Involvement of the Autophagy-ER Stress Axis in High Fat/Carbohydrate Diet-Induced Nonalcoholic Fatty Liver Disease


Abstract
:1. Introduction
2. How High Fat/High Carbohydrate Consumption Induces NAFLD

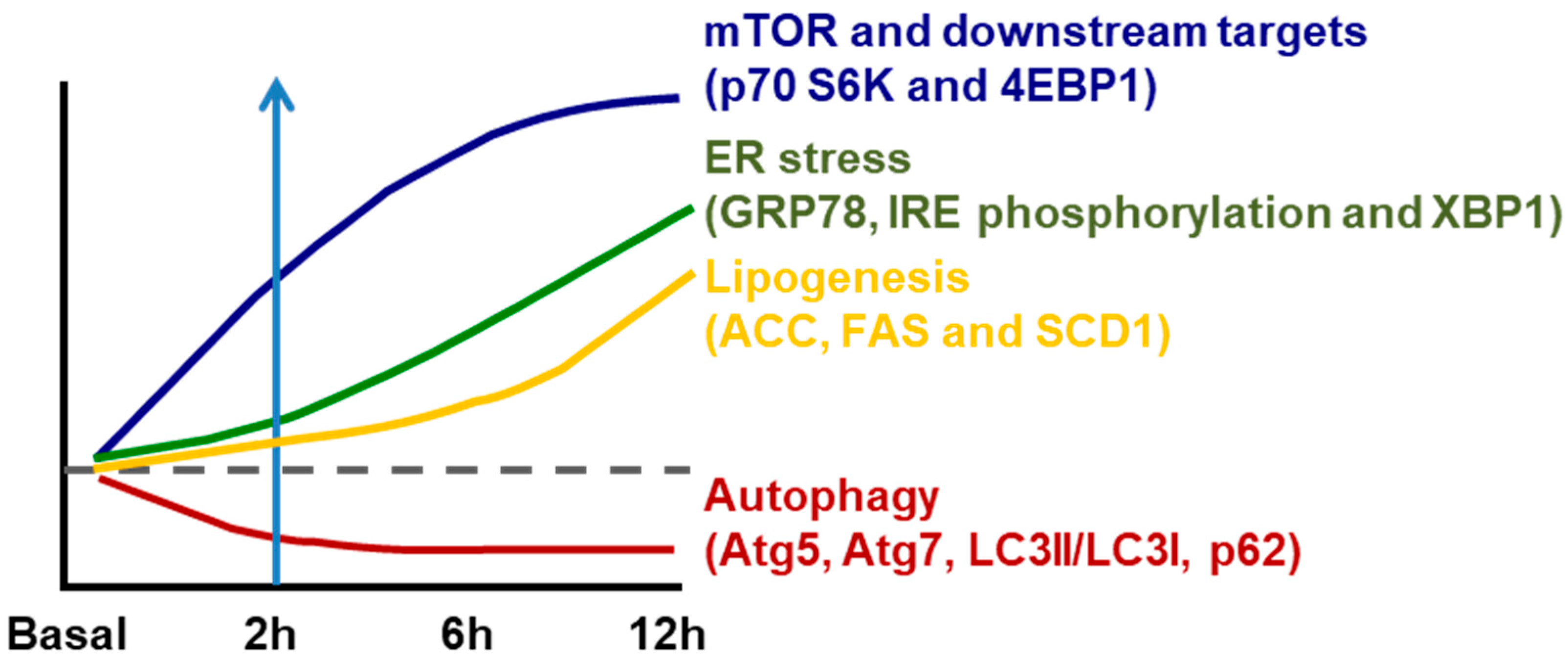{kind=link}
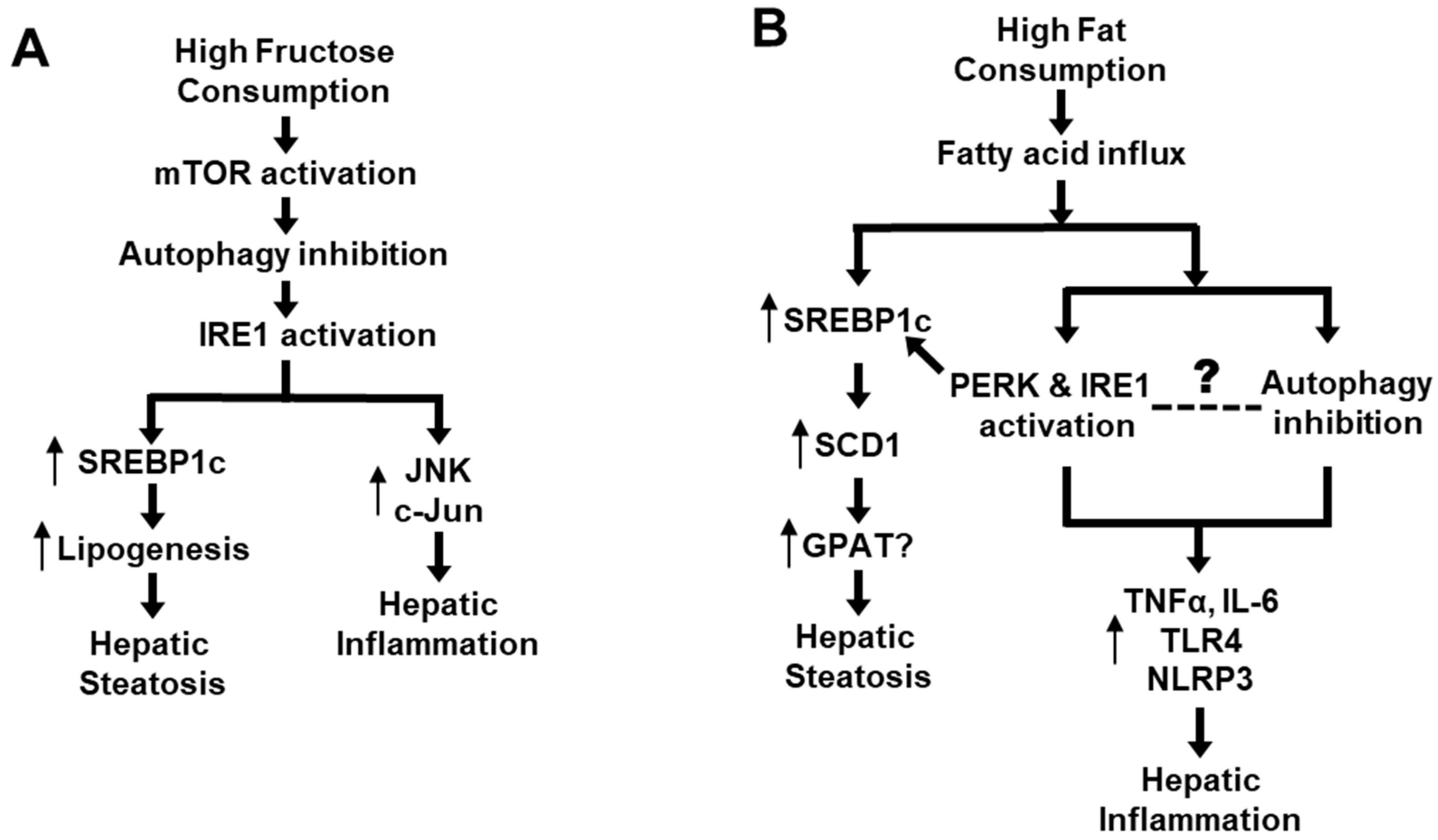{kind=link}
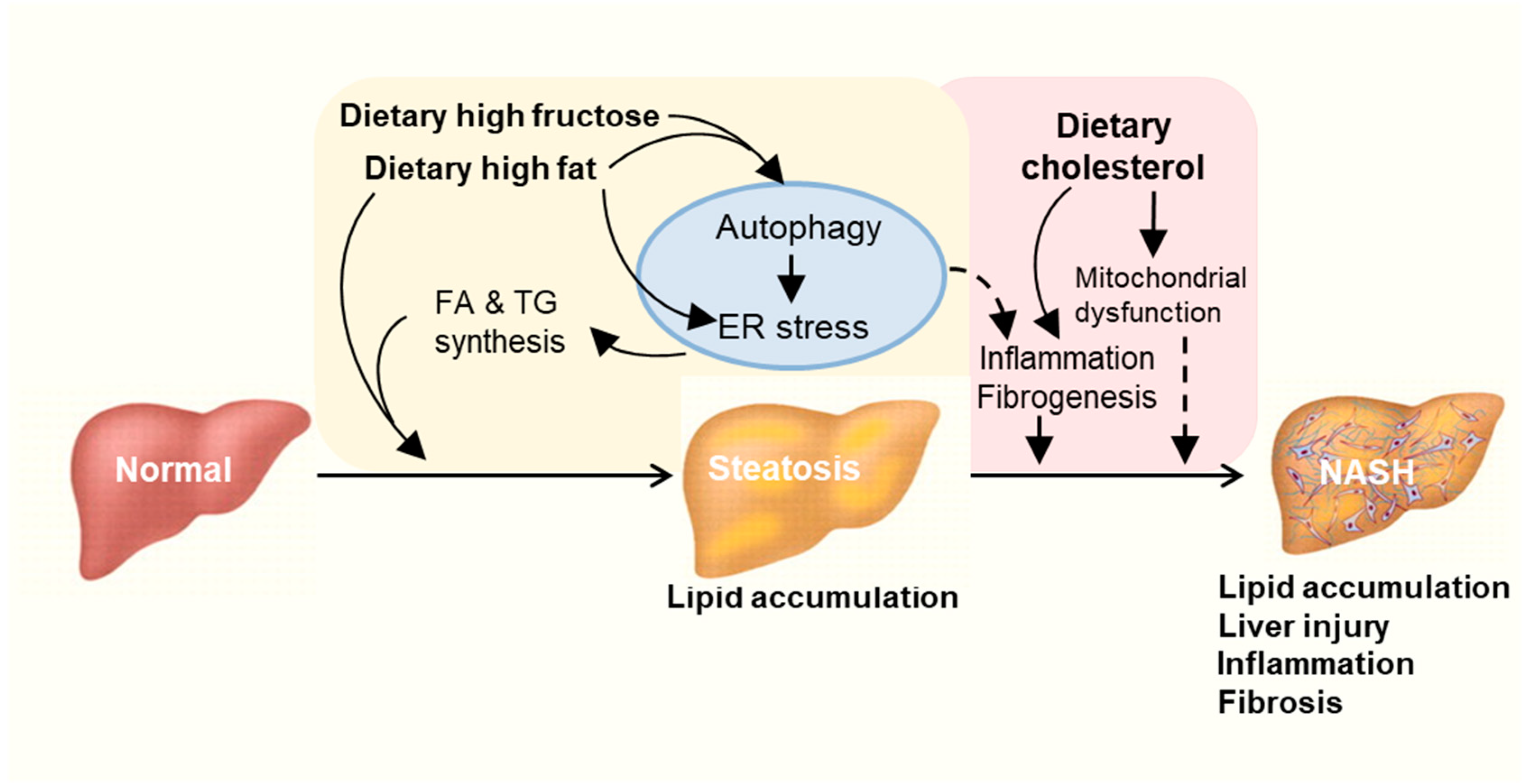{kind=link}
| Diet | Duration | Hepatic Steatosis | Hepatic Injury | Inflammation | Fibrosis | References |
|---|---|---|---|---|---|---|
| High fat (30–60% kcal) | 1–30 weeks | Bioassay TG content ↑ Histological steatosis ↑ | Serum ALT or AST ↑ Apoptotic markers ↑ Hepatocyte ballooning ↑ or none | Inflammatory markers ↑ No Histological inflammation | No change in fibrotic markers No histological fibrosis | [9,17,18,32,33] |
| 30–60 weeks | Bioassay TG content ↑ Histological steatosis ↑ | Serum ALT or AST ↑ Apoptotic markers ↑ Hepatocyte ballooning ↑ | Inflammatory markers ↑ Histological inflammation ↑ or none | Fibrotic markers ↑ Histological fibrosis ↑ | [15,16,32] | |
| High fructose (20–60% fructose) | Up to 25 weeks | Bioassay TG content ↑ Histological steatosis ↑ | Serum ALT or AST ↑ Apoptotic markers ↑ Hepatocyte ballooning ↑ | Inflammatory markers ↑ Histological inflammation ↑ | No change in fibrotic markers No histological fibrosis | [7,10,13,14,33] |
| High fat, high fructose | 12–25 weeks | Bioassay TG content ↑ Histological steatosis ↑ | Serum ALT or AST ↑ Apoptotic markers ↑ Hepatocyte ballooning ↑ | Inflammatory markers ↑ Histological inflammation ↑ | Fibrotic markers ↑ Histological fibrosis ↑ | [13,18,32,33,34,35] |
| High fat, high cholesterol (0.2–2% cholesterol) | 12–42 weeks | Bioassay TG content ↑ Histological steatosis ↑ | Serum ALT or AST ↑ Apoptotic markers ↑ Hepatocyte ballooning ↑ | Inflammatory markers ↑ Histological inflammation ↑ | Fibrotic markers ↑ Histological fibrosis ↑ or none | [19,20,21,22] |
| High fat, high fructose, high cholesterol | 25–30 weeks | Bioassay TG content ↑ Histological steatosis ↑ | Serum ALT or AST ↑ Apoptotic markers ↑ Hepatocyte ballooning ↑ | Inflammatory markers ↑ Histological inflammation ↑ | Fibrotic markers ↑ Histological fibrosis ↑ | [13,32,34,36] |
3. ER Stress and Its Possible Roles in the Pathogenesis of NAFLD
3.1. Involvement of ER Stress in the Development of Hepatic Steatosis
3.2. Involvement of ER Stress in the Progression to NASH
| Signalling Pathways | Molecular Mechanisms | Biological Effects | References | |
|---|---|---|---|---|
| UPR pathways (activation) | IRE1/XBP1 | ↑ SREBP1c, ↑ ACC, ↑ FAS, ↑ SCD1 | ↑ lipogenesis | [31,37,38,39] |
| ↑ IKK, ↑ NF-κB, ↑ JNK ↑ NLRP3 | ↑ inflammation | [29,45] [46] | ||
| ↑ CHOP | ↑ apoptosis | [46] | ||
| PERK/eIF2α | ↑ SREBP1c, ↑ ACC, ↑ FAS, ↑ SCD1 | ↑ lipogenesis | [7,9,31] | |
| ↑ IKK ↓ IκB, ↑ NFκB | ↑ inflammation | [29] [46] | ||
| ↑ CHOP, ↓ Bcl2 | ↑ apoptosis | [47] | ||
| ↑ ATF4, ↑ CHOP | ↓ autophagy | [48] | ||
| ATF6 | ↑ PPARα, ↑ CPT1, ↑ ACOX-1 | ↑ fatty acid β-oxidation | [41,42] | |
| ↓ SREBP2 | ↓ lipogenesis | [43] | ||
| ↑ SREBP1c, ↑ ACC, ↑ FAS, ↑ SCD1 | ↑ fatty acid synthesis | [7,49] | ||
| Autophagy (inhibition) | ↓ Atg7 | ↑ ER stress ↓ fibrosis | [7,50] [30] | |
| ↑ TNFα, ↑ IL-6 ↑ TLR4 ↑ NLRP3, ↑ IL-1β ↑ NF-κB | ↑ inflammation | [51,52] [52] [52,53,54] [52] | ||
4. Autophagy and Its Possible Role in the Pathogenesis of NAFLD
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chu, K.; Zhao, N.; Wu, J.; Ma, L.; Zhu, C.; Chen, X.; Wei, G.; Liao, M. Corilagin Alleviates Nonalcoholic Fatty Liver Disease in High-Fat Diet-Induced C57BL/6 Mice by Ameliorating Oxidative Stress and Restoring Autophagic Flux. Front. Pharmacol. 2019, 10, 1693. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, R.Q.; Zeng, X.Y.; Zhou, X.; Li, S.; Jo, E.; Molero, J.C.; Ye, J.M. Restoration of autophagy alleviates hepatic ER stress and impaired insulin signalling transduction in high fructose-fed male mice. Endocrinology 2015, 156, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.M.; Sun, R.Q.; Zeng, X.Y.; Choong, Z.H.; Wang, H.; Watt, M.J.; Ye, J.M. Activation of PPARalpha ameliorates hepatic insulin resistance and steatosis in high fructose-fed mice despite increased endoplasmic reticulum stress. Diabetes 2013, 62, 2095–2105. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.P.; Chan, S.M.; Zeng, X.Y.; Laybutt, D.R.; Iseli, T.J.; Sun, R.Q.; Kraegen, E.W.; Cooney, G.J.; Turner, N.; Ye, J.M. Differing endoplasmic reticulum stress response to excess lipogenesis versus lipid oversupply in relation to hepatic steatosis and insulin resistance. PLoS ONE 2012, 7, e30816. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.Q.; Wang, H.; Zeng, X.Y.; Chan, S.M.; Li, S.P.; Jo, E.; Leung, S.L.; Molero, J.C.; Ye, J.M. IRE1 impairs insulin signaling transduction of fructose-fed mice via JNK independent of excess lipid. Biochim. Biophys. Acta 2015, 1852, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.A.; Farah, B.L.; Singh, B.K.; Siddique, M.M.; Li, Y.; Wu, Y.; Ilkayeva, O.R.; Gooding, J.; Ching, J.; Zhou, J. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology 2014, 59, 1366–1380. [Google Scholar] [CrossRef] [PubMed]
- Recena Aydos, L.; Aparecida do Amaral, L.; Serafim de Souza, R.; Jacobowski, A.C.; Freitas Dos Santos, E.; Rodrigues Macedo, M.L. Nonalcoholic Fatty Liver Disease Induced by High-Fat Diet in C57bl/6 Models. Nutrients 2019, 11, 3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Kim, D.K.; Seo, W.; Gao, B.; Yoo, S.H.; Song, B.J. Fructose Promotes Leaky Gut, Endotoxemia, and Liver Fibrosis Through Ethanol-Inducible Cytochrome P450-2E1-Mediated Oxidative and Nitrative Stress. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Tajima, K.; Zolzaya, K.; Sato, K.; Inoue, R.; Yoneda, M.; Fujita, K.; Nozaki, Y.; Kubota, K.C.; Haga, H.; et al. Protection from non-alcoholic steatohepatitis and liver tumourigenesis in high fat-fed insulin receptor substrate-1-knockout mice despite insulin resistance. Diabetologia 2012, 55, 3382–3391. [Google Scholar] [CrossRef] [Green Version]
- Hill-Baskin, A.E.; Markiewski, M.M.; Buchner, D.A.; Shao, H.; DeSantis, D.; Hsiao, G.; Subramaniam, S.; Berger, N.A.; Croniger, C.; Lambris, J.D.; et al. Diet-induced hepatocellular carcinoma in genetically predisposed mice. Hum. Mol. Genet. 2009, 18, 2975–2988. [Google Scholar] [CrossRef] [Green Version]
- Kirpich, I.A.; Gobejishvili, L.N.; Bon Homme, M.; Waigel, S.; Cave, M.; Arteel, G.; Barve, S.S.; McClain, C.J.; Deaciuc, I.V. Integrated hepatic transcriptome and proteome analysis of mice with high-fat diet-induced nonalcoholic fatty liver disease. J. Nutr. Biochem. 2011, 22, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar] [CrossRef] [Green Version]
- Savard, C.; Tartaglione, E.V.; Kuver, R.; Haigh, W.G.; Farrell, G.C.; Subramanian, S.; Chait, A.; Yeh, M.M.; Quinn, L.S.; Ioannou, G.N. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013, 57, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Van Rooyen, D.M.; Larter, C.Z.; Haigh, W.G.; Yeh, M.M.; Ioannou, G.; Kuver, R.; Lee, S.P.; Teoh, N.C.; Farrell, G.C. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 2011, 141, 1393–1403. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N.; Subramanian, S.; Chait, A.; Haigh, W.G.; Yeh, M.M.; Farrell, G.C.; Lee, S.P.; Savard, C. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J. Lipid Res. 2017, 58, 1067–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zeng, X.Y.; Zhou, X.; Wang, H.; Jo, E.; Robinson, S.R.; Xu, A.; Ye, J.M. Dietary cholesterol induces hepatic inflammation and blunts mitochondrial function in the liver of high-fat-fed mice. J. Nutr. Biochem. 2016, 27, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aeberli, I.; Hochuli, M.; Gerber, P.A.; Sze, L.; Murer, S.B.; Tappy, L.; Spinas, G.A.; Berneis, K. Moderate amounts of fructose consumption impair insulin sensitivity in healthy young men: A randomized controlled trial. Diabetes Care 2013, 36, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Gambino, R. Non-alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov. 2016, 15, 249–274. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Jahn, D.; Kircher, S.; Hermanns, H.M.; Geier, A. Animal models of NAFLD from a hepatologist’s point of view. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Jun, D.W.; Kim, E.K.; Jeon, H.J.; Nam, H.H.; Saeed, W.K. Histologic and Metabolic Derangement in High-Fat, High-Fructose, and Combination Diet Animal Models. Sci. World J. 2015, 2015, 306326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlton, M.; Krishnan, A.; Viker, K.; Sanderson, S.; Cazanave, S.; McConico, A.; Masuoko, H.; Gores, G. Fast food diet mouse: Novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G825–G834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapper, J.R.; Hendricks, M.D.; Gu, G.; Wittmer, C.; Dolman, C.S.; Herich, J.; Athanacio, J.; Villescaz, C.; Ghosh, S.S.; Heilig, J.S.; et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G483–G495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [Green Version]
- Ning, J.; Hong, T.; Ward, A.; Pi, J.; Liu, Z.; Liu, H.Y.; Cao, W. Constitutive role for IRE1alpha-XBP1 signaling pathway in the insulin-mediated hepatic lipogenic program. Endocrinology 2011, 152, 2247–2255. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Wang, Z.V.; Tao, C.; Gao, N.; Holland, W.L.; Ferdous, A.; Repa, J.J.; Liang, G.; Ye, J.; Lehrman, M.A.; et al. The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. J. Clin. Investig. 2013, 123, 455–468. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takahara, K.; Oyadomari, S.; Okada, T.; Sato, T.; Harada, A.; Mori, K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 2010, 21, 2975–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, F.; Gong, Q.; Cui, A.; Zhuo, S.; Hu, Z.; Han, Y.; Gao, J.; Sun, Y.; Liu, Z.; et al. Hepatic ATF6 Increases Fatty Acid Oxidation to Attenuate Hepatic Steatosis in Mice Through Peroxisome Proliferator-Activated Receptor alpha. Diabetes 2016, 65, 1904–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Lu, M.; Mori, K.; Luo, S.; Lee, A.S.; Zhu, Y.; Shyy, J.Y. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004, 23, 950–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.M.; Kim, Y.; Lee, E.S.; Huh, J.H.; Chung, C.H. Caffeic acid ameliorates hepatic steatosis and reduces ER stress in high fat diet-induced obese mice by regulating autophagy. Nutrition 2018, 55-56, 63–70. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Kaczmarek, A.; Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. ER stress-induced inflammation: Does it aid or impede disease progression? Trends Mol. Med. 2012, 18, 589–598. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, J.L.; Cuervo, A.M. Liver autophagy: Much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, K.; Oe, S.; Honma, Y.; Izumi, H.; Baba, R.; Harada, M. Lipid-Induced Endoplasmic Reticulum Stress Impairs Selective Autophagy at the Step of Autophagosome-Lysosome Fusion in Hepatocytes. Am. J. Pathol. 2016, 186, 1861–1873. [Google Scholar] [CrossRef] [Green Version]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- El Kasmi, K.C.; Anderson, A.L.; Devereaux, M.W.; Fillon, S.A.; Harris, J.K.; Lovell, M.A.; Finegold, M.J.; Sokol, R.J. Toll-like receptor 4-dependent Kupffer cell activation and liver injury in a novel mouse model of parenteral nutrition and intestinal injury. Hepatology 2012, 55, 1518–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Chanda, D.; van Gorp, P.J.; Jeurissen, M.L.; Houben, T.; Walenbergh, S.M.; Debets, J.; Oligschlaeger, Y.; Gijbels, M.J.; Neumann, D.; et al. Macrophage Stimulating Protein Enhances Hepatic Inflammation in a NASH Model. PLoS ONE 2016, 11, e0163843. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wei, B.; de Assuncao, T.M.; Liu, Z.; Hu, X.; Ibrahim, S.; Cooper, S.A.; Cao, S.; Shah, V.H.; Kostallari, E. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J. Hepatol. 2020. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Fouda, S.; Li, D.; Zhang, K.; Ye, J.-M. Involvement of the Autophagy-ER Stress Axis in High Fat/Carbohydrate Diet-Induced Nonalcoholic Fatty Liver Disease. Nutrients 2020, 12, 2626. https://doi.org/10.3390/nu12092626
Zhou X, Fouda S, Li D, Zhang K, Ye J-M. Involvement of the Autophagy-ER Stress Axis in High Fat/Carbohydrate Diet-Induced Nonalcoholic Fatty Liver Disease. Nutrients. 2020; 12(9):2626. https://doi.org/10.3390/nu12092626
Chicago/Turabian StyleZhou, Xiu, Sherouk Fouda, Dongli Li, Kun Zhang, and Ji-Ming Ye. 2020. "Involvement of the Autophagy-ER Stress Axis in High Fat/Carbohydrate Diet-Induced Nonalcoholic Fatty Liver Disease" Nutrients 12, no. 9: 2626. https://doi.org/10.3390/nu12092626
APA StyleZhou, X., Fouda, S., Li, D., Zhang, K., & Ye, J. -M. (2020). Involvement of the Autophagy-ER Stress Axis in High Fat/Carbohydrate Diet-Induced Nonalcoholic Fatty Liver Disease. Nutrients, 12(9), 2626. https://doi.org/10.3390/nu12092626