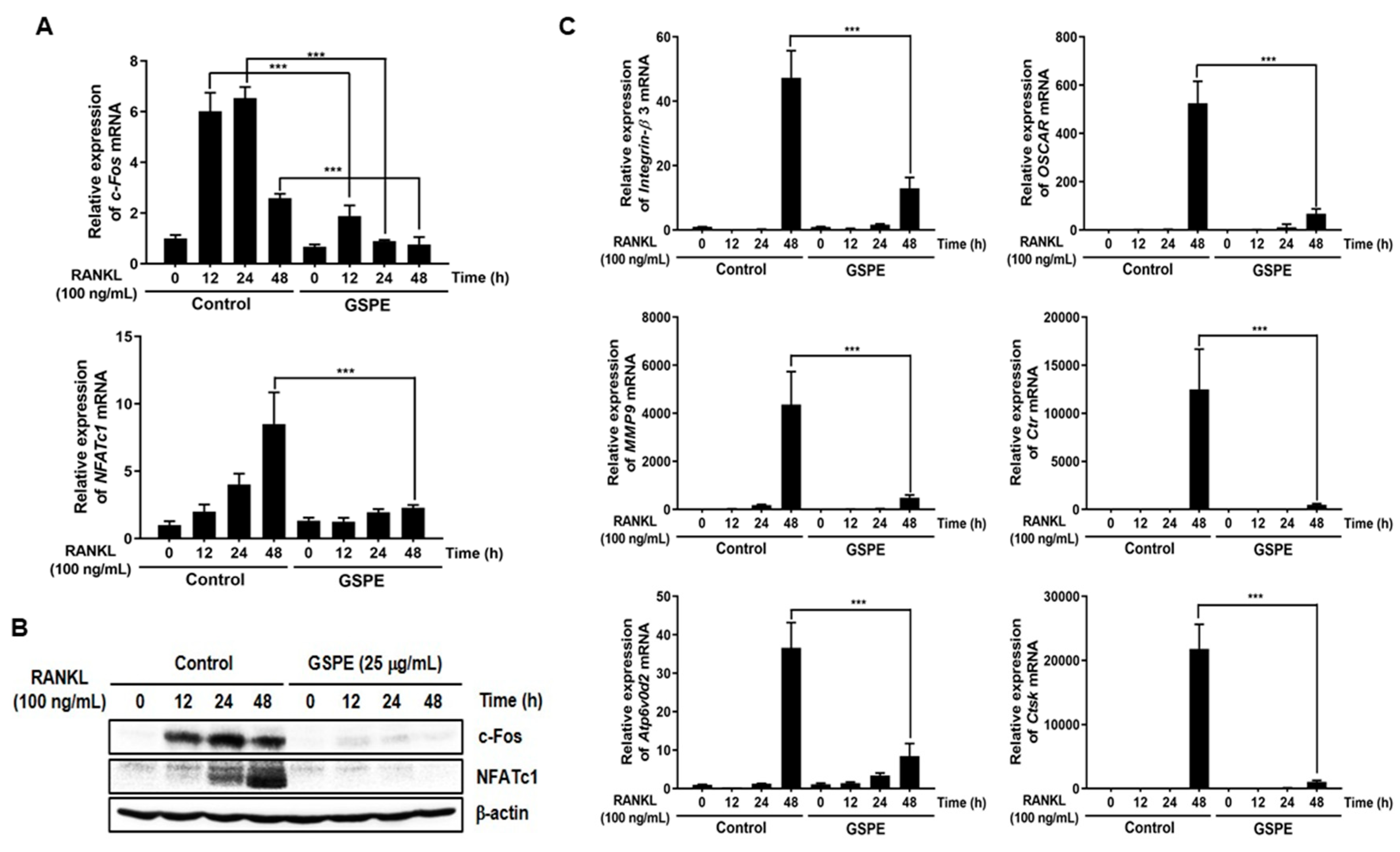
CtsK, which are required for osteoclast differentiation, fusion, and function. In our study, GSPE was found to inhibit the mRNA and protein expression of c-Fos and NFATc1 (
Figure 3A,B). Also, the inhibitory effect of GSPE via downregulation of c-Fos and NFATc1 was confirmed by changes in the mRNA expression levels of osteoclast-specific genes (
Figure 3C). The binding of RANK to RANKL activates NF-κB, which is one of the most important transcription factors in osteoclast differentiation [
11,
12,
27]. IκB binds to NF-κB, which prevents it from translocating to the nucleus and phosphorylation by the Ikk complex [
31]. Subsequently, the ubiquitination and proteasomal degradation of IκB induces NF-κB to translocate to the nucleus and stimulate the transcription of the target gene [
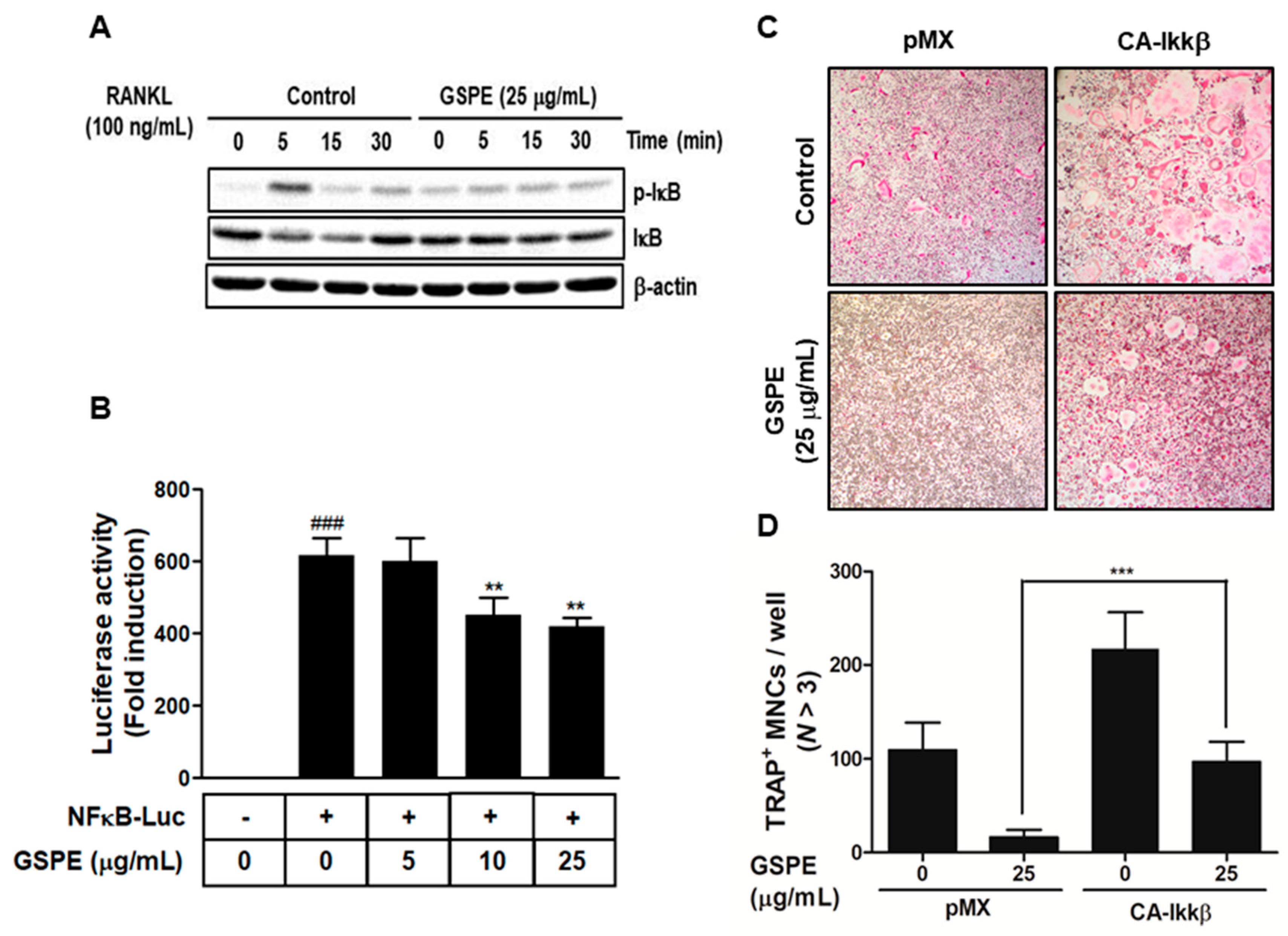
31]. In this study, GSPE was found to inhibit the phosphorylation and degradation of IκB (
Figure 4A), while Ikkβ overexpression partially reversed the GSPE-induced inhibition of osteoclastogenesis (
Figure 4C,D). Moreover, the luciferase assay confirmed that GSPE suppresses NF-κB transcription activity (
Figure 4B). These results indicate that GSPE inhibited the RANKL-induced transcription activity of NF-κB through increased phosphorylation and degradation of IκB by Ikkβ.
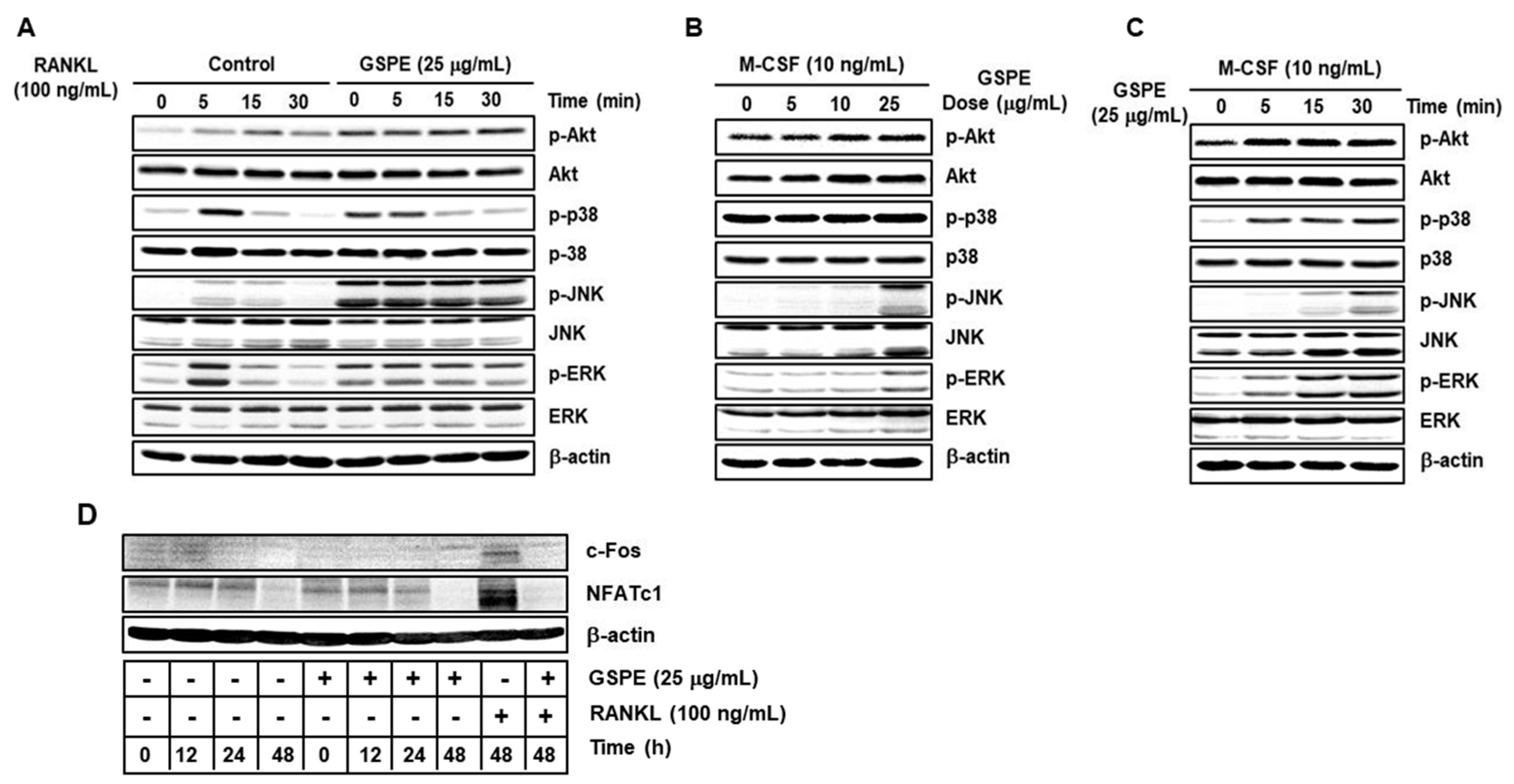
Akt and MAPK signaling pathways, including downstream signaling molecules p38, JNK, and ERK, are important for the regulation of osteoclastogenesis [
9,
10]. Contrary to expectations, our study showed that GSPE further activates RANKL-induced phosphorylation of Akt, p38, JNK, and ERK (
Figure 5A). Moreover, GSPE was found to increase the phosphorylation of Akt, p38, JNK, and ERK in a dose- or time-dependent manner in the presence of M-CSF, without RANKL stimulation (
Figure 5B,C). Therefore, we tested whether GSPE plays a role similar to RANKL; we found that GSPE did not induce c-Fos and NFATc1 activity by GSPE treatment without RANKL stimulation (
Figure 5D). Akt and MAPK signaling pathways play an important role not only in osteoclast differentiation, but also in the regulation of proliferation and apoptosis. It has been reported that M-CSF-activated Akt and MAPK signaling pathways are primarily involved in the proliferation and survival of osteoclast precursor, whereas RANKL-induced Akt and MAPK activation is mainly associated with osteoclast differentiation [
9]. However, in this study, it was confirmed that RANKL-induced Akt and MAPK activation in the presence of GSPE preferentially affects the pathways that promote osteoclast proliferation, while suppressing apoptosis rather than osteoclast differentiation (
Figure 6).
In keeping with its anti-osteoclastogenic effect in vitro, GSPE prevented LPS-induced inflammatory bone loss in vivo. LPS injection leads to inflammatory bone loss by recruiting inflammatory cells, increasing the number of osteoclasts, and activating osteoclast resorption. Micro-CT analysis performed in this study indicated that GSPE treatment for LPS-injected mice increased BV/TV, Tb.Th, and Tb.N, indicating that GSPE restored LPS-induced bone loss. Bone histomorphometry analysis showed that GSPE significantly reduced LPS-induced bone erosion and TRAP-positive osteoclast formation in vivo. Despite these promising results, our study has some limitations. Inflammatory bone loss caused by LPS in vivo is induced by a complex process of many cells, including osteoblasts and osteoclasts. In this study, the effect of GSPE on osteoclast proliferation, differentiation, survival and bone resorption was investigated. However, we cannot rule out the possibility that GSPE will affect osteoblast bone formation. Therefore, further research is needed to evaluate the effect of GSPE on osteoblast bone formation. Also, our study focused only on pathological bone loss due to LPS, which is dominated by excessive osteoclast activity. Therefore, to fully elucidate the effects and mechanisms of action of GSPE in bone remodeling, further studies are needed to evaluate the effects of GSPE on normal bone or other pathological conditions.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}