Vitamin C Transporters and Their Implications in Carcinogenesis


Abstract
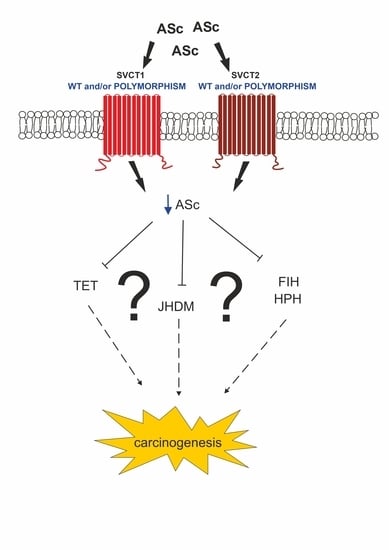
:
1. Vitamin C Properties and Redox Homeostasis
2. Vitamin C and Its Role in 2-OGDD Enzymes Activity
3. Vitamin C Transporters: Their Function and Distribution in the Human Body
4. Effects of Vitamin C on Cancer Cells
5. Vitamin C in Adjuvant Cancer Therapy
5.1. Hematological Malignancies
5.2. Breast Cancer
5.3. Melanoma
5.4. Glioma and Glioblastoma
5.5. Prostate Cancer
6. Impaired SVCT Function in Cancer
6.1. Polymorphisms of SVCT
6.2. Altered Expression of SVCT
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nishikimi, M.; Yagi, K. Biochemistry and molecular biology of ascorbic acid biosynthesis. Subcell. Biochem. 1996, 25, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Pan, Y.-H.; Zhang, Y.; Jones, G.; Zhang, S. Progressive pseudogenization: Vitamin C synthesis and its loss in bats. Mol. Biol. Evol. 2010, 28, 1025–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielski, B.H.J. Chemistry of ascorbic acid radicals. In Ascorbic Acid: Chemistry, Metabolism, and Uses; Advances in Chemistry; American Chemical Society: Washington, WA, USA, 1982; Volume 200, pp. 81–100. ISBN 978-0-8412-0632-8. [Google Scholar]
- Jackson, T.S.; Xu, A.; Vita, J.A.; Keaneyjr, J.F. Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations. Circ. Res. 1998, 83, 916–922. [Google Scholar] [CrossRef] [Green Version]
- Buettner, G.R. In the absence of catalytic metals ascorbate does not autoxidize at pH 7: Ascorbate as a test for catalytic metals. J. Biochem. Biophys. Methods 1988, 16, 27–40. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.-H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Ohtani, H.; Zhou, W.; Ørskov, A.D.; Charlet, J.; Zhang, Y.W.; Shen, H.; Baylin, S.B.; Liang, G.; Grønbæk, K.; et al. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc. Natl. Acad. Sci. USA 2016, 113, 10238–10244. [Google Scholar] [CrossRef] [Green Version]
- Clifton, I.J.; McDonough, M.A.; Ehrismann, D.; Kershaw, N.J.; Granatino, N.; Schofield, C.J. Structural studies on 2-oxoglutarate oxygenases and related double-stranded β-helix fold proteins. J. Inorg. Biochem. 2006, 100, 644–669. [Google Scholar] [CrossRef]
- A McDonough, M.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672. [Google Scholar] [CrossRef]
- Gorres, K.L.; Raines, R.T. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106–124. [Google Scholar] [CrossRef]
- Ono, R.; Taki, T.; Taketani, T.; Taniwaki, M.; Kobayashi, H.; Hayashi, Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res. 2002, 62, 4075–4080. [Google Scholar]
- Lorsbach, R.B.; Moore, J.; Mathew, S.; Raimondi, S.C.; Mukatira, S.T.; Downing, J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 2003, 17, 637–641. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate Induces Ten-Eleven Translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-Hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, R.; Mao, S.-Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic acid enhances tet-mediated 5-Methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef]
- Tsukada, Y.-I.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nat. Cell Biol. 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef]
- Miller, J.L.; Grant, P.A. The role of DNA methylation and histone modifications in transcriptional regulation in humans. Subcell. Biochem. 2013, 61, 289–317. [Google Scholar] [CrossRef]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef]
- Clark, S.; Melki, J. DNA methylation and gene silencing in cancer: Which is the guilty party? Oncogene 2002, 21, 5380–5387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffner, M.C.; Chaux, A.; Meeker, A.K.; Esopi, D.M.; Gerber, J.; Pellakuru, L.G.; Toubaji, A.; Argani, P.; Iacobuzio-Donahue, C.; Nelson, W.G.; et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011, 2, 627–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Asaoka, Y.; Ijichi, H.; Nagae, G.; Yoshida, H.; Aburatani, H.; Koike, K. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012, 103, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, O.; Li, R.; Hung, J.-H.; Chen, P.B.; Dong, X.; Ee, L.-S.; Weng, Z.; Rando, O.J.; Fazzio, T.G. Mbd3/NURD complex regulates expression of 5-Hydroxymethylcytosine marked genes in embryonic stem cells. Cell 2011, 147, 1498–1510. [Google Scholar] [CrossRef] [Green Version]
- Andricovich, J.; Kai, Y.; Tzatsos, A. Lysine-specific histone demethylases in normal and malignant hematopoiesis. Exp. Hematol. 2016, 44, 778–782. [Google Scholar] [CrossRef] [Green Version]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation inTET2in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Kosmider, O.; Gelsi-Boyer, V.; Ciudad, M.; Racoeur, C.; Jooste, V.; Vey, N.; Quesnel, B.; Fenaux, P.; Bastie, J.-N.; Beyne-Rauzy, O.; et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 2009, 94, 1676–1681. [Google Scholar] [CrossRef] [Green Version]
- Brahimi-Horn, M.C.; Pouysségur, J. HIF at a glance. J. Cell Sci. 2009, 122, 1055–1057. [Google Scholar] [CrossRef] [Green Version]
- Postovit, L.-M.; Abbott, D.E.; Payne, S.L.; Wheaton, W.W.; Margaryan, N.V.; Sullivan, R.; Jansen, M.K.; Csiszar, K.; Hendrix, M.J.; Kirschmann, D.A. Hypoxia/reoxygenation: A dynamic regulator of lysyl oxidase-facilitated breast cancer migration. J. Cell. Biochem. 2008, 103, 1369–1378. [Google Scholar] [CrossRef]
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal. 2007, 9, 1221–1236. [Google Scholar] [CrossRef]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Signal transduction to hypoxia-inducible factor 1. Biochem. Pharmacol. 2002, 64, 993–998. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Downes, N.L.; Laham-Karam, N.; Kaikkonen-Määttä, M.; Ylä-Herttuala, S. Differential but complementary HIF1α and HIF2α transcriptional regulation. Mol. Ther. 2018, 26, 1735–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Reevaluation of prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1978, 75, 4538–4542. [Google Scholar] [CrossRef] [Green Version]
- Creagan, E.T.; Moertel, C.G.; O’Fallon, J.R.; Schutt, A.J.; O’Connell, M.J.; Rubin, J.; Frytak, S. Failure of High-Dose Vitamin C (Ascorbic Acid) Therapy to Benefit Patients with Advanced Cancer. A controlled trial. N. Engl. J. Med. 1979, 301, 687–690. [Google Scholar] [CrossRef]
- Moertel, C.G.; Fleming, T.R.; Creagan, E.T.; Rubin, J.; O’Connell, M.J.; Ames, M.M. High-Dose Vitamin C versus Placebo in the Treatment of Patients with Advanced Cancer Who Have Had No Prior Chemotherapy. A randomized double-blind comparison. N. Engl. J. Med. 1985, 312, 137–141. [Google Scholar] [CrossRef]
- Levine, M.; Conry-Cantilena, C.; Wang, Y.; Welch, R.W.; Washko, P.W.; Dhariwal, K.R.; Park, J.B.; Lazarev, A.; Graumlich, J.F.; King, J.; et al. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc. Natl. Acad. Sci. USA 1996, 93, 3704–3709. [Google Scholar] [CrossRef] [Green Version]
- Graumlich, J.F.; Ludden, T.M.; Conry-Cantilena, C.; Cantilena, J.L.R.; Wang, Y.; Levine, M. Pharmacokinetic model of ascorbic acid in healthy male volunteers during depletion and repletion. Pharm. Res. 1997, 14, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Wang, Y.; Padayatty, S.J.; Morrow, J. A new recommended dietary allowance of vitamin C for healthy young women. Proc. Natl. Acad. Sci. USA 2001, 98, 9842–9846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukaguchi, H.; Tokui, T.; MacKenzie, B.; Berger, U.V.; Chen, X.-Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nat. Cell Biol. 1999, 399, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.X. Regulation of Vitamin C transport. Annu. Rev. Nutr. 2005, 25, 105–125. [Google Scholar] [CrossRef]
- Godoy, A.; Ormazabal, V.; Moraga-Cid, G.; Zúñiga, F.A.; Sotomayor, P.; Barra, V.; Vasquez, O.; Montecinos, V.; Mardones, L.; Guzmán, C.; et al. Mechanistic Insights and Functional Determinants of the Transport Cycle of the Ascorbic Acid Transporter SVCT2. Activation by sodium and absolute dependence on bivalent cations. J. Biol. Chem. 2006, 282, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Daruwala, R.; Song, J.; Koh, W.S.; Rumsey, S.C.; Levine, M. Cloning and functional characterization of the human sodium-dependent vitamin C transporters hSVCT1 and hSVCT2. FEBS Lett. 1999, 460, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Rivas, C.I.; Zúñiga, F.A.; Salas-Burgos, A.; Mardones, L.; Ormazabal, V.; Vera, J.C. Vitamin C transporters. J. Physiol. Biochem. 2008, 64, 357–375. [Google Scholar] [CrossRef]
- A Stratakis, C.; E Taymans, S.; Daruwala, R.; Song, J.; Levine, M. Mapping of the human genes (SLC23A2 and SLC23A1) coding for vitamin C transporters 1 and 2 (SVCT1 and SVCT2) to 5q23 and 20p12, respectively. J. Med. Genet. 2000, 37, E20. [Google Scholar] [CrossRef] [Green Version]
- Bürzle, M.; Suzuki, Y.; Ackermann, D.; Miyazaki, H.; Maeda, N.; Clémençon, B.; Burrier, R.; Hediger, M.A. The sodium-dependent ascorbic acid transporter family SLC23. Mol. Asp. Med. 2013, 34, 436–454. [Google Scholar] [CrossRef]
- Savini, I.; Rossi, A.; Pierro, C.; Avigliano, L.; Catani, M.V. SVCT1 and SVCT2: Key proteins for vitamin C uptake. Amino Acids 2007, 34, 347–355. [Google Scholar] [CrossRef]
- Corpe, C.P.; Tu, H.; Eck, P.K.; Wang, J.; Faulhaber-Walter, R.; Schnermann, J.; Margolis, S.; Padayatty, S.; Sun, H.; Wang, Y.; et al. Vitamin C transporter Slc23a1 links renal reabsorption, vitamin C tissue accumulation, and perinatal survival in mice. J. Clin. Investig. 2010, 120, 1069–1083. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Dutta, B.; Huang, W.; DeVoe, L.D.; Leibach, F.H.; Ganapathy, V.; Prasad, P.D. Human Na+-dependent vitamin C transporter 1 (hSVCT1): Primary structure, functional characteristics and evidence for a non-functional splice variant. Biochim. Biophys. Acta 1999, 1461, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.; Chen, A.; Hoogstraten-Miller, S.; Miller, G.F.; Kwon, O.; Levine, M.; Guttentag, S.H.; et al. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 2002, 8, 514–517. [Google Scholar] [CrossRef]
- Parker, W.H.; Qu, Z.-C.; May, J.M. Ascorbic acid transport in brain microvascular pericytes. Biochem. Biophys. Res. Commun. 2015, 458, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997, 272, 18982–18989. [Google Scholar] [CrossRef] [Green Version]
- Rumsey, S.C.; Daruwala, R.; Al-Hasani, H.; Zarnowski, M.J.; Simpson, I.A.; Levine, M. Dehydroascorbic acid transport by GLUT4 in xenopus oocytes and isolated rat adipocytes. J. Biol. Chem. 2000, 275, 28246–28253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corpe, C.P.; Eck, P.K.; Wang, J.; Al-Hasani, H.; Levine, M. Intestinal Dehydroascorbic Acid (DHA) transport mediated by the facilitative sugar transporters, GLUT2 and GLUT8*. J. Biol. Chem. 2013, 288, 9092–9101. [Google Scholar] [CrossRef] [Green Version]
- Linster, C.L.; Schaftingen, E.V. Vitamin C. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Washko, P.W.; Wang, Y.; Levine, M. Ascorbic acid recycling in human neutrophils. J. Biol. Chem. 1993, 268, 15531–15535. [Google Scholar]
- Litwack, G. Chapter 6—Insulin and sugars. In Human Biochemistry; Litwack, G., Ed.; Academic Press: Boston, MA, USA, 2018; pp. 131–160. ISBN 978-0-12-383864-3. [Google Scholar]
- Agus, D.B.; Gambhir, S.S.; Pardridge, W.M.; Spielholz, C.; Baselga, J.; Vera, J.C.; Golde, D.W. Vitamin C Crosses the Blood-Brain Barrier in the Oxidized Form through the Glucose Transporters. Available online: https://www.jci.org/articles/view/119832/pdf (accessed on 26 August 2020).
- Nualart, F.J.; Rivas, C.I.; Montecinos, V.P.; Godoy, A.S.; Guaiquil, V.H.; Golde, D.W.; Vera, J.C. Recycling of Vitamin C by a Bystander Effect. J. Biol. Chem. 2002, 278, 10128–10133. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.; Li, H.; Wang, Y.; Niyyati, M.; Wang, Y.; Leshin, J.; Levine, M. Low Red Blood Cell Vitamin C Concentrations Induce Red Blood Cell Fragility: A Link to Diabetes Via Glucose, Glucose Transporters, and Dehydroascorbic Acid. EBioMedicine 2015, 2, 1735–1750. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-R.; Park, S.-W.; Cho, H.-J.; Chae, K.-A.; Sung, J.-M.; Kim, J.-S.; Landowski, C.P.; Sun, D.; El-Aty, A.M.A.; Amidon, G.L.; et al. Comparative gene expression profiles of intestinal transporters in mice, rats and humans. Pharmacol. Res. 2007, 56, 224–236. [Google Scholar] [CrossRef]
- Peña, E.; Roa, F.J.; Inostroza, E.; Sotomayor, K.; González, M.; Gutierrez-Castro, F.A.; Maurin, M.; Sweet, K.; Labrousse, C.; Gatica, M.; et al. Increased expression of mitochondrial sodium-coupled ascorbic acid transporter-2 (mitSVCT2) as a central feature in breast cancer. Free. Radic. Biol. Med. 2019, 135, 283–292. [Google Scholar] [CrossRef]
- Roa, F.J.; Peña, E.; Inostroza, E.; Sotomayor, K.; González, M.; Gutierrez-Castro, F.A.; Maurin, M.; Sweet, K.; Labrousse, C.; Gatica, M.; et al. Data on SVCT2 transporter expression and localization in cancer cell lines and tissues. Data Brief 2019, 25, 103972. [Google Scholar] [CrossRef] [PubMed]
- Mayland, C.R.; Bennett, M.I.; Allan, K. Vitamin C deficiency in cancer patients. Palliat. Med. 2005, 19, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.-A.; Kim, B.-S.; Yu, R. Serum antioxidative vitamin levels and lipid peroxidation in gastric carcinoma patients. Cancer Lett. 1999, 136, 89–93. [Google Scholar] [CrossRef]
- Cha, J.; Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Ascorbate depletion increases growth and metastasis of melanoma cells in vitamin C deficient mice. Exp. Oncol. 2011, 33, 226–230. [Google Scholar] [PubMed]
- Cha, J.; Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Ascorbate supplementation inhibits growth and metastasis of B16FO melanoma and 4T1 breast cancer cells in vitamin C-deficient mice. Int. J. Oncol. 2013, 42, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Galloway, S.P.; McMillan, D.C.; Sattar, N. Effect of the inflammatory response on trace element and vitamin status. Ann. Clin. Biochem. 2000, 37, 289–297. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [Green Version]
- Takemura, Y.; Satoh, M.; Satoh, K.; Hamada, H.; Sekido, Y.; Kubota, S. High dose of ascorbic acid induces cell death in mesothelioma cells. Biochem. Biophys. Res. Commun. 2010, 394, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uetaki, M.; Tabata, S.; Nakasuka, F.; Soga, T.; Tomita, M. Metabolomic alterations in human cancer cells by vitamin C-induced oxidative stress. Sci. Rep. 2015, 5, 13896. [Google Scholar] [CrossRef] [Green Version]
- Otto, A.M. Warburg effect(s)—A biographical sketch of Otto Warburg and his impacts on tumor metabolism. Cancer Metab. 2016, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.V.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.D.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawada, K.; Nakamoto, Y.; Kawada, M.; Hida, K.; Matsumoto, T.; Murakami, T.; Hasegawa, S.; Togashi, K.; Sakai, Y. Relationship between 18F-Fluorodeoxyglucose accumulation and KRAS/BRAF mutations in colorectal cancer. Clin. Cancer Res. 2012, 18, 1696–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nat. Cell Biol. 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Sinnberg, T.; Noor, S.; Venturelli, S.; Berger, A.; Schuler, P.; Garbe, C.; Busch, C. The ROS-induced cytotoxicity of ascorbate is attenuated by hypoxia and HIF-1alpha in the NCI60 cancer cell lines. J. Cell. Mol. Med. 2014, 18, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nat. Cell Biol. 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, Q.; Zeng, F.; Li, W.; He, Z.; Chen, W.; Zhu, W.; Zhang, B. The prognostic value of global DNA hypomethylation in cancer: A meta-analysis. PLoS ONE 2014, 9, e106290. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Hou, L.; Zhou, M.-J.; Ma, Z.; Lin, D.-L.; Wu, L.; Ge, Y. Aberrant NDRG1 methylation associated with its decreased expression and clinicopathological significance in breast cancer. J. Biomed. Sci. 2013, 20, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starczak, M.; Zarakowska, E.; Modrzejewska, M.; Dziaman, T.; Szpila, A.; Linowiecka, K.; Guz, J.; Szpotan, J.; Gawronski, M.; Labejszo, A.; et al. In vivo evidence of ascorbate involvement in the generation of epigenetic DNA modifications in leukocytes from patients with colorectal carcinoma, benign adenoma and inflammatory bowel disease. J. Transl. Med. 2018, 16, 204. [Google Scholar] [CrossRef]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nat. Cell Biol. 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Gustafson, C.B.; Yang, C.; Dickson, K.M.; Shao, H.; Van Booven, D.; Harbour, J.W.; Liu, Z.-J.; Wang, G. Epigenetic reprogramming of melanoma cells by vitamin C treatment. Clin. Epigenet. 2015, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Modrzejewska, M.; Gawronski, M.; Skonieczna, M.; Zarakowska, E.; Starczak, M.; Foksinski, M.; Rzeszowska-Wolny, J.; Gackowski, D.; Olinski, R. Vitamin C enhances substantially formation of 5-hydroxymethyluracil in cellular DNA. Free. Radic. Biol. Med. 2016, 101, 378–383. [Google Scholar] [CrossRef]
- Peng, D.; Ge, G.; Gong, Y.; Zhan, Y.; He, S.; Guan, B.; Li, Y.; Xu, Z.; Hao, H.; He, Z.-S.; et al. Vitamin C increases 5-hydroxymethylcytosine level and inhibits the growth of bladder cancer. Clin. Epigenet. 2018, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hong, J.; Han, H.; Park, J.; Kim, D.; Park, H.; Ko, M.; Koh, Y.; Shin, D.-Y.; Yoon, S.-S. Decreased vitamin C uptake mediated by SLC2A3 promotes leukaemia progression and impedes TET2 restoration. Br. J. Cancer 2020, 122, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Zhu, H.; Huang, J.; Zhu, Y.; Hong, M.; Zhu, H.; Zhang, J.; Li, S.; Yang, L.; Lian, Y.; et al. The synergy of Vitamin C with decitabine activates TET2 in leukemic cells and significantly improves overall survival in elderly patients with acute myeloid leukemia. Leuk. Res. 2018, 66, 1–7. [Google Scholar] [CrossRef]
- Pollard, H.B.; A Levine, M.; Eidelman, O.; Pollard, M. Pharmacological ascorbic acid suppresses syngeneic tumor growth and metastases in hormone-refractory prostate cancer. In Vivo 2010, 24, 249–255. [Google Scholar]
- Polireddy, K.; Dong, R.; Reed, G.; Yu, J.; Chen, P.; Williamson, S.; Violet, P.-C.; Pessetto, Z.; Godwin, A.K.; Fan, F.; et al. High dose parenteral ascorbate inhibited pancreatic cancer growth and metastasis: Mechanisms and a phase I/IIa study. Sci. Rep. 2017, 7, 17188. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Camarena, V.; Mustafi, S.; Wang, G. Vitamin C inhibits triple-negative breast cancer metastasis by affecting the expression of YAP1 and synaptopodin 2. Nutrients 2019, 11, 2997. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Huang, K.; Zhu, Y.; Wang, T.; Shan, Y.; Long, B.; Li, Y.; Chen, Q.; Wang, P.; Zhao, S.; et al. Vitamin C–dependent lysine demethylase 6 (KDM6)-mediated demethylation promotes a chromatin state that supports the endothelial-to-hematopoietic transition. J. Biol. Chem. 2019, 294, 13657–13670. [Google Scholar] [CrossRef]
- Wang, T.; Chen, K.; Zeng, X.; Yang, J.; Wu, Y.; Shi, X.; Qin, B.; Zeng, L.; Esteban, M.A.; Pan, G.; et al. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a Vitamin-C-Dependent manner. Cell Stem Cell 2011, 9, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Ebata, K.; Mesh, K.; Liu, S.; Bilenky, M.; Fekete, A.; Acker, M.G.; Hirst, M.; Garcia, B.A.; Ramalho-Santos, M. Vitamin C induces specific demethylation of H3K9me2 in mouse embryonic stem cells via Kdm3a/b. Epigenet. Chromatin 2017, 10, 36. [Google Scholar] [CrossRef]
- Yong-Hee, R.; Kim, M.; Kim, S.-Y.; Yi, S.-H.; Rhee, Y.-H.; Kim, T.; Lee, E.-H.; Park, C.-H.; Dixit, S.; Harrison, F.E.; et al. Vitamin C Facilitates Dopamine Neuron Differentiation in Fetal Midbrain Through TET1- and JMJD3-Dependent Epigenetic Control Manner. STEM CELLS 2015, 33, 1320–1332. [Google Scholar] [CrossRef] [Green Version]
- Hoffer, L.J.; Levine, M.; Assouline, S.; Melnychuk, D.; Padayatty, S.J.; Rosadiuk, K.; Rousseau, C.; Robitaille, L.; Miller, W.H. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann. Oncol. 2008, 19, 1969–1974. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Vissers, M.C.M.; Cook, J.S. The effect of intravenous vitamin c on cancer- and chemotherapy-related fatigue and quality of life. Front. Oncol. 2014, 4, 283. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Kang, J.; Choi, J.; Heo, S.; Lee, D.-H. The effect of high dose intravenous Vitamin C during radiotherapy on breast cancer patients’ neutrophil–lymphocyte ratio. J. Altern. Complement. Med. 2020, 26, 1039–1046. [Google Scholar] [CrossRef]
- Carr, A.; Cook, J. Intravenous Vitamin C for cancer therapy—Identifying the current gaps in our knowledge. Front. Physiol. 2018, 9, 1182. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Kimler, B.F.; Yi, S.Y.; Park, S.H.; Kim, K.; Jung, C.W.; Kim, S.H.; Lee, E.R.; Rha, M.; Kim, S.; et al. Depletion of l-ascorbic acid alternating with its supplementation in the treatment of patients with acute myeloid leukemia or myelodysplastic syndromes. Eur. J. Haematol. 2009, 83, 108–118. [Google Scholar] [CrossRef]
- Xia, J.; Xu, H.; Zhang, X.; Allamargot, C.; Coleman, K.L.; Nessler, R.; Frech, I.; Tricot, G.; Zhan, F. Multiple myeloma tumor cells are selectively killed by pharmacologically-dosed ascorbic acid. EBioMedicine 2017, 18, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, N.; Bhagat, T.; Nieves, E.; Stenson, M.; Lawson, J.; Choudhary, G.S.; Habermann, T.; Nowakowski, G.; Singh, R.; Wu, X.; et al. Upregulation of TET activity with ascorbic acid induces epigenetic modulation of lymphoma cells. Blood Cancer J. 2017, 7, e587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.B.; Kakadia, P.M.; Wojcik, D.; Pemberton, L.; Browett, P.J.; Bohlander, S.; Vissers, M.C.M. Clinical remission following ascorbate treatment in a case of acute myeloid leukemia with mutations in TET2 and WT1. Blood Cancer J. 2019, 9, 82. [Google Scholar] [CrossRef]
- Foster, M.N.; Carr, A.; Antony, A.; Peng, S.; Fitzpatrick, M. Intravenous Vitamin C Administration improved blood cell counts and health-related quality of life of patient with history of relapsed acute myeloid leukaemia. Antioxidants 2018, 7, 92. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.J.; Dachs, G.U.; Morrin, H.R.; Davey, V.C.; Robinson, B.A.; Vissers, M.C.M. Activation of the hypoxia pathway in breast cancer tissue and patient survival are inversely associated with tumor ascorbate levels. BMC Cancer 2019, 19, 307. [Google Scholar] [CrossRef] [PubMed]
- Sant, D.W.; Mustafi, S.; Gustafson, C.B.; Chen, J.; Slingerland, J.M.; Wang, G. Vitamin C promotes apoptosis in breast cancer cells by increasing TRAIL expression. Sci. Rep. 2018, 8, 5306. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.-H.; Wang, Q.-M.; Feng, L.-Y.; Ke, Y.-D.; Xu, Q.-Z.; Wei, A.-Y.; Zhang, C.; Ying, R.-B. High-dose vitamin C suppresses the invasion and metastasis of breast cancer cells via inhibiting epithelial-mesenchymal transition. OncoTargets Ther. 2019, 12, 7405–7413. [Google Scholar] [CrossRef] [Green Version]
- Harris, H.R.; Orsini, N.; Wolk, A. Vitamin C and survival among women with breast cancer: A Meta-analysis. Eur. J. Cancer 2014, 50, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Cadeau, C.; Fournier, A.; Mesrine, S.; Clavel-Chapelon, F.; Fagherazzi, G.; Boutron-Ruault, M.-C. Vitamin C supplement intake and postmenopausal breast cancer risk: Interaction with dietary vitamin C. Am. J. Clin. Nutr. 2016, 104, 228–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Chen, Y.; Qu, C.; Pan, Z.; Qin, Y.; Zhang, X.; Liu, W.; Li, D.; Zheng, Q. Vitamin C induces human melanoma A375 cell apoptosis via Bax- and Bcl-2-mediated mitochondrial pathways. Oncol. Lett. 2019, 18, 3880–3886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Yan, Y.; Ma, Y.; Yang, Y. Vitamin C at high concentrations induces cytotoxicity in malignant melanoma but promotes tumor growth at low concentrations. Mol. Carcinog. 2017, 56, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- Zbytek, B.; Peacock, D.L.; Seagroves, T.N.; Slominski, A.T. Putative role of HIF transcriptional activity in melanocytes and melanoma biology. Derm. Endocrinol. 2013, 5, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.T.; Kim, T.-K.; Brożyna, A.; Janjetovic, Z.; Brooks, D.; Schwab, L.; Skobowiat, C.; Jóźwicki, W.; Seagroves, T. The role of melanogenesis in regulation of melanoma behavior: Melanogenesis leads to stimulation of HIF-1α expression and HIF-dependent attendant pathways. Arch. Biochem. Biophys. 2014, 563, 79–93. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.P.; Miles, S.L. Ascorbic acid, but not dehydroascorbic acid increases intracellular vitamin C content to decrease Hypoxia Inducible Factor -1 alpha activity and reduce malignant potential in human melanoma. Biomed. Pharmacother. 2017, 86, 502–513. [Google Scholar] [CrossRef]
- Miles, S.L.; Fischer, A.P.; Joshi, S.S.; Niles, R.M. Ascorbic acid and ascorbate-2-phosphate decrease HIF activity and malignant properties of human melanoma cells. BMC Cancer 2015, 15, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryszawy, D.; Pudełek, M.; Catapano, J.; Ciarach, M.; Setkowicz, Z.; Konduracka, E.; Madeja, Z.; Czyż, J. High doses of sodium ascorbate interfere with the expansion of glioblastoma multiforme cells in vitro and in vivo. Life Sci. 2019, 232, 116657. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, X.; Tan, Y.; Qiu, L.; Fang, H.; Li, W. Association between Vitamin C intake and glioma risk: Evidence from a meta-analysis. Neuroepidemiology 2015, 44, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Baillie, N.; Carr, A.; Peng, S. The Use of Intravenous Vitamin C as a supportive therapy for a patient with glioblastoma multiforme. Antioxidants 2018, 7, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Mikirova, N.; Hunnunghake, R.; Scimeca, R.C.; Chinshaw, C.; Ali, F.; Brannon, C.; Riordan, N. High-dose intravenous vitamin c treatment of a child with neurofibromatosis Type 1 and optic pathway glioma: A case report. Am. J. Case Rep. 2016, 17, 774–781. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.-Y.; Qu, X.; Jiang, X.; Xu, Z.; Yang, Y.; Su, Q.; Wang, M.; Wu, H. Association between dietary Vitamin C intake and risk of prostate cancer: A meta-analysis involving 103,658 subjects. J. Cancer 2015, 6, 913–921. [Google Scholar] [CrossRef]
- Parent, M.-E.; Richard, H.; Rousseau, M.-C.; Trudeau, K. Vitamin C intake and risk of prostate cancer: The montreal PROtEuS study. Front. Physiol. 2018, 9, 1218. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sesso, H.D.; Glynn, R.J.; Christen, W.G.; Bubes, V.; Manson, J.E.; Buring, J.E.; Gaziano, J.M. Vitamin E and C supplementation and risk of cancer in men: Posttrial follow-up in the Physicians’ Health Study II randomized trial. Am. J. Clin. Nutr. 2014, 100, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timpson, N.J.; Forouhi, N.G.; A Brion, M.-J.; Harbord, R.M.; Cook, D.G.; Johnson, P.; McConnachie, A.; Morris, R.; Rodriguez, S.; Luan, J.; et al. Genetic variation at the SLC23A1 locus is associated with circulating concentrations of l-ascorbic acid (vitamin C): Evidence from 5 independent studies with >15,000 participants. Am. J. Clin. Nutr. 2010, 92, 375–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duell, E.J.; Lujan-Barroso, L.; Llivina, C.; Muñoz, X.; Jenab, M.; Boutron-Ruault, M.-C.; Clavel-Chapelon, F.; Racine, A.; Boeing, H.; Buijsse, B.; et al. Vitamin C transporter gene (SLC23A1 and SLC23A2) polymorphisms, plasma vitamin C levels, and gastric cancer risk in the EPIC cohort. Genes Nutr. 2013, 8, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Wright, M.E.; Andreotti, G.; Lissowska, J.; Yeager, M.; Zatonski, W.; Chanock, S.J.; Chow, W.-H.; Hou, L. Genetic variation in sodium-dependent ascorbic acid transporters and risk of gastric cancer in Poland. Eur. J. Cancer 2009, 45, 1824–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waring, A.J.; Drake, I.M.; Schorah, C.J.; White, K.L.; A Lynch, D.; Axon, A.T.; Dixon, M.F. Ascorbic acid and total vitamin C concentrations in plasma, gastric juice, and gastrointestinal mucosa: Effects of gastritis and oral supplementation. Gut 1996, 38, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Skibola, C.F.; Bracci, P.M.; Halperin, E.; Nieters, A.; Hubbard, A.; Paynter, R.A.; Skibola, D.R.; Agana, L.; Becker, N.; Tressler, P.; et al. Polymorphisms in the estrogen receptor 1 and Vitamin C and matrix metalloproteinase gene families are associated with susceptibility to lymphoma. PLoS ONE 2008, 3, e2816. [Google Scholar] [CrossRef] [Green Version]
- Casabonne, D.; Gracia, E.; Espinosa, A.; Bustamante, M.; Benavente, Y.; Robles, C.; Costas, L.; Alonso, E.; Gonzalez-Barca, E.; Tardón, A.; et al. Fruit and vegetable intake and vitamin C transporter gene (SLC23A2) polymorphisms in chronic lymphocytic leukaemia. Eur. J. Nutr. 2017, 56, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, H.C.; Peters, U.; Eck, P.K.; Welch, R.; Schoen, R.E.; Yeager, M.; Levine, M.; Hayes, R.B.; Chanock, S. Genetic variation in sodium-dependent Vitamin C Transporters SLC23A1 and SLC23A2 and risk of advanced colorectal adenoma. Nutr. Cancer 2008, 60, 652–659. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.A.; Marsit, C.J.; Christensen, B.C.; Houseman, E.A.; McClean, M.; Smith, J.F.; Bryan, J.T.; Posner, M.R.; Nelson, H.H.; Kelsey, K. Genetic variation in the vitamin C transporter, SLC23A2, modifies the risk of HPV16-associated head and neck cancer. Carcinogenesis 2009, 30, 977–981. [Google Scholar] [CrossRef]
- Hong, S.-W.; Lee, S.-H.; Moon, J.-H.; Hwang, J.J.; Kim, D.E.; Ko, E.; Kim, H.-S.; Cho, I.J.; Kang, J.-S.; Kim, J.-E.; et al. SVCT-2 in breast cancer acts as an indicator for L-ascorbate treatment. Oncogene 2012, 32, 1508–1517. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Chae, J.S.; Shin, H.; Shin, Y.; Song, H.; Kim, Y.; Yoo, B.C.; Roh, K.; Cho, S.; Kil, E.-J.; et al. Hormetic dose response to L-ascorbic acid as an anti-cancer drug in colorectal cancer cell lines according to SVCT-2 expression. Sci. Rep. 2018, 8, 11372. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Wang, C.; Fang, T.; Li, T.; Lv, G.; Han, Q.; Yang, W.; Wang, H.-Y. Vitamin C preferentially kills cancer stem cells in hepatocellular carcinoma via SVCT-2. NPJ Precis. Oncol. 2018, 2, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Lv, H.; Yang, W.; Li, T.; Fang, T.; Lv, G.; Han, Q.; Dong, L.; Jiang, T.; Jiang, B.; et al. SVCT-2 determines the sensitivity to ascorbate-induced cell death in cholangiocarcinoma cell lines and patient derived xenografts. Cancer Lett. 2017, 398, 1–11. [Google Scholar] [CrossRef]
- Lièvre, A.; Bachet, J.-B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.-F.; Côté, J.-F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.-A.; Lee, D.-H.; Moon, J.-H.; Hong, S.-W.; Shin, J.-S.; Hwang, I.Y.; Shin, Y.J.; Kim, J.H.; Gong, E.-Y.; Kim, S.-M.; et al. L-Ascorbic acid can abrogate SVCT-2-dependent cetuximab resistance mediated by mutant KRAS in human colon cancer cells. Free. Radic. Biol. Med. 2016, 95, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Salazar, K.; Martinez, M.; Ulloa, V.; Bertinat, R.; Martínez, F.; Jara, N.; Espinoza, F.; Bongarzone, E.R.; Nualart, F. SVCT2 overexpression in neuroblastoma cells induces cellular branching that is associated with ERK signaling. Mol. Neurobiol. 2016, 53, 6668–6679. [Google Scholar] [CrossRef] [PubMed]
- Eck, P.K.; Erichsen, H.C.; Taylor, V.J.G.; Yeager, M.; Hughes, A.L.; Levine, M.; Chanock, S.J. Comparison of the genomic structure and variation in the two human sodium-dependent vitamin C transporters, SLC23A1 and SLC23A2. Qual. Life Res. 2004, 115, 285–294. [Google Scholar] [CrossRef]



{kind=link}
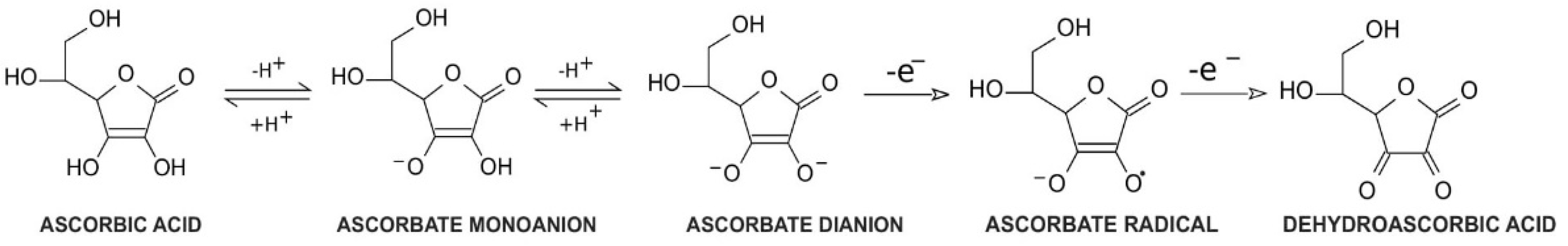{kind=link}
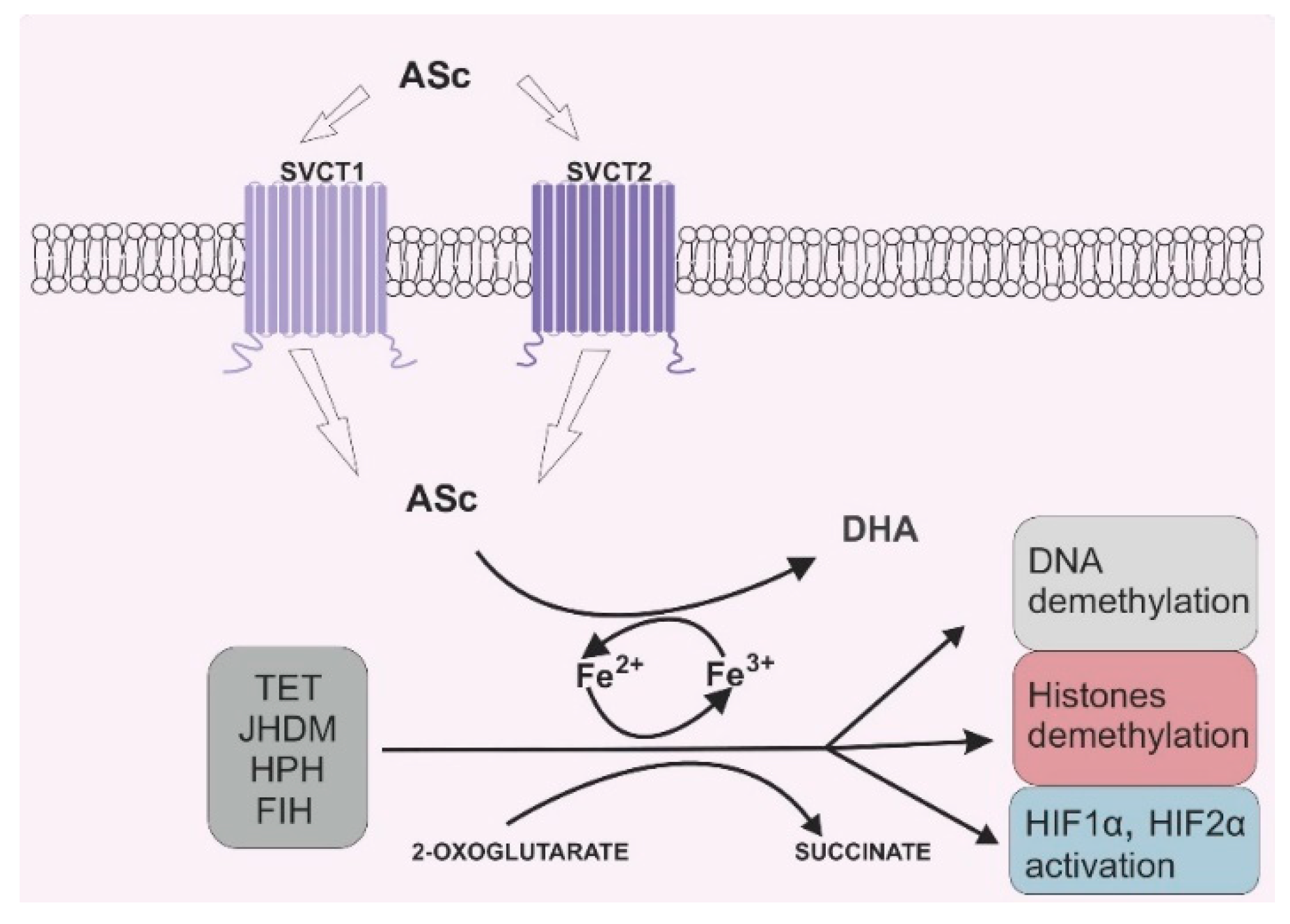{kind=link}
| SVCT Type | SNP ID | Effect | References |
|---|---|---|---|
| SVCT1 | SLC23A1 rs33972313 | decrease in circulating vitamin C | Timpson et al., 2010 [132] |
| 40–50% reduction in ascorbate accumulation in murine cells | Corpe et al., 2010 [52] | ||
| 24% decrease in vitamin C’s plasma concentration | Duell et al., 2013 [133] | ||
| SLC23A1 rs11950646 | 10–13% decrease in vitamin C’s plasma concentration | Duell et al., 2013 [133] | |
| SLC23A1 rs6596472 | higher risk of follicular lymphoma | Skibola et al., 2008 [136] | |
| SLC23A1 rs11950646 | higher risk of follicular lymphoma | Skibola et al., 2008 [136] | |
| SVCT2 | SLC23A2 rs6053005 | 24% increase in vitamin C’s plasma concentration | Duell et al., 2013 [133] |
| SLC23A2 rs6133175 | 24% increase in vitamin C’s plasma concentration | Duell et al., 2013 [133] | |
| SLC23A2 rs6116568 | higher risk of gastric cancer | Duell et al., 2013 [133] | |
| SLC23A2 rs12479919 | higher risk of gastric cancer | Wright et al., 2009 [134] | |
| SLC23A2 rs1776948 | higher risk of follicular lymphoma | Skibola et al., 2008 [136] | |
| higher risk of chronic lymphocytic leukemia (CLL) | Skibola et al., 2008 [136] Casabonne et al., 2017 [137] | ||
| SLC23A2 rs6133175 | higher risk of chronic lymphocytic leukemia (CLL) | Skibola et al., 2008 [136] Casabonne et al., 2017 [137] | |
| SLC23A2 rs1715364 | higher risk of chronic lymphocytic leukemia (CLL) | Skibola et al., 2008 [136] | |
| SLC23A2 rs4987219 | higher risk of colorectal adenoma | Erichsen et al., 2008 [138] | |
| plausible modifying factor of HPV16- associated head and neck cancer | Chen et al., 2009 [139] | ||
| SLC23A2 rs1110277 | higher risk of colorectal adenoma | Erichsen et al., 2008 [138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linowiecka, K.; Foksinski, M.; Brożyna, A.A. Vitamin C Transporters and Their Implications in Carcinogenesis. Nutrients 2020, 12, 3869. https://doi.org/10.3390/nu12123869
Linowiecka K, Foksinski M, Brożyna AA. Vitamin C Transporters and Their Implications in Carcinogenesis. Nutrients. 2020; 12(12):3869. https://doi.org/10.3390/nu12123869
Chicago/Turabian StyleLinowiecka, Kinga, Marek Foksinski, and Anna A. Brożyna. 2020. "Vitamin C Transporters and Their Implications in Carcinogenesis" Nutrients 12, no. 12: 3869. https://doi.org/10.3390/nu12123869
APA StyleLinowiecka, K., Foksinski, M., & Brożyna, A. A. (2020). Vitamin C Transporters and Their Implications in Carcinogenesis. Nutrients, 12(12), 3869. https://doi.org/10.3390/nu12123869