Diet in the Treatment of Epilepsy: What We Know So Far


Abstract
:1. Introduction
2. Different Dietary Regimens Inducing Ketosis
2.1. Classic Ketogenic Diet (KD)
2.2. Modified Atkins Diet (MAD)
2.3. Low-Glycemic Index Treatment (LGIT)
2.4. Medium-Chain Triglyceride Diet (MCTD)
3. Responders and Non-Responders to the Ketogenic Diet
4. Pathophysiology of Ketogenic Diet and Epilepsy
5. Clinical Efficacy against Epilepsy
5.1. Pediatric Population
5.1.1. Refractory Epilepsy
5.1.2. Other Childhood Epilepsies
5.2. Adult Population
6. Tolerability
6.1. General Comment
6.2. Short Term Adverse Events
6.3. Long Term Adverse Events
6.4. Deaths, Retention Rates, and Reasons for Diet Discontinuation
7. Special Considerations
7.1. Exclusion Criteria of the Ketogenic Diet
7.2. Variation of Ketogenic Diet Regimen per Age Group
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Michael-Titus, A.T.; Revest, P.A.; Shortland, P.J. Epilepsy. In The Nervous System, 2nd ed.; Horne, T., Stader, L., Eds.; Elsevier Ltd., Churchill Livingstone: London, UK, 2010; pp. 237–250. [Google Scholar] [CrossRef]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Allen Hauser, W.; Mathem, G.; Moshé, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef]
- D’Andrea Meira, I.; Romão, T.T.; Pires do Prado, H.J.; Krüger, L.T.; Pires, M.E.P.; da Conceição, P.O. Ketogenic Diet and Epilepsy: What We Know So Far. Front. Neurosci. 2019, 13, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergqvist, A.G.C.; Schall, J.I.; Gallagher, P.R.; Cnaan, A.; Stallings, V.A. Fasting versus gradual initiation of the ketogenic diet: A prospective, randomized clinical trial of efficacy. Epilepsia 2005, 46, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Martin-McGill, K.J.; Jackson, C.F.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2018, 11, CD001903. [Google Scholar] [CrossRef] [PubMed]
- Schoeler, N.E.; Cross, J.H. Ketogenic dietary therapies in adults with epilepsy: A practical guide. Pract. Neurol. 2016, 16, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Payne, N.E.; Cross, J.H.; Sander, J.W.; Sisodiya, S.M. The ketogenic and related diets in adolescents and adults—A review. Epilepsia 2018, 52, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Barzegar, M.; Afghan, M.; Tarmahi, V.; Behtari, M.; Rahimi Khamaneh, S.; Raeisi, S. Ketogenic diet: Overview, types, and possible anti-seizure mechanisms. Nutr. Neurosci. 2019, 26, 1–10. [Google Scholar] [CrossRef]
- Miranda, M.J.; Turner, Z.; Magrath, G. Alternative diets to the classical ketogenic diet—Can we be more liberal? Epilepsy Res. 2012, 100, 278–285. [Google Scholar] [CrossRef]
- Wilder, R.M. The effect of ketonemia on the course of epilepsy. Mayo Clin. Bull. 1921, 2, 307–308. [Google Scholar]
- Rho, J.M. How does the ketogenic diet induce anti-seizure effects? Neurosci. Lett. 2017, 637, 4–10. [Google Scholar] [CrossRef]
- Rubenstein, J.E. Use of the ketogenic diet in neonates and infants. Epilepsia 2008, 49, 30–32. [Google Scholar] [CrossRef] [PubMed]
- Armeno, M.; Caraballo, R.; Vaccarezza, M.; Alberti, M.J.; Ríos, V.; Galicchio, S.; de Grandis, E.S.; Mestre, G.; Escobal, N.; Matarrese, P.; et al. National consensus on the ketogenic diet. Rev. Neurol. 2014, 59, 213–223. (In Spanish) [Google Scholar]
- Kossoff, E.H.; Krauss, G.L.; Mc Grogan, J.R.; Freeman, J.M. Efficacy of the Atkins diet as therapy for intractable epilepsy. Neurology 2003, 61, 1789–1791. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Dorward, J.L. The modified Atkins diet. Epilepsia 2008, 49, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H. More fat and fewer seizures: Dietary therapies for epilepsy. Lancet Neurol. 2004, 3, 415–420. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Zupec-Kania, B.A.; Amark, P.E.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Helen Cross, J.; Dahlin, M.G.; et al. Optimal clinical management of children receiving the ketogenic diet: Recommendations of the International Ketogenic Diet Study Group. Epilepsia 2009, 50, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Carrette, E.; Vonck, K.; de Herdt, V.; Dewaele, I.; Raedt, R.; Goossens, L.; Van Zandijcke, M.; Wadman, W.; Thadani, V.; Boon, P. A pilot trial with modified Atkins’ diet in adult patients with refractory epilepsy. Clin. Neurol. Neurosurg. 2008, 110, 797–803. [Google Scholar] [CrossRef]
- Pfeifer, H.H.; Thiele, E.A. Low-glycemic-index treatment: A liberalized ketogenic diet for treatment of intractable epilepsy. Neurology 2005, 65, 1810–1812. [Google Scholar] [CrossRef]
- Coppola, G.; D’Aniello, A.; Messana, T.; Di Pasquale, F.; della Corte, R.; Pascotto, A.; Verrotti, A. Low glycemic index diet in children and young adults with refractory epilepsy: First Italian experience. Seizure 2011, 20, 526–528. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, H.H.; Lyczkowski, D.A.; Thiele, E.A. Low glycemic index treatment: Implementation and new insights into efficacy. Epilepsia 2008, 49, 42–45. [Google Scholar] [CrossRef]
- Huttenlocher, P.R.; Wilbourn, A.J.; Signore, J.M. Mediumchain triglycerides as a therapy for intractable childhood epilepsy. Neurology 1971, 21, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The ketogenic diet for the treatment of childhood epilepsy: A randomized controlled trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef]
- Liu, Y.M. Medium-chain triglyceride (MCT) ketogenic therapy. Epilepsia 2008, 49, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.; Swaminathan, A.; Paseka, J.; Hanson, C. Efficacy and Safety of a Ketogenic Diet in Children and Adolescents with Refractory Epilepsy—A Review. Nutrients 2020, 12, 1809. [Google Scholar] [CrossRef]
- Schoeler, N.E.; Leu, C.; Balestrini, S.; Mudge, J.M.; Steward, C.A.; Frankish, A.; Leung, M.A.; Mackay, M.; Scheffer, I.; Williams, R.; et al. Genome-wide association study: Exploring the genetic basis for responsiveness to ketogenic dietary therapies for drug-resistant epilepsy. Epilepsia 2018, 59, 1557–1566. [Google Scholar] [CrossRef]
- Ko, A.; Jung, D.E.; Kim, S.H.; Kang, H.C.; Lee, J.S.; Lee, S.T.; Choi, J.R.; Kim, H.D. The Efficacy of Ketogenic Diet for Specific Genetic Mutation in Developmental and Epileptic Encephalopathy. Front. Neurol. 2018, 9, 530. [Google Scholar] [CrossRef] [Green Version]
- Bough, K.J.; Rho, J.M. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 2007, 48, 43–58. [Google Scholar] [CrossRef]
- Maalouf, M.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 293–315. [Google Scholar] [CrossRef] [Green Version]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The gut microbiota mediates the anti-seizure effects of the ketogenic diet. Cell 2018, 173, 1728–1741. [Google Scholar] [CrossRef] [Green Version]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Ketogenic Diet and Epilepsy. Nutrients 2019, 11, 2510. [Google Scholar] [CrossRef] [Green Version]
- Calderón, N.; Betancourt, L.; Hernández, L.; Rada, P. A ketogenic diet modifies glutamate, gammaaminobutyric acid and agmatine levels in the hippocampus of rats: A microdialysis study. Neurosci. Lett. 2017, 642, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Bergqvist, C.; Hunter, J.V.; Jin, D.; Wang, D.J.; Wehrli, S.; Zimmerman, R.A. In Vivo Measurement of Brain Metabolites Using Two-Dimensional Double-Quantum MR Spectroscopy—Exploration of GABA Levels in a Ketogenic Diet. Magn. Reson. Med. 2003, 49, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, M.; Elfving, A.; Ungerstedt, U.; Amark, P. The ketogenic diet influences the levels of excitatory and inhibitory amino acids in the CSF in children with refractory epilepsy. Epilepsy Res. 2005, 64, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.; Zhang, K.; Yang, W.; Li, B. The Anticonvulsant Effects of Ketogenic Diet on Epileptic Seizures and Potential Mechanisms. Curr. Neuropharmacol. 2018, 16, 66–70. [Google Scholar] [CrossRef]
- Yudkoff, M.; Daikhin, Y.; Horyn, O.; Nissim, I.; Nissim, I. Ketosis and brain handling of glutamate, glutamine, and GABA. Epilepsia 2008, 49, 73–75. [Google Scholar] [CrossRef] [Green Version]
- Masino, S.A.; Kawamura, M.; Wasser, C.D.; Pomeroy, L.T.; Ruskin, D.N. Adenosine, ketogenic diet and epilepsy: The emerging therapeutic relationship between metabolism and brain activity. Curr. Neuropharmacol. 2009, 7, 257–268. [Google Scholar] [CrossRef]
- Dahlin, M.; Månsson, J.E.; Åmark, P. CSF levels of dopamine and serotonin, but not norepinephrine, metabolitesare influenced by the ketogenic diet in children with epilepsy. Epilepsy Res. 2012, 99, 132–138. [Google Scholar] [CrossRef]
- Baraban, S. Neuropeptide Y and epilepsy: Recent progress, prospects and controversies. Neuropeptides 2004, 38, 261–265. [Google Scholar] [CrossRef]
- Weinshenker, D. The contribution of norepinephrine and orexigenic neuropeptides to the anticonvulsant effect of the ketogenic diet. Epilepsia 2008, 49, 104–107. [Google Scholar] [CrossRef]
- Mazarati, A.M. Galanin and galanin receptors in epilepsy. Neuropeptides 2004, 38, 331–343. [Google Scholar] [CrossRef]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Milder, J.B.; Liang, L.P.; Patel, M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol. Dis. 2010, 40, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, E.J.; Jeong, J.H.; Chung, Y.H.; Kim, W.K.; Ko, K.H.; Bach, J.H.; Hong, J.S.; Yoneda, Y.; Kim, H.C. Role of oxidative stress in epileptic seizures. Neurochem. Int. 2011, 59, 122–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, G.R.; Lutas, A.; Martínez-François, J.R.; Yellen, G. Single K ATP channel opening in response to action potential firing in mouse dentate granule neurons. J. Neurosci. 2011, 31, 8689–8696. [Google Scholar] [CrossRef] [Green Version]
- Yellen, G. Ketone bodies, glycolysis, and KATP channels in the mechanism of the ketogenic diet. Epilepsia 2008, 49, 80–82. [Google Scholar] [CrossRef] [Green Version]
- Vamecq, J.; Vallée, L.; Lesage, F.; Gressens, P.; Stables, J.P. Antiepileptic popular ketogenic diet: Emerging twists in an ancient story. Prog. Neurobiol. 2005, 75, 1–28. [Google Scholar] [CrossRef]
- Franks, N.P.; Honoré, E. The TREK K2P channels and their role in general anaesthesia and neuroprotection. Trends Pharmacol. Sci. 2004, 25, 601–608. [Google Scholar] [CrossRef]
- Simeone, T.A.; Matthews, S.A.; Samson, K.K.; Simeone, K.A. Regulation of brain PPARgamma2 contributes to ketogenic diet anti-seizure efficacy. Exp. Neurol. 2017, 287, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.R.; Lee, E.J.; Kim, H.D.; Lee, J.H.; Kang, H.C. Polyunsaturated fatty acid-enriched diet therapy for a child with epilepsy. Brain Dev. 2014, 36, 163–166. [Google Scholar] [CrossRef]
- Fraser, D.D.; Whiting, S.; Andrew, R.D.; Macdonald, E.A.; Musa-Veloso, K.; Cunnane, S.C. Elevated polyunsaturated fatty acids in blood serum obtained from children on the ketogenic diet. Neurology 2003, 60, 1026–1029. [Google Scholar] [CrossRef]
- Taha, A.; Ryan, M.A.A.; Cunnane, S.C. Despite transient ketosis, the classic high-fat ketogenic diet induces marked changes in fatty acid metabolism in rats. Metab. Clin. Exp. 2005, 54, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Dingledine, R.; Varvel, N.H.; Dudek, F.E. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv. Exp. Med. Biol. 2014, 813, 109–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.G.; Wang, H.D.; Jin, W.; Yin, H.X. Ketogenic diet reduces cytochrome c release and cellular apoptosis following traumatic brain injury in juvenile rats. Ann. Clin. Lab. Sci. 2009, 39, 76–83. [Google Scholar]
- Noh, H.S.; Kang, S.S.; Kim, D.W.; Kim, Y.H.; Park, C.H.; Han, J.Y.; Cho, G.J.; Choi, W.S. Ketogenic diet increases calbindin-D28k in the hippocampi of male ICR mice with kainic acid seizures. Epilepsy Res. 2005, 65, 153–159. [Google Scholar] [CrossRef]
- Noh, H.S.; Kim, D.W.; Kang, S.S.; Cho, G.J.; Choi, W.S. Ketogenic diet prevents clusterin accumulation induced by kainic acid in the hippocampus of male ICR mice. Brain Res. 2005, 1042, 114–118. [Google Scholar] [CrossRef]
- Dupuis, N.; Curatolo, N.; Benoist, J.F.; Auvin, S. Ketogenic diet exhibits anti-inflammatory properties. Epilepsia 2015, 56, 95–98. [Google Scholar] [CrossRef]
- Vining, E.P.; Freeman, J.M.; Ballaban-Gil, K.; Camfield, C.S.; Camfield, P.R.; Holmes, G.L.; Shinnar, S.; Shuman, R.; Trevathan, E.; Wheless, J.W. A multicenter study of the efficacy of the ketogenic diet. Arch. Neurol. 1998, 55, 1433–1437. [Google Scholar] [CrossRef] [Green Version]
- Freeman, J.M.; Vining, E.P.; Pillas, D.J.; Pyzik, P.L.; Casey, J.C.; Kelly, L.M. The efficacy of the ketogenic diet 1998: A prospective evaluation of intervention in 150 children. Pediatrics 1998, 102, 1358–1363. [Google Scholar] [CrossRef]
- Freitas, A.; Paz, J.A.; Casella, E.B.; Marques-Dias, M.J. Ketogenic diet for the treatment of refractory epilepsy: A 10 year experience in children. Arq. Neuro Psiquiatr. 2007, 65, 381–384. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.J.; Zhang, L.M.; Chai, Y.M.; Wang, J.; Yu, L.F.; Li, W.H.; Zhou, Y.F.; Zhou, S.Z. Six-month efficacy of the Ketogenic diet is predicted after 3 months and is unrelated to clinical variables. Epilepsy Behav. 2016, 55, 165–169. [Google Scholar] [CrossRef] [PubMed]
- El-Rashidy, O.F.; Nassar, M.F.; Abdel-Hamid, I.A.; Shatla, R.H.; Abdel-Hamid, M.H.; Gabr, S.S.; Mohamed, S.G.; El-Sayed, W.S.; Shaaban, S.Y. Modified Atkins diet vs. classic ketogenic formula in intractable epilepsy. Acta Neurol. Scand. 2013, 128, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Sankhyan, N.; Gulati, S.; Agarwala, A. Use of the modified Atkins diet for treatment of refractory childhood epilepsy: A randomized controlled trial. Epilepsia 2013, 54, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Goel, S.; Jain, P.; Agarwala, A.; Aneja, S. Evaluation of a simplified modified Atkins diet for use by parents with low levels of literacy in children with refractory epilepsy: A randomized controlled trial. Epilepsy Res. 2016, 127, 152–159. [Google Scholar] [CrossRef]
- Lambrechts, D.A.J.E.; de Kinderen, R.J.A.; Vles, J.S.H.; de Louw, A.J.; Aldenkamp, A.P.; Majoie, H.J. A randomized controlled trial of the ketogenic diet in refractory childhood epilepsy. Acta Neurol. Scand. 2017, 135, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Wijnen, B.F.M.; de Kinderen, R.J.A.; Lambrechts, D.A.J.E.; Postulart, D.; Aldenkamp, A.P.; Majoie, M.H.J.M.; Evers, S.M.A.A. Long-term clinical outcomes and economic evaluation of the ketogenic diet versus care as usual in children and adolescents with intractable epilepsy. Epilepsy Res. 2017, 132, 91–99. [Google Scholar] [CrossRef]
- Sourbron, J.; Klinkenberg, S.; van Kuijk, S.M.J.; Lagae, L.; Lambrechts, D.; Braakman, H.M.H.; Majoie, M. Ketogenic diet for the treatment of pediatric epilepsy: Review and meta-analysis. Child Nerv. Syst. 2020, 36, 1099–1109. [Google Scholar] [CrossRef]
- Than, K.D.; Kossoff, E.H.; Rubenstein, J.E.; Pyzik, P.L.; McGrogan, J.R.; Vining, E.P. Can you predict an immediate, complete, and sustained response to the ketogenic diet? Epilepsia 2005, 46, 580–582. [Google Scholar] [CrossRef]
- Eun, S.H.; Kang, H.C.; Kim, D.W.; Kim, H.D. Ketogenic diet for treatment of infantile spasms. Brain Dev. 2006, 28, 566–571. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Hedderick, E.F.; Turner, Z.; Freeman, J.M. A case-control evaluation of the ketogenic diet versus ACTH for new-onset infantile spasms. Epilepsia 2008, 49, 1504–1509. [Google Scholar] [CrossRef]
- Hong, A.M.; Turner, Z.; Hamdy, R.F.; Kossoff, E.H. Infantile spasms treated with the ketogenic diet: Prospective single-center experience in 104 consecutive infants. Epilepsia 2010, 51, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, R.; Vaccarezza, M.; Cersósimo, R.; Rios, V.; Soraru, A.; Arroyo, H.; Agosta, G.; Escobal, N.; Demartini, M.; Maxit, C.; et al. Long-term follow-up of the ketogenic diet for refractory epilepsy: Multicenter Argentinean experience in 216 pediatric patients. Seizure 2011, 20, 640–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numis, A.L.; Yellen, M.B.; Chu-Shore, C.J.; Pfeifer, H.H.; Thiele, E.A. The relationship of ketosis and growth to the efficacy of the ketogenic diet in infantile spasms. Epilepsy Res. 2011, 96, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, J.H.; Yu, H.J.; Lee, M. Prognostic factors of infantile spasms: Role of treatment options including a ketogenic diet. Brain Dev. 2013, 35, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tong, L.; Jia, G.; Sun, R. Effects of ketogenic diet on the clinical and electroencephalographic features of children with drug therapy-resistant epilepsy. Exp. Ther. Med. 2013, 5, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Pires, M.E.; Ilea, A.; Bourel, E.; Bellavoine, V.; Merdariu, D.; Berquin, P.; Auvin, S. Ketogenic diet for infantile spasms refractory to first-line treatments: An open prospective study. Epilepsy Res. 2013, 105, 189–194. [Google Scholar] [CrossRef]
- Kayyali, H.R.; Gustafson, M.; Myers, T.; Thompson, L.; Williams, M.; Abdelmoity, A. Ketogenic diet efficacy in the treatment of intractable epileptic spasms. Pediatr. Neurol. 2014, 50, 224–227. [Google Scholar] [CrossRef]
- Hirano, Y.; Oguni, H.; Shiota, M.; Nishikawa, A.; Osawa, M. Ketogenic diet therapy can improve ACTH-resistant West syndrome in Japan. Brain Dev. 2015, 37, 18–22. [Google Scholar] [CrossRef]
- Ville, D.; Chiron, C.; Laschet, J.; Dulac, O. The ketogenic diet can be used successfully in combination with corticosteroids for epileptic encephalopathies. Epilepsy Behav. 2015, 48, 61–65. [Google Scholar] [CrossRef]
- Hussain, S.A.; Shin, J.H.; Shih, E.J.; Murata, K.K.; Sewak, S.; Kezele, M.E.; Sankar, R.; Matsumoto, J.H. Limited efficacy of the ketogenic diet in the treatment of highly refractory epileptic spasms. Seizure 2016, 35, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, S.; Abdurahman, A.A.; Saghazadeh, A.; Badv, R.S.; Mahmoudi, M. Short-term and long-term efficacy of classical ketogenic diet and modified Atkins diet in children and adolescents with epilepsy: A systematic review and meta-analysis. Nutr. Neurosci. 2019, 22, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Korff, C.; Laux, L.; Kelley, K.; Goldstein, J.; Koh, S.; Nordli, D., Jr. Dravet syndrome (severe myoclonic epilepsy in infancy): A retrospective study of 16 patients. J. Child Neurol. 2007, 22, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, R.H. Nonpharmacologic treatments of Dravet syn-drome: Focus on the ketogenic diet. Epilepsia 2011, 52, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Laux, L.; Blackford, R. The ketogenic diet in Dravet syn-drome. J. Child Neurol. 2013, 28, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Chen, J.; Zhang, J.; Yang, X.; Ji, T.; Zhang, Y.; Wu, Y.; Fang, F.; Wu, X.; Zhang, Y. The Efficacy of Ketogenic Diet in 60 Chinese Patients With Dravet Syndrome. Front. Neurol. 2019, 10, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabbout, R.; Copioli, C.; Chipaux, M.; Chemaly, N.; Desguerre, I.; Dulac, O.; Chiron, C. Ketogenic diet also benefits Dravet syndrome patients receiving stiripentol: A prospectivepilot study. Epilepsia 2011, 52, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Dressler, A.; Trimmel-Schwahofer, P.; Reithofer, E.; Mühlebner, A.; Gröppel, G.; Reiter-Fink, E.; Benninger, F.; Grassl, R.; Feucht, M. Efficacy and tolerability of the ketogenic diet in Dravet syndrome—Comparison with various standard antiepileptic drug regimen. Epilepsy Res. 2015, 109, 81–89. [Google Scholar] [CrossRef]
- Dutton, S.B.; Sawyer, N.T.; Kalume, F.; Jumbo-Lucioni, P.; Borges, K.; Catterall, W.A.; Escayg, A. Protective effect of the ketogenic diet in Scn1a mutant mice. Epilepsia 2011, 52, 2050–2056. [Google Scholar] [CrossRef] [Green Version]
- Yan, N.; Xin-Hua, W.; Lin-Mei, Z.; Yi-Ming, C.; Wen-Hui, L.; Yuan-Feng, Z.; Shui-Zhen, Z. Prospective study of the efficacy of a ketogenic diet in 20 patients with Dravet syndrome. Seizure 2018, 60, 144–148. [Google Scholar] [CrossRef] [Green Version]
- Teran, F.A.; Kim, Y.; Crotts, M.S.; Bravo, E.; Emaus, K.J.; Richerson, G.B. Time of Day and a Ketogenic Diet Influence Susceptibility to SUDEP in Scn1a R1407X/+Mice. Front. Neurol. 2019, 10, 278. [Google Scholar] [CrossRef] [Green Version]
- Oguni, H.; Tanaka, T.; Hayashi, K.; Funatsuka, M.; Sakauchi, M.; Shirakawa, S.; Osawa, M. Treatment and long-term prognosis of myoclonic–astatic epilepsy of early childhood. Neuropediatrics 2002, 33, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, R.H.; Cerosimo, R.O.; Sakr, D.; Cresta, A.; Escobal, N.; Fejerman, N. Ketogenic diet in patients with myoclonic–astatic epilepsy. Epileptic Disord. 2006, 8, 151–155. [Google Scholar] [PubMed]
- Kilaru, S.; Bergqvist, A.G. Current treatment of myoclonic astatic epilepsy: Clinical experience at the Children’s Hospital of Philadelphia. Epilepsia 2007, 48, 1703–1707. [Google Scholar] [CrossRef] [PubMed]
- Prezioso, G.; Carlone, G.; Zaccara, G.; Verrotti, A. Efficacy of ketogenic diet for infantile spasms: A systematic review. Acta Neurol. Scand. 2017, 137, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Dressler, A.; Benninger, F.; Trimmel-Schwahofer, P.; Gröppel, G.; Porsche, B.; Abraham, K.; Mühlebner, A.; Samueli, S.; Male, C.; Feucht, M. Efficacy and tolerability of the ketogenic diet versus high-dose adrenocorticotropic hormone for infantile spasms: A single-center parallel-cohort randomized controlled trial. Epilepsia 2019, 60, 441–451. [Google Scholar] [CrossRef]
- Nabbout, R.; Mazzuca, M.; Hubert, P.; Peudennier, S.; Allaire, C.; Flurin, V.; Aberastury, M.; Silva, W.; Dulac, O. Efficacy of ketogenic diet in severe refractory status epilepticus initiating Fever Induced Refractory Epileptic Encephalopathy (FIRES) in school age children. Epilepsia 2010, 51, 2033–2037. [Google Scholar] [CrossRef]
- Appavu, B.; Vanatta, L.; Condie, J.; Kerrigan, J.F.; Jarrar, R. Ketogenic diet treatment for pediatric super-refractory status epilepticus. Seizure 2016, 41, 62–65. [Google Scholar] [CrossRef] [Green Version]
- Fox, K.; Wells, M.E.; Tennison, M.; Vaughn, B. Febrile Infection Related Epilepsy Syndrome (FIRES): A literature review and case study. Neurodiagn. J. 2017, 57, 224–233. [Google Scholar] [CrossRef]
- Van Baalen, A.; Vezzani, A.; Häusler, M.; Kluger, G. Febrile infection–related epilepsy syndrome: Clinical review and hypotheses of epileptogenesis. Neuropediatrics 2017, 48, 5–18. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Joshi, S.M.; Potter, D.M.; Leber, S.M.; Carlson, M.D.; Shellhaas, R.A. Cognitive outcomes in febrile infection-related epilepsy syndrome treated with the ketogenic diet. Pediatrics 2014, 134, 1431–1435. [Google Scholar] [CrossRef] [Green Version]
- Peng, P.; Peng, J.; Yin, F.; Deng, X.; Chen, C.; He, F.; Wang, X.; Guang, S.; Mao, L. Ketogenic Diet as a Treatment for Super-Refractory Status Epilepticus in Febrile Infection-Related Epilepsy Syndrome. Front. Neurol. 2019, 10, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caraballo, R.; Darra, F.; Reyes, G.; Armeno, M.; Cresta, A.; Mestre, G.; Bernardina, B.D. The ketogenic diet in patients with myoclonic status in non-progressive encephalopathy. Seizure 2017, 51, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Yang, Y.; Wang, Y.; Tang, H.; Zhang, F.; Zhang, Y.; Zhao, Y. Ketogenic diet for treatment of intractable epilepsy in adults: A meta-analysis of observational studies. Epilepsia Open 2018, 3, 9–17. [Google Scholar] [CrossRef]
- Poorshiri, B.; Barzegar, M.; Tahmasebi, S.; Shiva, S.; Raeisi, S.; Ebadi, Z. The efficacy comparison of classic ketogenic diet and modified Atkins diet in children with refractory epilepsy: A clinical trial. Acta Neurol. Belg. 2019, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.Y.; Zhou, Z.J.; Luo, R.; Gan, J.; Li, S.P.; Mu, D.Z.; Wan, C.M. Safety and tolerability of the ketogenic diet used for the treatment of refractory childhood epilepsy: A systematic review of published prospective studies. World J. Pediatr. 2017, 13, 528–536. [Google Scholar] [CrossRef]
- Furth, S.L.; Casey, J.C.; Pyzik, P.L.; Neu, A.M.; Docimo, S.G.; Vining, E.P.; Freeman, J.M.; Fivush, B.A. Risk factors for urolithiasis in children on the ketogenic diet. Pediatr. Nephrol. 2000, 15, 125–128. [Google Scholar] [CrossRef]
- Bergqvist, A.G.; Schall, J.I.; Stallings, V.A.; Zemel, B.S. Progressive bone mineral content loss in children with intractable epilepsy treated with the ketogenic diet. Am. J. Clin. Nutr. 2008, 88, 1678–1684. [Google Scholar] [CrossRef] [Green Version]
- Kwiterovich, P.O., Jr.; Vining, E.P.; Pyzik, P.; Skolasky, R., Jr.; Freeman, J.M. Effect of a high-fat ketogenic diet on plasma levels of lipids, lipoproteins, and apolipoproteins in children. JAMA 2003, 290, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Nizamuddin, J.; Turner, Z.; Rubenstein, J.E.; Pyzik, P.L.; Kossoff, E.H. Management and risk factors for dyslipidemia with the ketogenic diet. J. Child Neurol. 2008, 23, 758–761. [Google Scholar] [CrossRef]
- Christodoulides, S.S.; Neal, E.G.; Fitzsimmons, G.; Chaffe, H.M.; Jeanes, Y.M.; Aitkenhead, H.; Cross, J.H. The effect of the classical and medium chain triglyceride ketogenic diet on vitamin and mineral levels. J. Hum. Nutr. Diet. 2012, 25, 16–26. [Google Scholar] [CrossRef]
- Liu, Y.M.; Williams, S.; Basualdo-Hammond, C.; Stephens, D.; Curtis, R. A prospective study: Growth and nutritional status of children treated with the ketogenic diet. J. Am. Diet. Assoc. 2003, 103, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Kapetanakis, M.; Liuba, P.; Odermarsky, M.; Lundgren, J.; Hallböök, T. Effects of ketogenic diet on vascular function. Eur. J. Paediatr. Neurol. 2014, 18, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Doksöz, Ö.; Güzel, O.; Yılmaz, Ü.; İşgüder, R.; Çeleğen, K.; Meşe, T.; Uysal, U. The short-term effect of ketogenic diet on carotid intimamedia thickness and elastic properties of the carotid artery and the aorta in epileptic children. J. Child Neurol. 2015, 30, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Doksöz, Ö.; Çeleğen, K.; Güzel, O.; Yılmaz, Ü.; Uysal, U.; İşgüder, R.; Çeleğen, M.; Meşe, T. The short-term effects of ketogenic diet on cardiac ventricular functions in epileptic children. Pediatr. Neurol. 2015, 53, 233–237. [Google Scholar] [CrossRef]
- Masood, W.; Annamaraju, P.; Uppaluri, K.R. Ketogenic Diet. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Armeno, M.; Araujo, C.; Sotomontesano, B.; Caraballo, R.H. Update on the adverse effects during therapy with a ketogenic diet in paediatric refractory epilepsy. Rev. Neurol. 2018, 66, 193–200. (In Spanish) [Google Scholar]
- Riantarini, I.; Kim, H.D.; Ko, A.; Kim, S.H.; Kang, H.C.; Lee, J.S.; Jung, D.E. Short- and long-term seizure-free outcomes of dietary treatment in infants according to etiology. Seizure 2019, 71, 100–104. [Google Scholar] [CrossRef]
- Elia, M.; Klepper, J.; Leiendecker, B.; Hartmann, H. Ketogenic Diets in the Treatment of Epilepsy. Curr. Pharm. Des. 2017, 23, 5691–5701. [Google Scholar] [CrossRef]
- Verrotti, A.; Tambucci, R.; Di Francesco, L.; Pavone, P.; Iapadre, G.; Altobelli, E.; Matricardi, S.; Farello, G.; Belcastro, V. The role of polytherapy in the management of epilepsy: Suggestions for rational antiepileptic drug selection. Expert Rev. Neurother. 2020, 20, 167–173. [Google Scholar] [CrossRef]
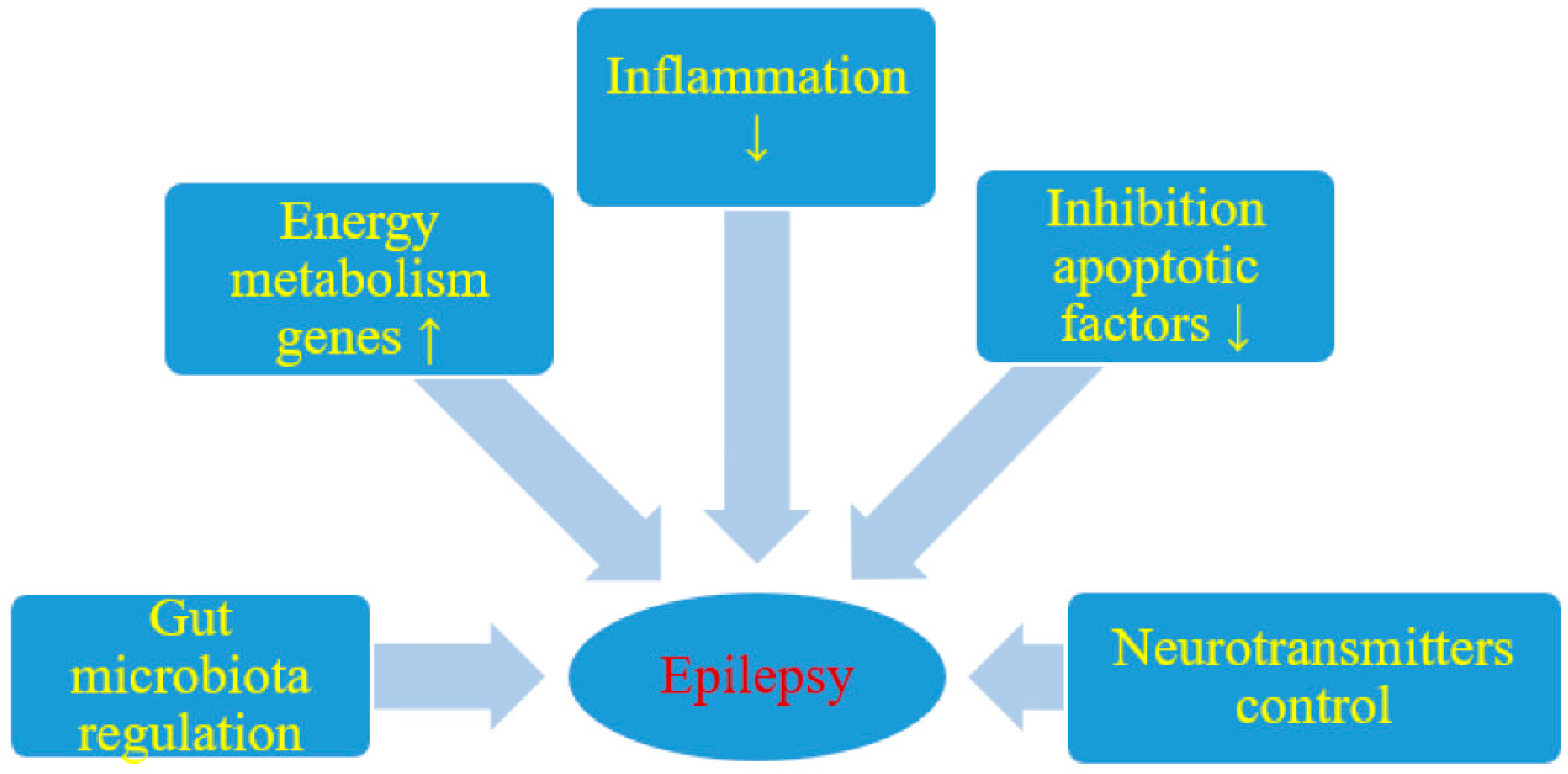

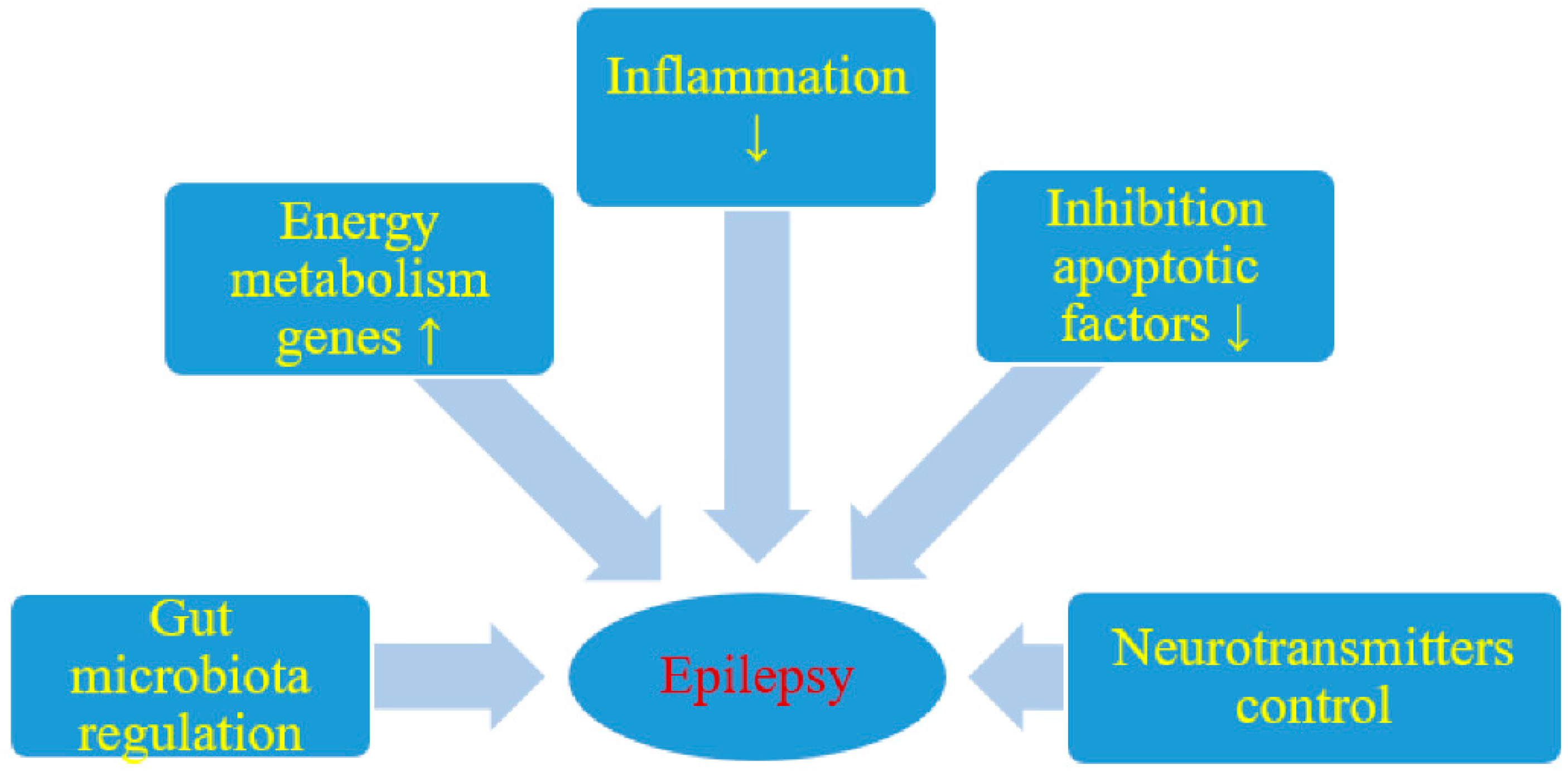{kind=link}
| Ketogenic Diets | Macronutrients (% Total Daily Calories) | Ketogenic Ratio | ||
|---|---|---|---|---|
| Fat | Protein | Carbohydrate | ||
| Classic | 80–90 | 6–8 | 2–4 | 4:1 |
| MAD | 65 | 25 | 10 | 1:1 |
| LGIT | 60 | 20–30 | 10 | 1:0.6 |
| MCTD | 30–60 | 10 | 15–19 | 1:1 or 2:1 |
| Primary Physiological Alteration | Possible Anti-Seizure Mechanisms |
|---|---|
|
|
|
|
|
|
|
|
|
|
| References | Type of Study | Participants | KD Ratio | Fasting | Outcomes | Follow-Up | Statistical Significance | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [] | (Months) | ||||||||||||
| Number | Age (Years) | KD Group | Control Group | ||||||||||
| Number | SFR >50%(n) | SFR >50% | Number | SFR >50%(n) | SFR >50% | ||||||||
| Vining EP, et al. 1998 [59] | Observational | 51 | 8–1 years | Classic KD/4:1 | Yes | 45 | 54 | 28 | / | / | / | 3 | p < 0.001 |
| 35 | 53 | 27 | / | / | / | 6 | NA | ||||||
| 24 | 40 | 20 | / | / | / | 12 | NA | ||||||
| Freeman JM, et al. 1998 [60] | Observational | 150 | 0.3–16 years | Classic KD/3–4:1 | Yes | 125 | 89 | 60 | / | / | / | 3 | NA |
| 106 | 77 | 51 | / | / | / | 6 | NA | ||||||
| 83 | 75 | 50 | / | / | / | 12 | NA | ||||||
| Freitas A, et al. 2007 [61] | Observational | 54 | 1–12 years | Classic KD/4:1 | Yes | 54 | 47 | 88.9 | / | / | / | 2 | NA |
| 49 | 44 | 89.4 | / | / | / | 6 | Efficacy for generalized epilepsy p < 0.001 | ||||||
| 39 | 38 | 97.5 | / | / | / | 12 | NA | ||||||
| 29 | 29 | 100 | / | / | / | 24 | NA | ||||||
| Wu YJ, et al. 2016 [62] | Observational | 87 | 0.5–16 years | Classic KD/4:1 | Yes | 87 | 47 | 41 | / | / | / | 1 | NA |
| 87 | 51 | 44 | / | / | / | 3 | NA | ||||||
| 87 | 52 | 45 | / | / | / | 6 | Positive correlation between increased cognition and KD efficacy after 3 months (p = 0.003) | ||||||
| Neal EG, et al. 2008 [23] | RCT | 145 | 2–16 years | Classic KD or MCT/2–5:1 or 40−60% | No | 54 | 20 | 38 | 49 | 3 | 6 | 12 | p < 0.0001 |
| El-Rashidy OF, et al. 2013 [63] | RCT | 10 | 26 ± 0.9 mon | Classic KD/4:1 | NA | 25 | 12 | 49.41 | 15 | 1 | 8.31 | 6 | p < 0.005 |
| Sharma S, et al. 2013 [64] | RCT | 102 | 2–14 years | MAD (carbohydrate 10 g/die) | NA | 50 | 26 | 52 | 52 | 6 | 11.5 | 3 | p < 0.001 |
| Sharma S, et al. 2016 [65] | RCT | 81 | 2–14 years | MAD | NA | 41 | 23 | 56.1 | 40 | 3 | 7.5 | 3 | p < 0.0001 |
| Lambrechts DAJE, et al. 2017 [66] | RCT | 48 | 1–17 years | prevalent MCT | No | 26 | 13 | 50 | 22 | 4 | 18.2 | 4 | p = 0.024 |
| Wijnen BFM, et al. 2017 [67] | RCT | 48 | 1–18 years | Classic KD or MCT | NA | 26 | 9 | 35 | 22 | 4 | 18 | 16 | p = 0.171 |
| Sourbron J, et al. 2020 [68] | Review-meta-analysis 5 RCTs | 472 | 35–56.1 | 6–18.2 | p < 0.001 | ||||||||
| References | Type of Study | Participants | KD Ratio | Fasting | Outcomes | Follow-Up | Statistical Significance | |||
|---|---|---|---|---|---|---|---|---|---|---|
| [] | (Months) | |||||||||
| Number | Age | Pt on KD | SFR >50% | SFR >50% | ||||||
| (Months) | (n) | (%) | ||||||||
| Than KD, et al. 2005 [69] | Retrospective observational | 25 | NA | 3–4:1 | Yes | 25 | NA | NA | NA | NA |
| Eun SH, et al. 2006 [70] | Retrospective observational | 43 | 1–14 | 3–4:1 | Yes | 43 | 12 | 27,9 | NA | NA |
| 35 | 30 | 85,7 | 3 | NA | ||||||
| 25 | 23 | 92 | 6 | NA | ||||||
| Kossoff EH, et al. 2008 [71] | Retrospective observational | 13 | Median (range): 5 (2–10) | 3–4:1 | Yes | 13 | 8 | 62 | 1 | p = 0.06 |
| Hong AM, et al. 2010 [72] | Prospective observational | 104 | Median: 4.8 | 3–4:1 | Yes | 86 | 66 | 63 | 3 | NA |
| 76 | 67 | 64 | 6 | p = 0.84 | ||||||
| No | 53 | 76 | 73 | 9 | NA | |||||
| 47 | 80 | 77 | 12 | NA | ||||||
| 28 | 80 | 77 | 24 | NA | ||||||
| Caraballo R, et al. 2011 [73] | Retrospective observational | 12 | NA | 3–4:1 | NA | 12 | 9 | 75 | NA | NA |
| Numis AL, et al. 2011 [74] | Retrospective observational | 26 | Mean ± SD: 19.5 ± 2.2 | 3–4:1 | No | 26 | 12 | 46.2 | 3 | p = 0.02 |
| 21 | 11 | 52.4 | 6 | p = 0.02 | ||||||
| 19 | 12 | 63.2 | 12 | p = 0.02 | ||||||
| Lee J, et al. 2013 [75] | Retrospective observational | 14 | NA | NA | NA | 14 | 11 | 78,6 | NA | NA |
| Li B, et al. 2013 [76] | Prospective observational | 31 | 7–84 | 4:01 | Yes | 31 | 21 | 67,74 | 1 | NA |
| 22 | 70,97 | 3 | NA | |||||||
| Pires ME, et al. 2013 [77] | Prospective observational | 17 | Mean ± SD: 9.4 ± 1.1 | 3–4:1 | No | 17 | 15 | 88.2 | 3 | NA |
| 16 | 14 | 87.5 | 6 | NA | ||||||
| Kayyali HR, et al. 2014 [78] | Prospective observational | 20 | 4–35 | 3–3.5:1 | No | 20 | 14 | 70 | 3 | NA |
| 20 | 13 | 72.2 | 6 | NA | ||||||
| 17 | 13 | 76.5 | 12 | NA | ||||||
| 8 | 8 | 100 | 24 | NA | ||||||
| Hirano Y, et al. 2015 [79] | Retrospective observational | 6 | 9–40 | 3–4:1 | NA | 6 | 5 | 83,3 | 3 | NA |
| Ville D, et al. 2015 [80] | Retrospective observational | 23 | NA | 4:01 | Yes | 23 | 4 | 17,4 | NA | NA |
| Hussain SA, et al. 2016 [81] | Retrospective observational | 22 | 12.5–38.7 | 3–4:1 | No | 22 | 7 | 35 | 3 | NS |
| 20 | 6 | 35.3 | 6 | NS | ||||||
| 17 | 2 | 20 | 12 | NS | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verrotti, A.; Iapadre, G.; Di Francesco, L.; Zagaroli, L.; Farello, G. Diet in the Treatment of Epilepsy: What We Know So Far. Nutrients 2020, 12, 2645. https://doi.org/10.3390/nu12092645
Verrotti A, Iapadre G, Di Francesco L, Zagaroli L, Farello G. Diet in the Treatment of Epilepsy: What We Know So Far. Nutrients. 2020; 12(9):2645. https://doi.org/10.3390/nu12092645
Chicago/Turabian StyleVerrotti, Alberto, Giulia Iapadre, Ludovica Di Francesco, Luca Zagaroli, and Giovanni Farello. 2020. "Diet in the Treatment of Epilepsy: What We Know So Far" Nutrients 12, no. 9: 2645. https://doi.org/10.3390/nu12092645
APA StyleVerrotti, A., Iapadre, G., Di Francesco, L., Zagaroli, L., & Farello, G. (2020). Diet in the Treatment of Epilepsy: What We Know So Far. Nutrients, 12(9), 2645. https://doi.org/10.3390/nu12092645