β-hydroxybutyrate Impedes the Progression of Alzheimer’s Disease and Atherosclerosis in ApoE-Deficient Mice

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Animals
2.3. Cell Culture
2.4. Quantitative Real-Time PCR
2.5. Immunohistochemistry
2.6. Immunofluoresence and Thioflavin-S Staining
2.7. Oil Red O (ORO) Staining
2.8. Low Density Lipoprotein (LDL) Isolation and Oxidation
2.9. Lipid Profiles
2.10. Analysis of Leptin and Resistin Levels
2.11. Statistical Analysis
3. Results
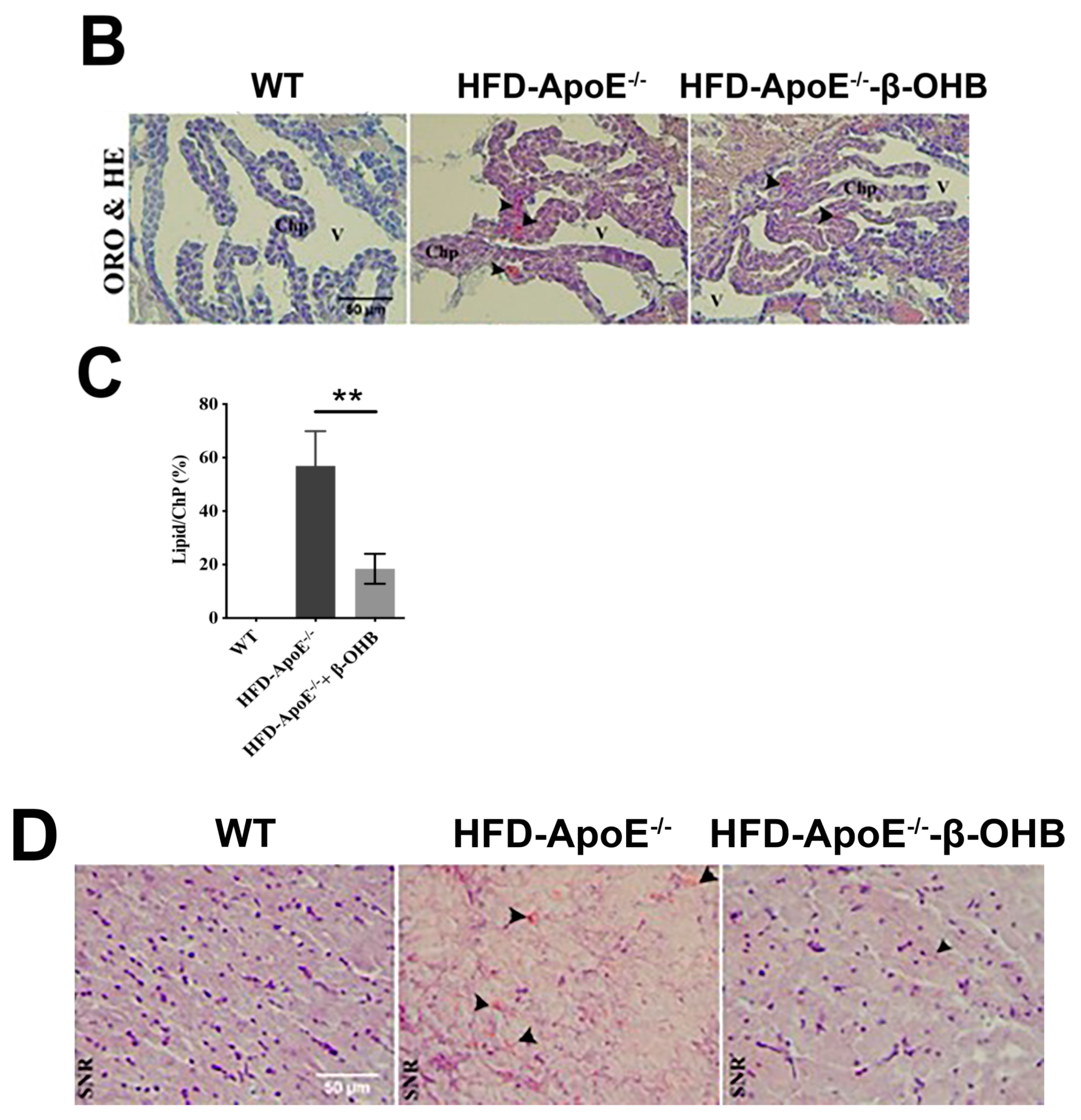
3.1. β-OHB Attenuated HFD-Induced Lipid Deposition, Amyloid Plaque Formation in the ChP, and Tau Accumulation in the Hippocampal Region of ApoE−/− Mice
3.2. β-OHB Treatment Reduced IgG Extravasation in ApoE−/− Mice
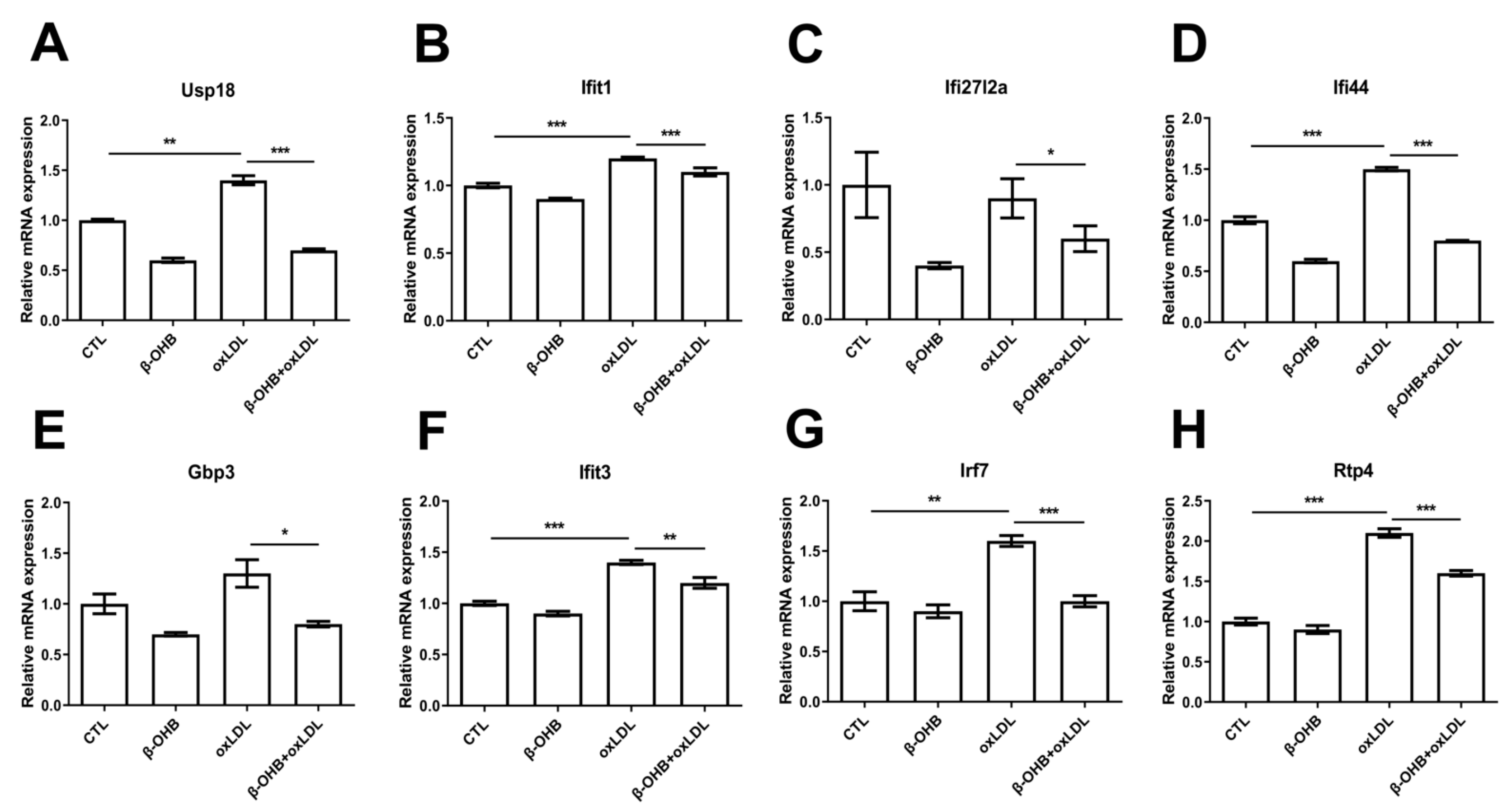
3.3. β-OHB Inhibited Inflammation in the Brain of ApoE−/− Mice
3.4. β-OHB Treatment Reduced Atherogenic Plaque Formation in ApoE−/− Mice
3.5. β-OHB Treatment Reduced Serum Resistin Levels in ApoE−/− Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mendiola-Precoma, J.; Berumen, L.C.; Padilla, K.; Garcia-Alcocer, G. Therapies for Prevention and Treatment of Alzheimer’s Disease. Biomed. Res. Int. 2016, 2016, 2589276. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Abeta in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Amarenco, P.; Lavallee, P.C.; Monteiro Tavares, L.; Labreuche, J.; Albers, G.W.; Abboud, H.; Anticoli, S.; Audebert, H.; Bornstein, N.M.; Caplan, L.R.; et al. Five-Year Risk of Stroke after TIA or Minor Ischemic Stroke. N. Engl. J. Med. 2018, 378, 2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Linton, M.F.; Atkinson, J.B.; Fazio, S. Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science 1995, 267, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Maatta, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef]
- Poff, A.M.; Rho, J.M.; D’Agostino, D.P. Ketone Administration for Seizure Disorders: History and Rationale for Ketone Esters and Metabolic Alternatives. Front. Neurosci. 2019, 13, 1041. [Google Scholar] [CrossRef]
- Rho, J.M.; Shao, L.R.; Stafstrom, C.E. 2-Deoxyglucose and Beta-Hydroxybutyrate: Metabolic Agents for Seizure Control. Front. Cell Neurosci. 2019, 13, 172. [Google Scholar] [CrossRef]
- Taylor, M.K.; Swerdlow, R.H.; Sullivan, D.K. Dietary Neuroketotherapeutics for Alzheimer’s Disease: An Evidence Update and the Potential Role for Diet Quality. Nutrients 2019, 11, 1910. [Google Scholar] [CrossRef]
- Fortier, M.; Castellano, C.A.; Croteau, E.; Langlois, F.; Bocti, C.; St-Pierre, V.; Vandenberghe, C.; Bernier, M.; Roy, M.; Descoteaux, M.; et al. A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement. 2019, 15, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D. Real-time quantitative PCR. Methods 2001, 25, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Menter, T.; Bachmann, M.; Grieshaber, S.; Tzankov, A. A More Accurate Approach to Amyloid Detection and Subtyping: Combining in situ Congo Red Staining and Immunohistochemistry. Pathobiology 2017, 84, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Q.; Chen, Q.; Liu, N.Q.; Li, F.L.; Lu, Z.B.; Qin, C.; Zhu, H.; Huang, Y.Y.; He, W.; et al. Nicotine attenuates beta-amyloid-induced neurotoxicity by regulating metal homeostasis. FASEB J. 2006, 20, 1212–1214. [Google Scholar] [CrossRef] [PubMed]
- Paigen, B.; Morrow, A.; Holmes, P.A.; Mitchell, D.; Williams, R.A. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis 1987, 68, 231–240. [Google Scholar] [CrossRef]
- Walker, L.C.; Parker, C.A.; Lipinski, W.J.; Callahan, M.J.; Carroll, R.T.; Gandy, S.E.; Smith, J.D.; Jucker, M.; Bisgaier, C.L. Cerebral lipid deposition in aged apolipoprotein-E-deficient mice. Am. J. Pathol. 1997, 151, 1371–1377. [Google Scholar]
- Van Ree, J.H.; Gijbels, M.J.; van den Broek, W.J.; Hofker, M.H.; Havekes, L.M. Atypical xanthomatosis in apolipoprotein E-deficient mice after cholesterol feeding. Atherosclerosis 1995, 112, 237–243. [Google Scholar] [CrossRef]
- Brkic, M.; Balusu, S.; Van Wonterghem, E.; Gorle, N.; Benilova, I.; Kremer, A.; Van Hove, I.; Moons, L.; De Strooper, B.; Kanazir, S.; et al. Amyloid beta Oligomers Disrupt Blood-CSF Barrier Integrity by Activating Matrix Metalloproteinases. J. Neurosci. 2015, 35, 12766–12778. [Google Scholar] [CrossRef]
- Yin, C.; Ackermann, S.; Ma, Z.; Mohanta, S.K.; Zhang, C.; Li, Y.; Nietzsche, S.; Westermann, M.; Peng, L.; Hu, D.; et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat. Med. 2019, 25, 496–506. [Google Scholar] [CrossRef]
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93. [Google Scholar] [CrossRef]
- Gu, T.; Lu, L.; An, C.; Zhang, Y.; Wu, X.; Xu, Q.; Chen, G. Negative regulation of the RLR-mediated IFN signaling pathway by duck ubiquitin-specific protease 18 (USP18). J. Cell Physiol. 2019, 234, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Sun, L.; Zheng, Y.; Yu, S.; Ou-Yang, J.; Han, H.; Dai, X.; Yu, X.; Li, M.; Lan, Q. GBP3 promotes glioma cell proliferation via SQSTM1/p62-ERK1/2 axis. Biochem. Biophys. Res. Commun. 2018, 495, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.M.; Meston, N.M.; Gardner, S.G.; Matthews, D.R. The hospital and home use of a 30-second hand-held blood ketone meter: Guidelines for clinical practice. Diabet. Med. 2001, 18, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Jordao, M.J.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef]
- Lun, M.P.; Monuki, E.S.; Lehtinen, M.K. Development and functions of the choroid plexus-cerebrospinal fluid system. Nat. Rev. Neurosci. 2015, 16, 445–457. [Google Scholar] [CrossRef]
- Moore, G.R.; Laule, C.; Leung, E.; Pavlova, V.; Morgan, B.P.; Esiri, M.M. Complement and Humoral Adaptive Immunity in the Human Choroid Plexus: Roles for Stromal Concretions, Basement Membranes, and Epithelium. J. Neuropathol. Exp. Neurol. 2016, 75, 415–428. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef]
- Schwartz, M.; Baruch, K. The resolution of neuroinflammation in neurodegeneration: Leukocyte recruitment via the choroid plexus. EMBO J. 2014, 33, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Grundke-Iqbal, I.; Zaidi, T.; Merz, P.A.; Wen, G.Y.; Shaikh, S.S.; Wisniewski, H.M.; Alafuzoff, I.; Winblad, B. Defective brain microtubule assembly in Alzheimer’s disease. Lancet 1986, 2, 421–426. [Google Scholar] [CrossRef]
- Kopke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [PubMed]
- Misiak, B.; Leszek, J.; Kiejna, A. Metabolic syndrome, mild cognitive impairment and Alzheimer’s disease--the emerging role of systemic low-grade inflammation and adiposity. Brain Res. Bull. 2012, 89, 144–149. [Google Scholar] [CrossRef]
- Reilly, M.P.; Lehrke, M.; Wolfe, M.L.; Rohatgi, A.; Lazar, M.A.; Rader, D.J. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation 2005, 111, 932–939. [Google Scholar] [CrossRef]
- Hu, W.T.; Chen-Plotkin, A.; Arnold, S.E.; Grossman, M.; Clark, C.M.; Shaw, L.M.; Pickering, E.; Kuhn, M.; Chen, Y.; McCluskey, L.; et al. Novel CSF biomarkers for Alzheimer’s disease and mild cognitive impairment. Acta Neuropathol. 2010, 119, 669–678. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Toledo, J.B.; Nefedov, A.; Polikar, R.; Raghavan, N.; Xie, S.X.; Farnum, M.; Schultz, T.; Baek, Y.; Deerlin, V.V.; et al. Identifying amyloid pathology-related cerebrospinal fluid biomarkers for Alzheimer’s disease in a multicohort study. Alzheimers Dement. 2015, 1, 339–348. [Google Scholar] [CrossRef]
- Bednarska-Makaruk, M.; Graban, A.; Wisniewska, A.; Lojkowska, W.; Bochynska, A.; Gugala-Iwaniuk, M.; Slawinska, K.; Lugowska, A.; Ryglewicz, D.; Wehr, H. Association of adiponectin, leptin and resistin with inflammatory markers and obesity in dementia. Biogerontology 2017, 18, 561–580. [Google Scholar] [CrossRef]
- Stubb, B.J.; Cox, P.J.; Evans, R.D.; Cyranka, M.; Clarke, K.; de Wet, H. A ketone ester drink lowers human ghrelin and appetite. Obesity 2018, 26, 269–273. [Google Scholar] [CrossRef]
- Izumi, Y.; Ishii, K.; Katsuki, H.; Benz, A.M.; Zorumski, C.F. beta-Hydroxybutyrate fuels synaptic function during development. Histological and physiological evidence in rat hippocampal slices. J. Clin. Investig. 1998, 101, 1121–1132. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.L.; Lee, S.M.; Fujima, L.S.; Hovda, D.A. Increased cerebral uptake and oxidation of exogenous betaHB improves ATP following traumatic brain injury in adult rats. J. Neurochem. 2004, 90, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L.; Heal, D.J.; Martin, K.F. KTX 0101: A potential metabolic approach to cytoprotection in major surgery and neurological disorders. CNS Drug Rev. 2005, 11, 113–140. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnan, M.; Hwang, J.S.; Kim, M.; Kim, Y.J.; Seo, J.H.; Jung, J.; Ha, E. β-hydroxybutyrate Impedes the Progression of Alzheimer’s Disease and Atherosclerosis in ApoE-Deficient Mice. Nutrients 2020, 12, 471. https://doi.org/10.3390/nu12020471
Krishnan M, Hwang JS, Kim M, Kim YJ, Seo JH, Jung J, Ha E. β-hydroxybutyrate Impedes the Progression of Alzheimer’s Disease and Atherosclerosis in ApoE-Deficient Mice. Nutrients. 2020; 12(2):471. https://doi.org/10.3390/nu12020471
Chicago/Turabian StyleKrishnan, Manigandan, Jong Su Hwang, Mikyung Kim, Yun Jin Kim, Ji Hae Seo, Jeeyoun Jung, and Eunyoung Ha. 2020. "β-hydroxybutyrate Impedes the Progression of Alzheimer’s Disease and Atherosclerosis in ApoE-Deficient Mice" Nutrients 12, no. 2: 471. https://doi.org/10.3390/nu12020471
APA StyleKrishnan, M., Hwang, J. S., Kim, M., Kim, Y. J., Seo, J. H., Jung, J., & Ha, E. (2020). β-hydroxybutyrate Impedes the Progression of Alzheimer’s Disease and Atherosclerosis in ApoE-Deficient Mice. Nutrients, 12(2), 471. https://doi.org/10.3390/nu12020471