Multi-Tissue DNA Methylation Remodeling at Mitochondrial Quality Control Genes According to Diet in Rat Aging Models

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. DNA and mRNA Extraction
2.3. Primer Design for EpiTYPER Assay
2.4. Bisulfite Treatment and PCR Conditions
2.5. Dephosphorylation of Unincorporated Deoxynucleosidetriphosphates and in vitro Transcription and RNaseA Cleavage
2.6. Mass Spectrometry
2.7. Expression Profile Analysis of rat Polg, Polg2, Tfam, Fis1, and Opa1 Genes
2.8. Quantification of mtDNA Copy Number
2.9. Statistical Analyses
3. Results
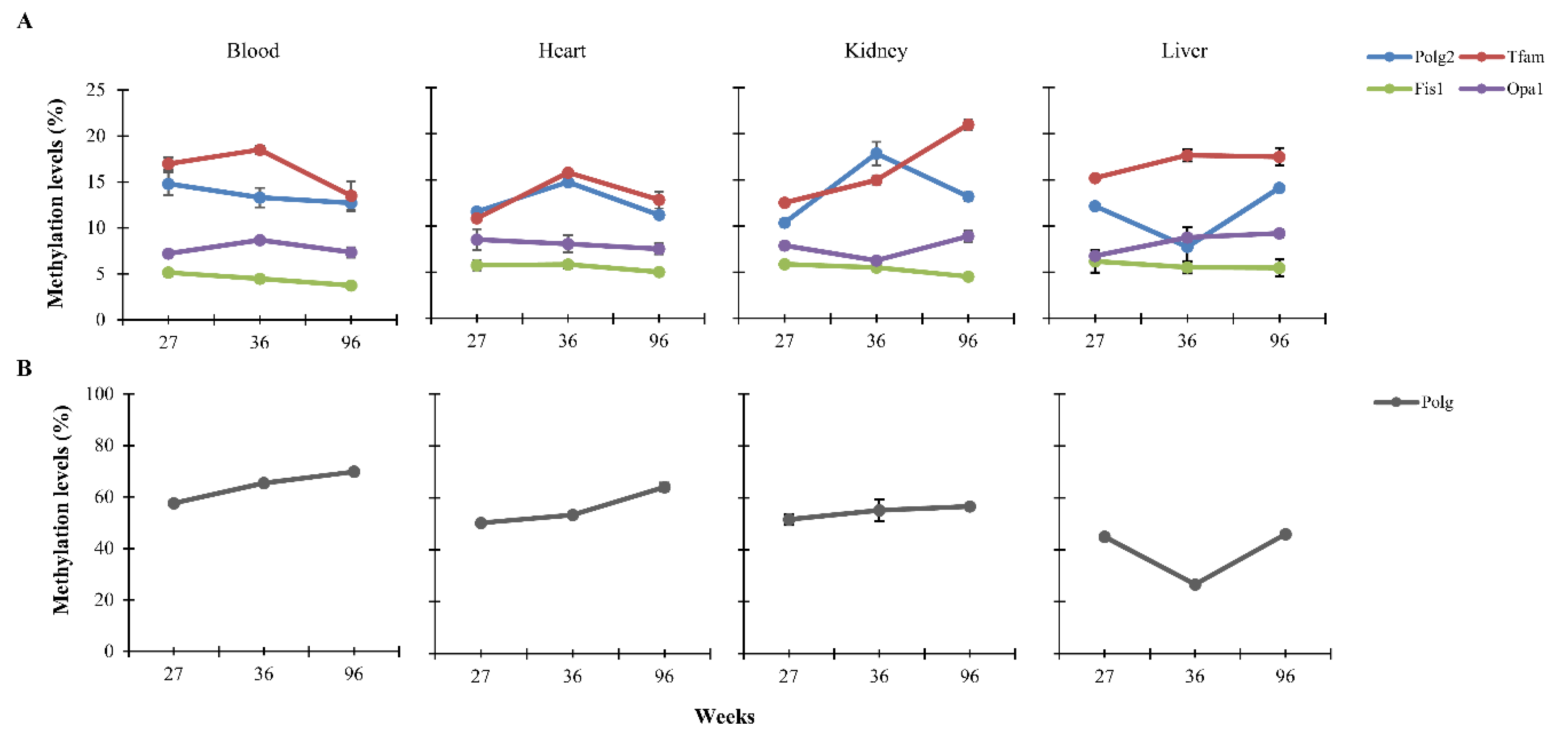
3.1. CpG Methylation Levels of Polg, Polg2, Tfam, Fis1, and Opa1 in Tissues from Rats of Different Ages
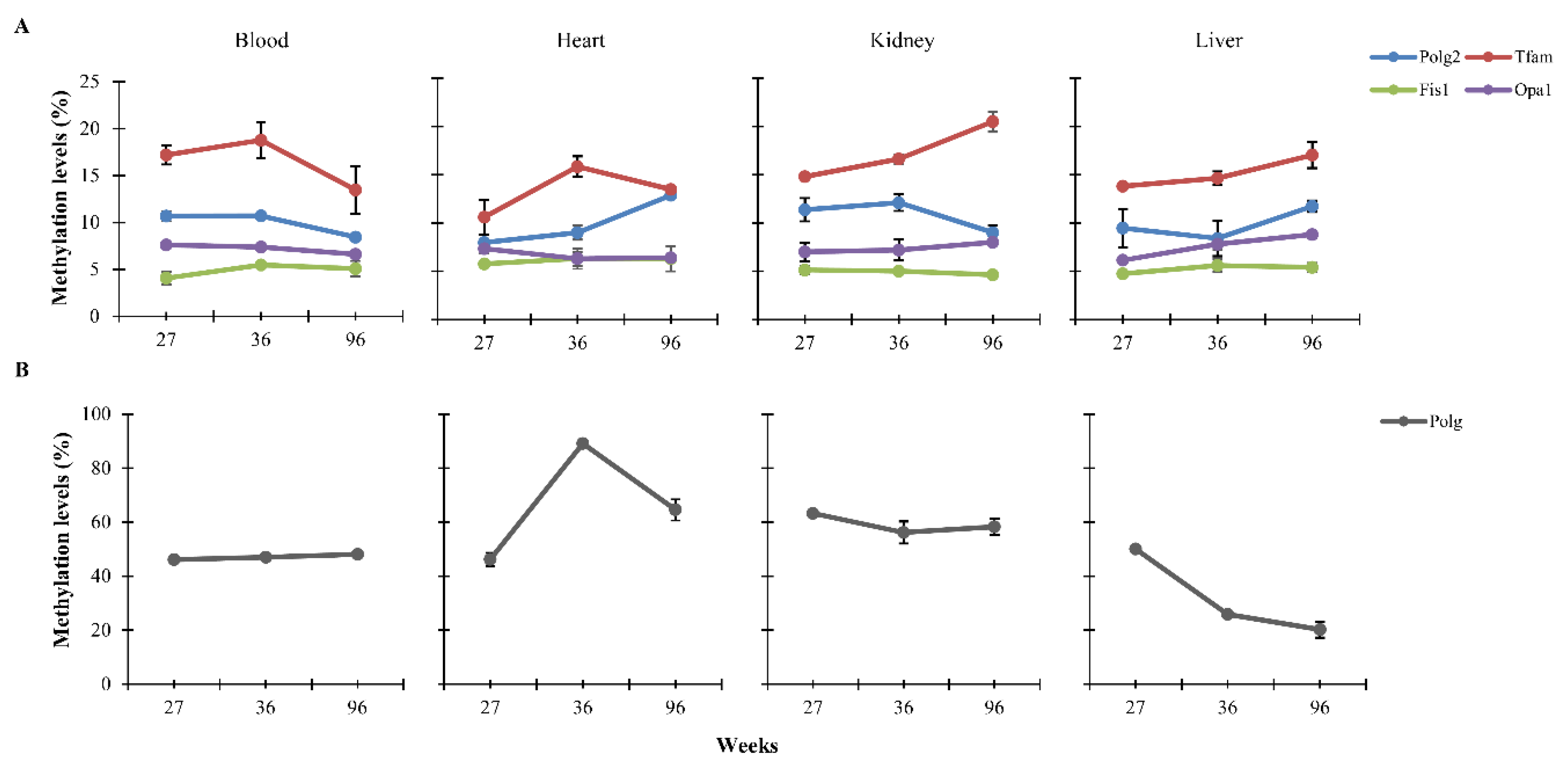
3.2. Dietary Effects on CpG Methylation Levels of Polg, Polg2, Tfam, Fis1, and Opa1 in Tissues from Rats of Different Ages
3.3. Correlation among Methylation, mRNA Levels and mtDNA Copy Number
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Loeb, L.A.; Wallace, D.C.; Martin, G.M. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 18769–18770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Trabucco, S.E.; Zhang, H. Oxidative stress, mitochondrial dysfunction and the mitochondria theory of aging. Interdiscip. Top. Gerontol. 2014, 39, 86–107. [Google Scholar] [PubMed]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial quality control: An integrated network of pathways. Trends Biochem. Sci. 2012, 37, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Yu, S.B.; Pekkurnaz, G. Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. J. Mol. Biol. 2018, 430, 3922–3941. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.N.; Zhang, Y.; Ren, J. Mitophagy, Mitochondrial Dynamics, and Homeostasis in Cardiovascular Aging. Oxid Med. Cell Longev. 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Molina, A.J.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.C.; Di, B.G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Las, G.; Serada, S.B.; Wikstrom, J.D.; Twig, G.; Shirihai, O.S. Fatty acids suppress autophagic turnover in beta-cells. J. Biol. Chem. 2011, 286, 42534–42544. [Google Scholar] [CrossRef] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Hesselink, M.K.; Schrauwen-Hinderling, V.; Schrauwen, P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 266–271. [Google Scholar] [CrossRef]
- Richardson, D.K.; Kashyap, S.; Bajaj, M.; Cusi, K.; Mandarino, S.J.; Finlayson, J.; DeFronzo, R.A.; Jenkinson, C.P.; Mandarino, L.J. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J. Biol. Chem. 2005, 280, 10290–10297. [Google Scholar] [CrossRef] [Green Version]
- Sparks, L.M.; Xie, H.; Koza, R.A.; Mynatt, R.; Hulver, M.W.; Bray, G.A.; Smith, S.R. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes 2005, 54, 1926–1933. [Google Scholar] [CrossRef] [Green Version]
- Hoeks, J.; Hesselink, M.; Russell, A.; Mensink, M.; Saris, W.; Mensink, R.; Saris, W.H.; Mensink, R.P.; Schrauwen, P. Peroxisome proliferator-activated receptor-γ coactivator-1 and insulin resistance: Acute effect of fatty acids. Diabetologia 2006, 49, 2419–2426. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Jiang, Z.; Sun, Z.; Wang, L.; Zhao, G.; Hassan, H.M.; Fan, S.; Zhou, W.; Han, S.; Zhang, L.; et al. Mitochondrial dysfunction and inhibition of myoblast differentiation in mice with high-fat-diet-induced pre-diabetes. J. Cell. Physiol. 2019, 234, 7510–7523. [Google Scholar] [CrossRef]
- Palacios, O.M.; Carmona, J.J.; Michan, S.; Chen, K.Y.; Manabe, Y.; Ward, J.L., III; Goodyear, L.J.; Tong, Q. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging 2009, 1, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.E.; Tippetts, T.S.; Brassfield, E.S.; Tucker, B.J.; Ockey, A.; Swensen, A.C.; Anthonymuthu, T.S.; Washburn, T.D.; Kane, D.A.; Prince, J.T.; et al. Mitochondrial fission mediates ceramide-induced metabolic disruption in skeletal muscle. Biochem. J. 2013, 456, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motawi, T.M.K.; Hashem, R.M.; Rashed, L.A.; El-Razek, S.M.A. Comparative study between the effect of the peroxisome proliferator activated receptor-α ligands fenofibrate and n-3 polyunsaturated fatty acids on activation of 5′-AMP-activated protein kinase-α1 in high-fat fed rats. J. Pharm. Pharmacol. 2009, 61, 1339–1346. [Google Scholar] [CrossRef]
- Lanza, I.R.; Blachnio-Zabielska, A.; Johnson, M.L.; Schimke, J.M.; Jakaitis, D.R.; Lebrasseur, N.K.; Jensen, M.D.; Sreekumaran Nair, K.; Zabielski, P. Author informationInfluence of fish oil on skeletal muscle mitochondrial energetics and lipid metabolites during high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1391–E1403. [Google Scholar] [CrossRef]
- Bach, D.; Naon, D.; Pich, S.; Soriano, F.X.; Vega, N.; Rieusset, J.; Laville, M.; Guillet, C.; Boirie, Y.; Wallberg-Henriksson, H.; et al. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: Effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes 2005, 54, 2685–2693. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, L.; Sica, R.; Mollica, M.P.; Putti, R. High-lard and high-fish oil diets differ in their effects on insulin resistance development, mitochondrial morphology and dynamic behaviour in rat skeletal muscle. Food Nutr. Sci. 2013, 4, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, L.; Mollica, M.P.; Donizzetti, I.; Gifuni, G.; Sica, R.; Pignalosa, A.; Cavaliere, G.; Gaita, M.; De Filippo, C.; Zorzano, A.; et al. High-lard and high-fish-oil diets differ in their effects on function and dynamic behaviour of rat hepatic mitochondria. PLoS ONE 2014, 9, e92753. [Google Scholar] [CrossRef] [Green Version]
- Casanova, E.; Baselga-Escudero, L.; Ribas-Latre, A.; Arola-Arnal, A.; Bladé, C.; Arola, L.; Salvadó, M.J. Epigallocatechin gallate counteracts oxidative stress in docosahexaenoxic acid-treated myocytes. Biochim. Biophys. Acta 2014, 1837, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef]
- Lopez-Lluch, G.; Hunt, N.; Jones, B.; Zhu, M.; Jamieson, H.; Hilmer, S.; Cascajo, M.V.; Allard, J.; Ingram, D.K.; Navas, P.; et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. USA 2006, 103, 1768–1773. [Google Scholar] [CrossRef] [Green Version]
- Civitarese, A.E.; Carling, S.; Heilbronn, L.K.; Hulver, M.H.; Ukropcova, B.; Deutsch, W.A.; Smith, S.R.; Ravussin, E.; CALERIE Pennington Team. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007, 4, e76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, C.R.; Han, D.; Higashida, K.; Kim, S.H.; Holloszy, J.O. Does calorie restriction induce mitochondrial biogenesis? A reevaluation. FASEB J. 2011, 25, 785–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khraiwesh, H.; López-Domínguez, J.A.; López-Lluch, G.; Navas, P.; De Cabo, R.; Ramsey, J.J.; Villalba, J.M.; González-Reyes, J.A. Alterations of ultrastructural and fission/fusion markers in hepatocyte mitochondria from mice following calorie restriction with different dietary fats. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1023–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, L.M.; Redman, L.M.; Conley, K.E.; Harper, M.; Yi, F.; Hodges, A.; Eroshkin, A.; Costford, S.R.; Gabriel, M.E.; Shook, C.; et al. Effects of 12 months of caloric restriction on muscle mitochondrial function in healthy individuals. J. Clin. Endocrinol. Metabol. 2016, 102, 111–121. [Google Scholar] [CrossRef]
- Menshikova, E.V.; Ritov, V.B.; Dube, J.J.; Amati, F.; Stefanovic-Racic, M.; Toledo, F.G.; Coen, P.M.; Goodpaster, B.H. Calorie restriction-induced weight loss and exercise have differential effects on skeletal muscle mitochondria despite similar effects on insulin sensitivity. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 73, 81–87. [Google Scholar] [CrossRef]
- Mulvey, L.; Sands, W.A.; Salin, K.; Carr, A.E.; Selman, C. Disentangling the effect of dietary restriction on mitochondrial function using recombinant inbred mice. Mol. Cell. Endocrinol. 2017, 455, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Ruetenik, A.; Barrientos, A. Dietary restriction, mitochondrial function and aging: From yeast to humans. Biochim. Biophys Acta. 2015, 1847, 1434–1447. [Google Scholar] [CrossRef] [Green Version]
- Soultoukis, G.A.; Partridge, L. Dietary Protein, Metabolism, and Aging. Annu Rev. Biochem. 2016, 85, 5–34. [Google Scholar] [CrossRef] [Green Version]
- Cummings, N.E.; Lamming, D.W. Regulation of metabolic health and aging by nutrient-sensitive signaling pathways. Mol. Cell Endocrinol. 2017, 455, 13–22. [Google Scholar] [CrossRef]
- Guarasci, F.; D’Aquila, P.; Mandalà, M.; Garasto, S.; Lattanzio, F.; Corsonello, A.; Passarino, G.; Bellizzi, D. Aging and nutrition induce tissue-specific changes on global DNA methylation status in rats. Mech Ageing Dev. 2018, 174, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Gensous, N.; Franceschi, C.; Santoro, A.; Milazzo, M.; Garagnani, P.; Bacalini, M.G. The Impact of Caloric Restriction on the Epigenetic Signatures of Aging. Int. J. Mol. Sci. 2019, 20, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taormina, G.; Russo, A.; Latteri, M.A.; Mirisola, M.G. Mitochondrion at the Crossroad Between Nutrients and Epigenome. Front. Endocrinol. (Lausanne) 2019, 4. [Google Scholar] [CrossRef]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Ibebunjo, C.; Chick, J.M.; Kendall, T.; Eash, J.K.; Li, C.; Zhang, Y.; Vickers, C.; Wu, Z.; Clarke, B.A.; Shi, J.; et al. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell Biol. 2013, 33, 194–212. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Pesce, V.; Fracasso, F.; Joseph, A.M.; Leeuwenburgh, C.; Lezza, A.M. Aging and calorie restriction oppositely affect mitochondrial biogenesis through TFAM binding at both origins of mitochondrial DNA replication in rat liver. PLoS ONE 2013, 8, e74644. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed. Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Youle, R.J.; Finkel, T. The mitochondrial basis of aging. Mol. Cell. 2016, 61, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian mitochondria and aging: An update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef] [Green Version]
- Barazzoni, R.; Short, K.R.; Nair, K.S. Effects of aging on mitochondrial DNA copy number and cytochrome c oxidase gene expression in rat skeletal muscle, liver, and heart. J. Biol. Chem. 2000, 275, 3343–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffann, J.; Pouliet, A.; Adjal, H.; Bole, C.; Fourrage, C.; Martinovic, J.; Rolland-Galmiche, L.; Rotig, A.; Tores, F.; Munnich, A.; et al. No correlation between mtDNA amount and methylation levels at the CpG island of POLG exon 2 in wild-type and mutant human differentiated cells. J. Med. Genet. 2017, 54, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.D.; Mahmud, A.; McKenzie, M.; Trounce, I.A.; St John, J.C. Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear-encoded DNA polymerase gamma A. Nucleic Acids Res. 2012, 40, 10124–10138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sziráki, A.; Tyshkovskiy, A.; Gladyshev, V.N. Global remodeling of the mouse DNA methylome during aging and in response to calorie restriction. Aging Cell 2018, 17, e12738. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.J.; Robertson, N.A.; Rather, M.I.; Thomson, J.P.; McBryan, T.; Sproul, D.; Wang, T.; Brock, C.; Clark, W.; Ideker, T.; et al. Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol. 2017, 18, 58. [Google Scholar] [CrossRef] [Green Version]
- Hahn, O.; Grönke, S.; Stubbs, T.M.; Ficz, G.; Hendrich, O.; Krueger, F.; Andrews, S.; Zhang, Q.; Wakelam, M.J.; Beyer, A.; et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 2017, 18, 56. [Google Scholar] [CrossRef] [Green Version]
- D’Aquila, P.; Montesanto, A.; De Rango, F.; Guarasci, F.; Passarino, G.; Bellizzi, D. Epigenetic signature: Implications for mitochondrial quality control in human aging. Aging (Albany NY) 2019, 11, 1240–1251. [Google Scholar] [CrossRef]

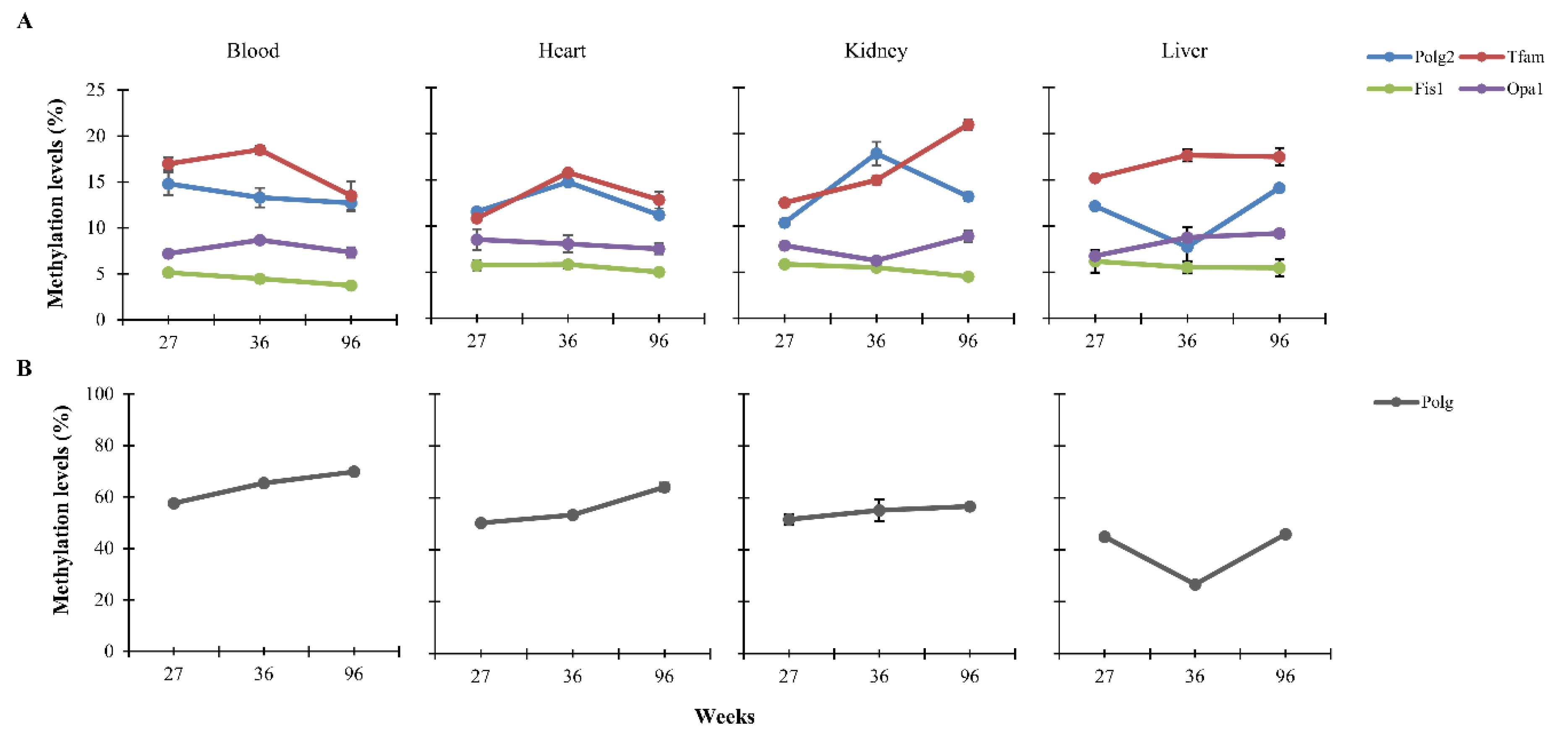


{kind=link}
{kind=link}
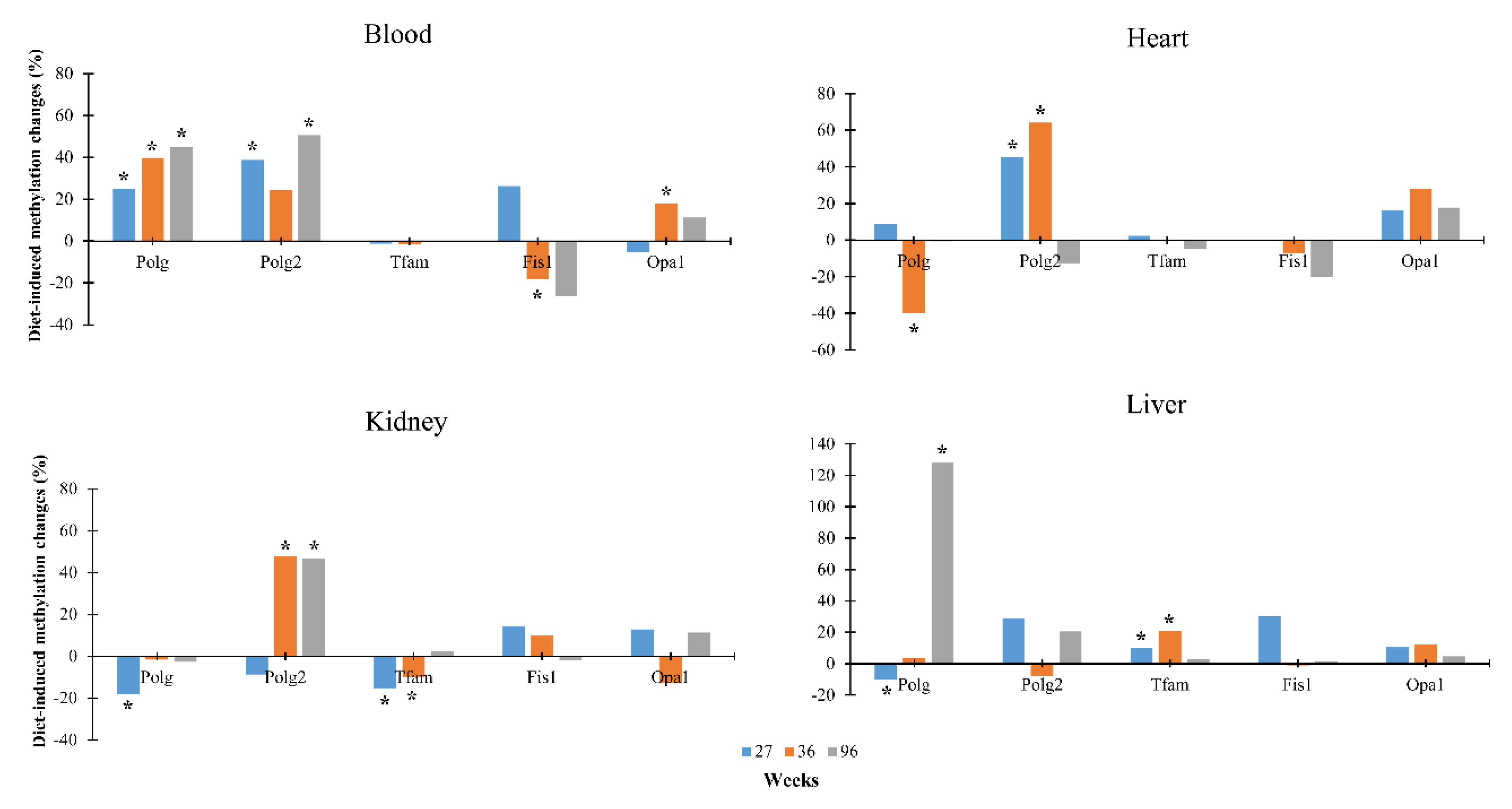{kind=link}
| Standard Diet | Low-Calorie Diet | ||||||
|---|---|---|---|---|---|---|---|
| Weeks | Pearson′s R Correlation | p | 95% CI | Pearson′s R Correlation | p | 95% CI | |
| 36 | Polg methylation-Polg expression | −0.799 | 0.002 | −0.941, −0.415 | −0.877 | <0.001 | −0.965, −0.611 |
| Polg methylation-mtDNA copy number | −0.549 | 0.065 | −0.854, 0.036 | −0.835 | <0.001 | −0.953, −0.502 | |
| Polg2 methylation-Polg2 expression | −0.258 | 0.418 | −0.725, 0.371 | −0.672 | 0.017 | −0.899, −0.160 | |
| Polg2 methylation−mtDNA copy number | −0.090 | 0.782 | −0.631, 0.511 | −0.794 | 0.002 | −0.940, −0.405 | |
| Tfam methylation-Tfam expression | 0.708 | 0.010 | 0.225, 0.911 | −0.748 | 0.005 | −0.925, −0.306 | |
| Tfam methylation-mtDNA copy number | −0.473 | 0.121 | −0.823, 0.139 | 0.140 | 0.664 | −0.472, 0.661 | |
| Fis1 methylation-Fis1 expression | −0.179 | 0.578 | −0.683, 0.440 | −0.170 | 0.598 | −0.678, 0.448 | |
| Fis1 methylation-mtDNA copy number | −0.600 | 0.059 | −0.873, −0.039 | 0.178 | 0.579 | −0.441, 0.682 | |
| Opa1 methylation-Opa1 expression | −0.448 | 0.144 | −0.813, 0.169 | −0.404 | 0.193 | −0.794, 0.221 | |
| Opa1 methylation-mtDNA copy number | 0.258 | 0.418 | −0.371, 0.725 | 0.388 | 0.212 | −0.239, 0.787 | |
| 96 | Polg methylation-Polg expression | −0.925 | <0.001 | −0.979, −0.748 | −0.857 | <0.001 | −0.959, −0.558 |
| Polg methylation-mtDNA copy number | 0.114 | 0.725 | −0.492, 0.645 | −0.171 | 0.595 | −0.678, 0.447 | |
| Polg2 methylation-Polg2 expression | −0.513 | 0.088 | −0.840, 0.086 | 0.057 | 0.859 | −0.534, 0.611 | |
| Polg2 methylation-mtDNA copy number | 0.774 | 0.003 | 0.360, 0.933 | 0.565 | 0.056 | −0.013, 0.860 | |
| Tfam methylation-Tfam expression | −0.746 | 0.005 | −0.924, −0.302 | −0.941 | <0.001 | −0.984, −0.798 | |
| Tfam methylation-mtDNA copy number | 0.565 | 0.055 | −0.012, 0.860 | −0.054 | 0.866 | −0.609, 0.536 | |
| Fis1 methylation-Fis1 expression | −0.294 | 0.354 | −0.743, 0.337 | −0.294 | 0.354 | −0.743, 0.337 | |
| Fis1 methylation-mtDNA copy number | 0.457 | 0.135 | −0.804, 0.194 | 0.522 | 0.082 | −0.074, 0.843 | |
| Opa1 methylation-Opa1 expression | −0.799 | 0.002 | −0.941, −0.416 | −0.358 | 0.253 | −0.773, 0.272 | |
| Opa1 methylation-mtDNA copy number | 0.009 | 0.977 | −0.568, 0.580 | 0.284 | 0.371 | −0.347, 0.738 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Aquila, P.; De Rango, F.; Guarasci, F.; Mandalà, M.; Corsonello, A.; Bellizzi, D.; Passarino, G. Multi-Tissue DNA Methylation Remodeling at Mitochondrial Quality Control Genes According to Diet in Rat Aging Models. Nutrients 2020, 12, 460. https://doi.org/10.3390/nu12020460
D’Aquila P, De Rango F, Guarasci F, Mandalà M, Corsonello A, Bellizzi D, Passarino G. Multi-Tissue DNA Methylation Remodeling at Mitochondrial Quality Control Genes According to Diet in Rat Aging Models. Nutrients. 2020; 12(2):460. https://doi.org/10.3390/nu12020460
Chicago/Turabian StyleD’Aquila, Patrizia, Francesco De Rango, Francesco Guarasci, Maurizio Mandalà, Andrea Corsonello, Dina Bellizzi, and Giuseppe Passarino. 2020. "Multi-Tissue DNA Methylation Remodeling at Mitochondrial Quality Control Genes According to Diet in Rat Aging Models" Nutrients 12, no. 2: 460. https://doi.org/10.3390/nu12020460
APA StyleD’Aquila, P., De Rango, F., Guarasci, F., Mandalà, M., Corsonello, A., Bellizzi, D., & Passarino, G. (2020). Multi-Tissue DNA Methylation Remodeling at Mitochondrial Quality Control Genes According to Diet in Rat Aging Models. Nutrients, 12(2), 460. https://doi.org/10.3390/nu12020460