Sulforaphane Diminishes the Formation of Mammary Tumors in Rats Exposed to 17β-Estradiol


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Study
2.2. Ethics Statement
2.3. Blood, Tumors and Organ Collections
2.4. RNA Isolation from Tissue and Real-Time PCR
2.5. NQO1 Activity
2.6. Immunohistochemistry
2.7. UPLC-MS/MS Analysis of Depurinating DNA Adducts
2.8. Sample Preparation and Analysis of Free Fatty Acids and Triglyceride
2.9. Untargeted Lipidomics
2.10. Statistical Analysis
3. Results
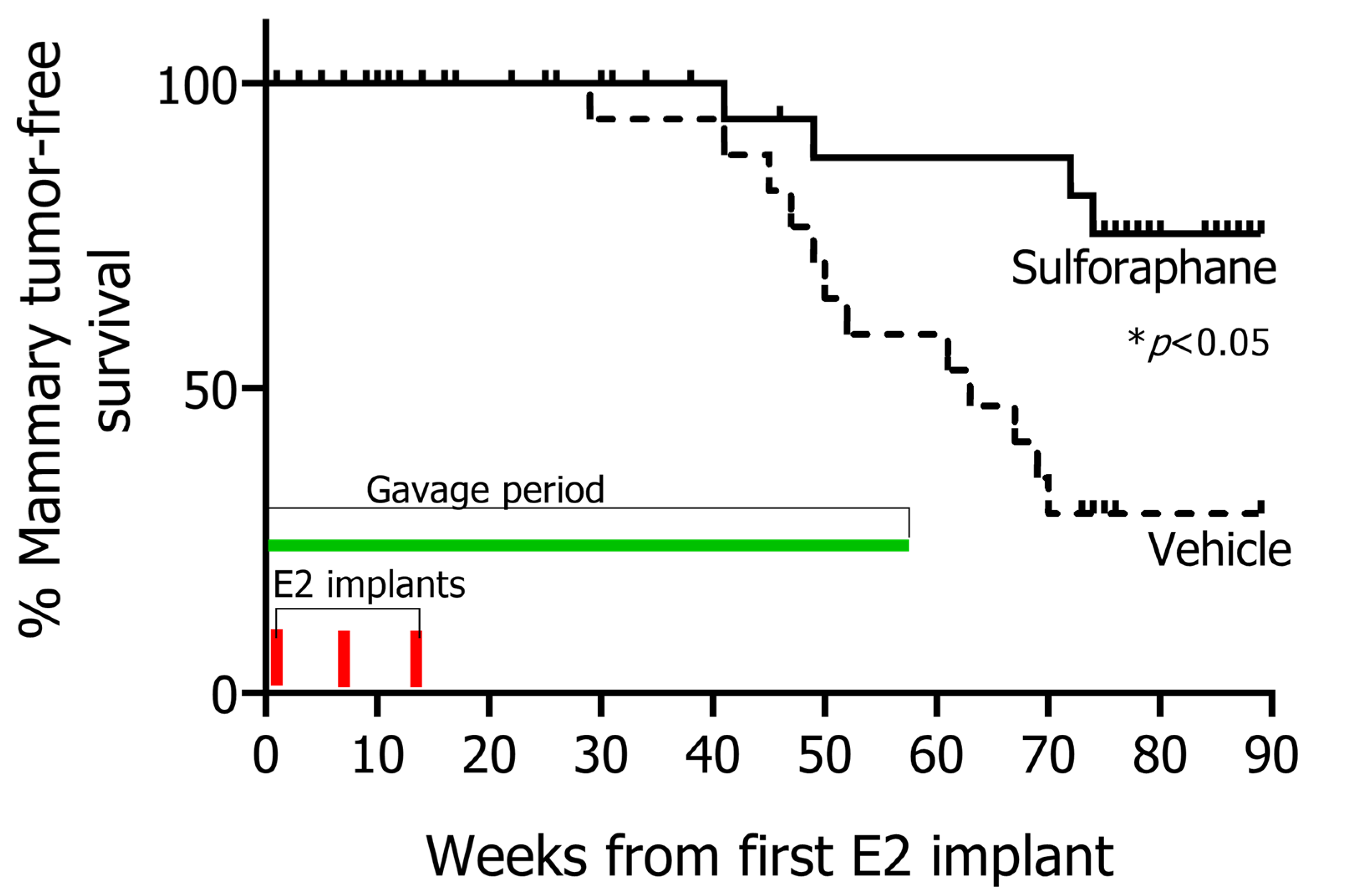
3.1. SFN Treatment Leads to Significant Prevention of Mammary Tumor Formation of Rats Exposed to E2
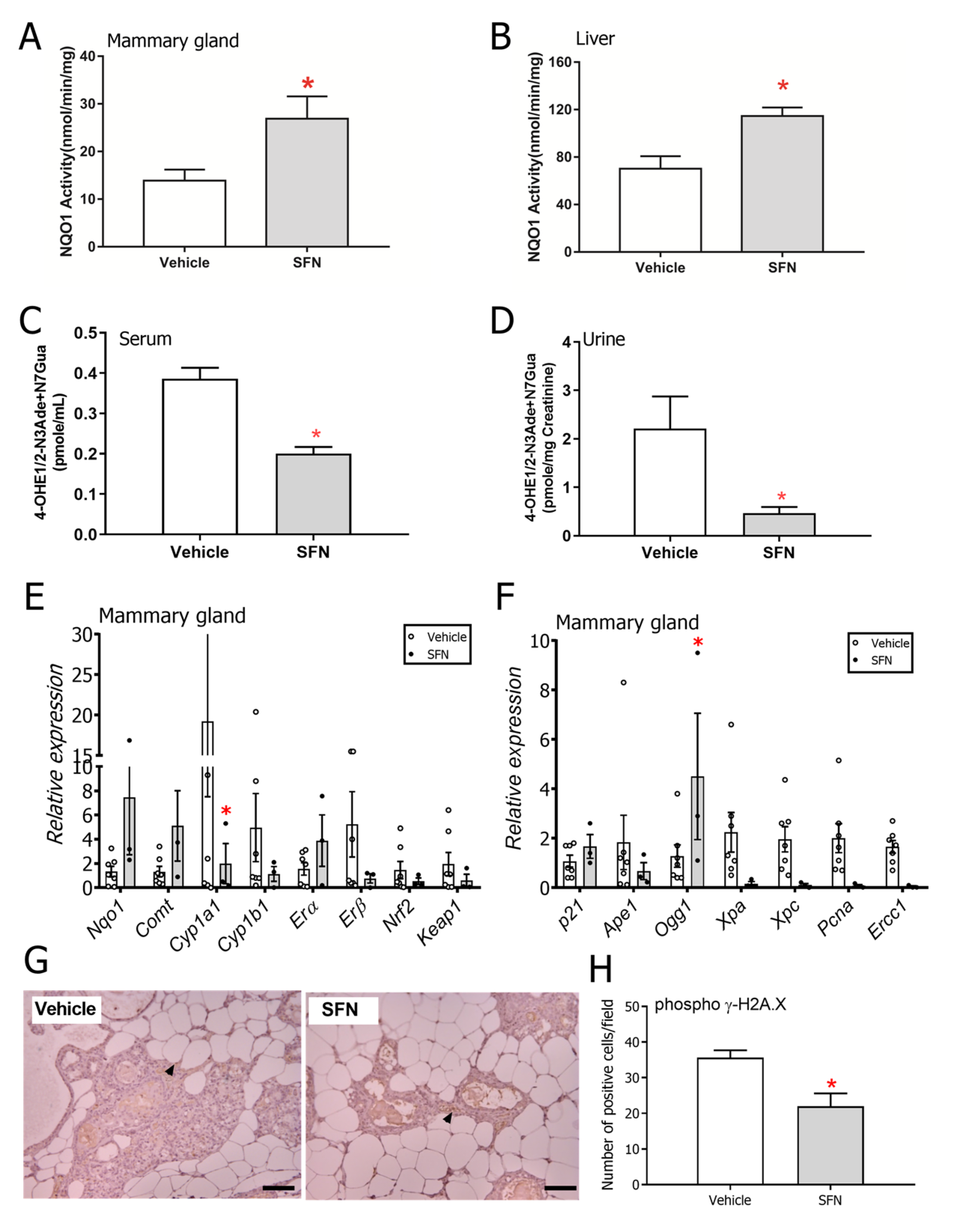
3.2. SFN-Treated Rats Show Altered DNA Damage and Estrogen Metabolism Profiles
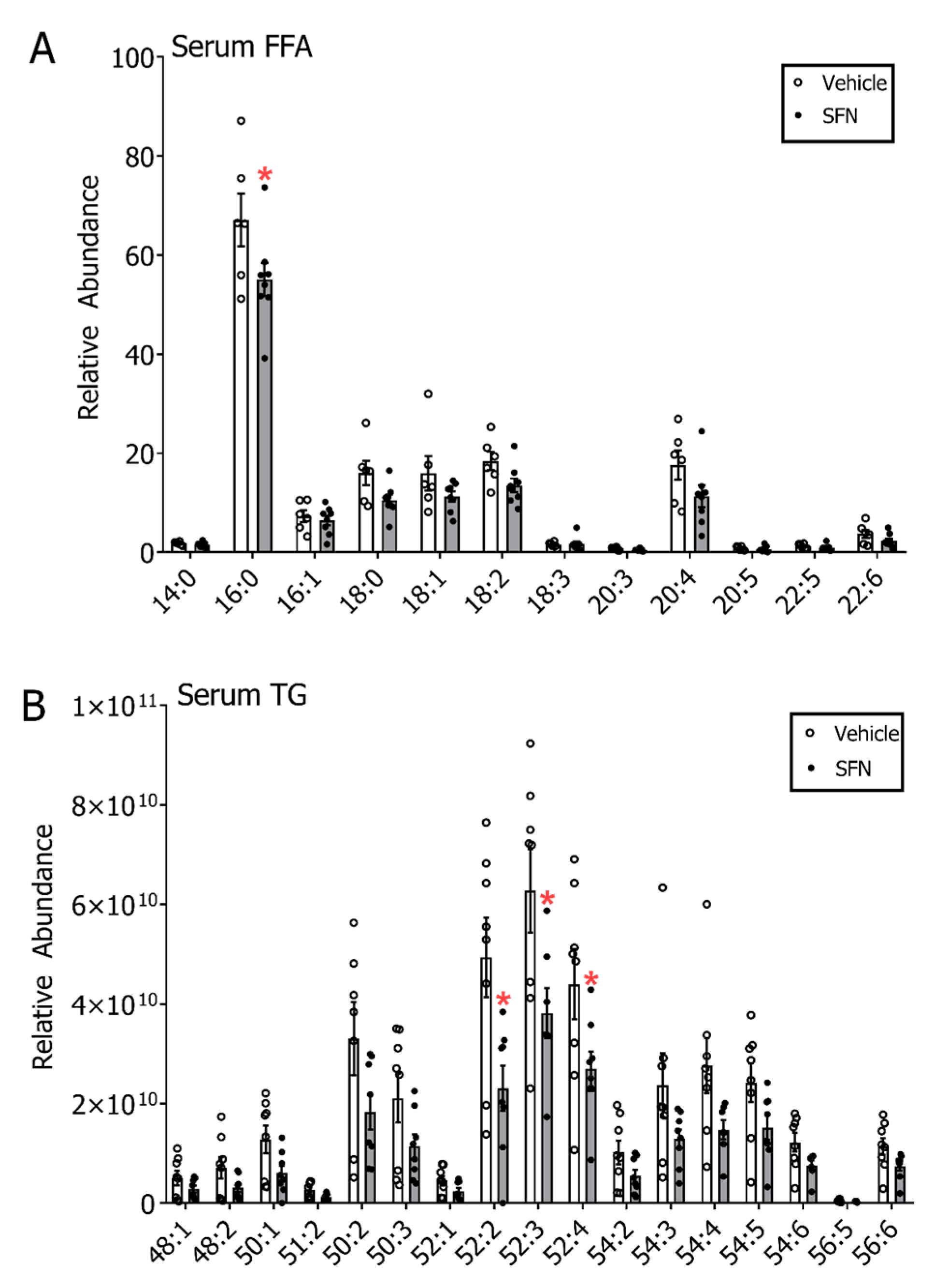
3.3. SFN Treatment Leads to Decreased Free Fatty Acids and Triglycerides in Serum but not in Mammary Gland and Liver of E2-Treated Rats
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.C.; Matthews, C.E.; Shu, X.O.; Yu, K.; Gail, M.H.; Xu, X.; Ji, B.T.; Chow, W.H.; Cai, Q.; Li, H.; et al. Endogenous Estrogens, Estrogen Metabolites, and Breast Cancer Risk in Postmenopausal Chinese Women. J. Natl. Cancer Inst. 2016, 108, djw103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santen, R.J.; Yue, W.; Wang, J.P. Estrogen metabolites and breast cancer. Steroids 2015, 99, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Hilvo, M.; Denkert, C.; Lehtinen, L.; Muller, B.; Brockmoller, S.; Seppanen-Laakso, T.; Budczies, J.; Bucher, E.; Yetukuri, L.; Castillo, S.; et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011, 71, 3236–3245. [Google Scholar] [CrossRef] [Green Version]
- Palmisano, B.T.; Zhu, L.; Eckel, R.H.; Stafford, J.M. Sex differences in lipid and lipoprotein metabolism. Mol. Metab 2018, 15, 45–55. [Google Scholar] [CrossRef]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Brown, K. Breast cancer chemoprevention: Risk-benefit effects of the antioestrogen tamoxifen. Expert Opin. Drug Saf. 2002, 1, 253–267. [Google Scholar] [CrossRef]
- Visvanathan, K.; Fabian, C.J.; Bantug, E.; Brewster, A.M.; Davidson, N.E.; DeCensi, A.; Floyd, J.D.; Garber, J.E.; Hofstatter, E.W.; Khan, S.A.; et al. Use of Endocrine Therapy for Breast Cancer Risk Reduction: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2019, 37, 3152–3165. [Google Scholar] [CrossRef]
- Harris, H.R.; Willett, W.C.; Vaidya, R.L.; Michels, K.B. An Adolescent and Early Adulthood Dietary Pattern Associated with Inflammation and the Incidence of Breast Cancer. Cancer Res. 2017, 77, 1179–1187. [Google Scholar] [CrossRef] [Green Version]
- Bosetti, C.; Filomeno, M.; Riso, P.; Polesel, J.; Levi, F.; Talamini, R.; Montella, M.; Negri, E.; Franceschi, S.; La, V.C. Cruciferous vegetables and cancer risk in a network of case-control studies. Ann. Oncol. 2012, 23, 2198–2203. [Google Scholar] [CrossRef]
- Ambrosone, C.B.; McCann, S.E.; Freudenheim, J.L.; Marshall, J.R.; Zhang, Y.; Shields, P.G. Breast cancer risk in premenopausal women is inversely associated with consumption of broccoli, a source of isothiocyanates, but is not modified by GST genotype. J. Nutr. 2004, 134, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Atwell, L.L.; Farris, P.E.; Ho, E.; Shannon, J. Associations between cruciferous vegetable intake and selected biomarkers among women scheduled for breast biopsies. Public Health Nutr. 2016, 19, 1288–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Kensler, T.W.; Cho, C.G.; Posner, G.H.; Talalay, P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc. Natl. Acad. Sci. USA 1994, 91, 3147–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahey, J.W.; Zhang, Y.; Talalay, P. Broccoli sprouts: An exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc. Natl. Acad. Sci. USA 1997, 94, 10367–10372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornblatt, B.S.; Ye, L.; Dinkova-Kostova, A.T.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.S.; Stierer, T.; Garrett-Mayer, E. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 2007, 28, 1485–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, T.; Korkaya, H.; Liu, S.; Lee, H.F.; Newman, B.; Yu, Y.; Clouthier, S.G.; Schwartz, S.J.; Wicha, M.S.; et al. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin. Cancer Res. 2010, 16, 2580–2590. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.J.; Singletary, K.W. Sulforaphane: A naturally occurring mammary carcinoma mitotic inhibitor, which disrupts tubulin polymerization. Carcinogenesis 2004, 25, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Atwell, L.L.; Zhang, Z.; Mori, M.; Farris, P.; Vetto, J.T.; Naik, A.M.; Oh, K.Y.; Thuillier, P.; Ho, E.; Shannon, J. Sulforaphane Bioavailability and Chemopreventive Activity in Women Scheduled for Breast Biopsy. Cancer Prev. Res. (Phila) 2015, 8, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or Sulforaphane: Is It the Source or Dose That Matters? Molecules 2019, 24, 3593. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zahid, M.; Liao, Y.; Rogan, E.G.; Cavalieri, E.L.; Davidson, N.E.; Yager, J.D.; Visvanathan, K.; Groopman, J.D.; Kensler, T.W. Reduced formation of depurinating estrogen-DNA adducts by sulforaphane or KEAP1 disruption in human mammary epithelial MCF-10A cells. Carcinogenesis 2013, 34, 2587–2592. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Tian, S.; Teng, C.; Huang, L.; Liu, X.; Wang, J.; Zhang, Y.; Li, B.; Shan, Y. Sulforaphane Improves Lipid Metabolism by Enhancing Mitochondrial Function and Biogenesis In Vivo and In Vitro. Mol. Nutr. Food Res. 2019, 63, e1800795. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Lei, P.; Teng, C.; Sun, Y.; Song, X.; Li, B.; Shan, Y. Targeting PLIN2/PLIN5-PPARgamma: Sulforaphane Disturbs the Maturation of Lipid Droplets. Mol. Nutr. Food Res. 2019, 79, e1900183. [Google Scholar] [CrossRef] [PubMed]
- Valli, V.; Heilmann, K.; Danesi, F.; Bordoni, A.; Gerhauser, C. Modulation of Adipocyte Differentiation and Proadipogenic Gene Expression by Sulforaphane, Genistein, and Docosahexaenoic Acid as a First Step to Counteract Obesity. Oxid. Med. Cell. Longev. 2018, 2018, 1617202. [Google Scholar] [CrossRef] [PubMed]
- Gould, K.A.; Tochacek, M.; Schaffer, B.S.; Reindl, T.M.; Murrin, C.R.; Lachel, C.M.; VanderWoude, E.A.; Pennington, K.L.; Flood, L.A.; Bynote, K.K.; et al. Genetic determination of susceptibility to estrogen-induced mammary cancer in the ACI rat: Mapping of Emca1 and Emca2 to chromosomes 5 and 18. Genetics 2004, 168, 2113–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shull, J.D.; Spady, T.J.; Snyder, M.C.; Johansson, S.L.; Pennington, K.L. Ovary-intact, but not ovariectomized female ACI rats treated with 17beta-estradiol rapidly develop mammary carcinoma. Carcinogenesis 1997, 18, 1595–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, E.W.; Wong, P.Y.; Lakshmanaswamy, R.; Guzman, R.; Nandi, S. Both ovarian hormones estrogen and progesterone are necessary for hormonal mammary carcinogenesis in ovariectomized ACI rats. Proc. Natl. Acad. Sci. USA 2008, 105, 3527–3532. [Google Scholar] [CrossRef] [Green Version]
- Aiyer, H.S.; Srinivasan, C.; Gupta, R.C. Dietary berries and ellagic acid diminish estrogen-mediated mammary tumorigenesis in ACI rats. Nutr. Cancer 2008, 60, 227–234. [Google Scholar] [CrossRef]
- Gaikwad, N.W.; Yang, L.; Muti, P.; Meza, J.L.; Pruthi, S.; Ingle, J.N.; Rogan, E.G.; Cavalieri, E.L. The molecular etiology of breast cancer: Evidence from biomarkers of risk. Int. J. Cancer 2008, 122, 1949–1957. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Frankem, A.A. Improved LC-MS method for the determination of fatty acids in red blood cells by LC-orbitrap MS. Anal. Chem. 2011, 83, 3192–3198. [Google Scholar] [CrossRef] [Green Version]
- Dubuc, P.U. Effects of estradiol implants on body weight regulation in castrated and intact female rats. Endocrinology 1974, 95, 1733–1736. [Google Scholar] [CrossRef]
- Xu, Y.; Lopez, M. Central regulation of energy metabolism by estrogens. Mol. Metab. 2018, 15, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Li, S.A. Estrogen carcinogenesis in Syrian hamster tissues: Role of metabolism. Fed. Proc. 1987, 46, 1858–1863. [Google Scholar] [PubMed]
- Liehr, J.G.; Fang, W.F.; Sirbasku, D.A.; Ari-Ulubelen, A. Carcinogenicity of catechol estrogens in Syrian hamsters. J. Steroid Biochem. 1986, 24, 353–356. [Google Scholar] [CrossRef]
- Smith, S.; Sepkovic, D.; Bradlow, H.L.; Auborn, K.J. 3,3’-Diindolylmethane and genistein decrease the adverse effects of estrogen in LNCaP and PC-3 prostate cancer cells. J. Nutr. 2008, 138, 2379–2385. [Google Scholar] [CrossRef]
- Singh, K.B.; Kim, S.H.; Hahm, E.R.; Pore, S.K.; Jacobs, B.L.; Singh, S.V. Prostate cancer chemoprevention by sulforaphane in a preclinical mouse model is associated with inhibition of fatty acid metabolism. Carcinogenesis 2018, 39, 826–837. [Google Scholar] [CrossRef]
- Kay, H.Y.; Kim, W.D.; Hwang, S.J.; Choi, H.S.; Gilroy, R.K.; Wan, Y.J.; Kim, S.G. Nrf2 inhibits LXRalpha-dependent hepatic lipogenesis by competing with FXR for acetylase binding. Antioxid Redox Signal 2011, 15, 2135–2146. [Google Scholar] [CrossRef]
- Bianchini, F.; Kaaks, R.; Vainio, H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002, 3, 565–574. [Google Scholar] [CrossRef]
- Neuhouser, M.L.; Aragaki, A.K.; Prentice, R.L.; Manson, J.E.; Chlebowski, R.; Carty, C.L.; Ochs-Balcom, H.M.; Thomson, C.A.; Caan, B.J.; Tinker, L.F.; et al. Overweight, Obesity, and Postmenopausal Invasive Breast Cancer Risk: A Secondary Analysis of the Women’s Health Initiative Randomized Clinical Trials. JAMA Oncol. 2015, 1, 611–621. [Google Scholar] [CrossRef]
- Schoemaker, M.J.; Nichols, H.B.; Wright, L.B.; Brook, M.N.; Jones, M.E.; O’Brien, K.M.; Adami, H.O.; Baglietto, L.; Bernstein, L.; Bertrand, K.A.; et al. Association of Body Mass Index and Age With Subsequent Breast Cancer Risk in Premenopausal Women. JAMA Oncol. 2018, 4, e181771. [Google Scholar] [CrossRef]
- Simpson, E.R. Sources of estrogen and their importance. J. Steroid Biochem. Mol. Biol. 2003, 86, 225–230. [Google Scholar] [CrossRef]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Obesity and cancer--mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Blucher, C.; Stadler, S.C. Obesity and Breast Cancer: Current Insights on the Role of Fatty Acids and Lipid Metabolism in Promoting Breast Cancer Growth and Progression. Front. Endocrinol. (Lausanne) 2017, 8, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madak-Erdogan, Z.; Band, S.; Zhao, Y.C.; Smith, B.P.; Kulkoyluoglu-Cotul, E.; Zuo, Q.; Santaliz, C.A.; Wrobel, K.; Rossi, G.; Smith, R.L.; et al. Free fatty acids rewire cancer metabolism in obesity-associated breast cancer via estrogen receptor and mTOR signaling. Cancer Res. 2019, 79, 2494–2510. [Google Scholar] [CrossRef] [Green Version]
- Milgraum, L.Z.; Witters, L.A.; Pasternack, G.R.; Kuhajda, F.P. Enzymes of the fatty acid synthesis pathway are highly expressed in in situ breast carcinoma. Clin. Cancer Res. 1997, 3, 2115–2120. [Google Scholar] [PubMed]
- Sukocheva, O.; Wadham, C. Role of sphingolipids in oestrogen signalling in breast cancer cells: An update. J. Endocrinol. 2014, 220, R25–R35. [Google Scholar] [CrossRef] [Green Version]
- Bjorndal, B.; Alteras, E.K.; Lindquist, C.; Svardal, A.; Skorve, J.; Berge, R.K. Associations between fatty acid oxidation, hepatic mitochondrial function, and plasma acylcarnitine levels in mice. Nutr. Metab. (Lond.) 2018, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Vazquez-Martin, A.; Ortega, F.J.; Fernandez-Real, J.M. Fatty acid synthase: Association with insulin resistance, type 2 diabetes, and cancer. Clin. Chem. 2009, 55, 425–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, M.A.; Birnbaum, M.J. De-meaning of metabolism. Science 2012, 33, 1651–1652. [Google Scholar] [CrossRef] [PubMed]
- Quevedo-Coli, S.; Crespi, C.; Benito, E.; Palou, A.; Roca, P. Alterations in circulating fatty acids and the compartmentation of selected metabolites in women with breast cancer. Biochem. Mol. Biol. Int. 1997, 41, 1–10. [Google Scholar] [CrossRef]
- Chen, H.; Krishnamachari, S.; Guo, J.; Yao, L.; Murugan, P.; Weight, C.J.; Turesky, R.J. Quantitation of Lipid Peroxidation Product DNA Adducts in Human Prostate by Tandem Mass Spectrometry: A Method that Mitigates Artifacts. Chem. Res. Toxicol. 2019, 32, 1850–1862. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palliyaguru, D.L.; Yang, L.; Chartoumpekis, D.V.; Wendell, S.G.; Fazzari, M.; Skoko, J.J.; Liao, Y.; Oesterreich, S.; Michalopoulos, G.K.; Kensler, T.W. Sulforaphane Diminishes the Formation of Mammary Tumors in Rats Exposed to 17β-Estradiol. Nutrients 2020, 12, 2282. https://doi.org/10.3390/nu12082282
Palliyaguru DL, Yang L, Chartoumpekis DV, Wendell SG, Fazzari M, Skoko JJ, Liao Y, Oesterreich S, Michalopoulos GK, Kensler TW. Sulforaphane Diminishes the Formation of Mammary Tumors in Rats Exposed to 17β-Estradiol. Nutrients. 2020; 12(8):2282. https://doi.org/10.3390/nu12082282
Chicago/Turabian StylePalliyaguru, Dushani L., Li Yang, Dionysios V. Chartoumpekis, Stacy G. Wendell, Marco Fazzari, John J. Skoko, Yong Liao, Steffi Oesterreich, George K. Michalopoulos, and Thomas W. Kensler. 2020. "Sulforaphane Diminishes the Formation of Mammary Tumors in Rats Exposed to 17β-Estradiol" Nutrients 12, no. 8: 2282. https://doi.org/10.3390/nu12082282
APA StylePalliyaguru, D. L., Yang, L., Chartoumpekis, D. V., Wendell, S. G., Fazzari, M., Skoko, J. J., Liao, Y., Oesterreich, S., Michalopoulos, G. K., & Kensler, T. W. (2020). Sulforaphane Diminishes the Formation of Mammary Tumors in Rats Exposed to 17β-Estradiol. Nutrients, 12(8), 2282. https://doi.org/10.3390/nu12082282